國 立 交 通 大 學
應 用 化 學 研 究 所
碩 士 論 文
末端修飾聚三-己烷噻吩高分子之合成、自組裝調
控與其在太陽電池之應用
Synthesis and Controlled Self-Assembly of
End-Capped Modified Poly(3-hexylthiophene) and
Their Application in Organic Solar Cell
研 究 生:吳昀錚
指導教授:許千樹 博士
末端修飾聚三-己烷噻吩高分子之合成、自組裝調
控與其在太陽電池之應用
Synthesis and Controlled Self-Assembly of
End-Capped Modified Poly(3-hexylthiophene) and
Their Application in Organic Solar Cell
研 究 生:吳 昀 錚 Student: Yun-Chen Wu 指導教授:許 千 樹 博士 Advisor: Dr. Chain-Shu Hsu
國立交通大學 應用化學研究所
碩士論文
A Thesis
Submitted to Department of Applied Chemistry College of Science
National Chiao-Tung University in Partial Fulfillment of the Requirements
for the Degree of Master in
Applied Chemistry June 2008
Hsinchu, Taiwan, Republic of China
ii
末端修飾聚三-己烷噻吩高分子之合成、自組裝調控與其在
太陽電池之應用
學生:吳昀錚 指導教授:許千樹 博士 國立交通大學 應用化學系研究所 碩士班 摘要 有機太陽能電池(Organic Photovoltaics,OPV)為以有機化合物作為 太陽能電池的材料,符合低價格且製程簡易的優點。本論之研究目的在於 合成新穎聚噻吩高分子,利用其高分子之特性製作成元件。 第一部份為聚噻吩鏈段共聚物之合成與光電性質研究,本研究之高分 子的構思源自於 Ho, R. M. et al.將polystyrene-block-poly(l-lactide) (PS-PLLA) 的鏈段共聚物製作成具有自組裝現象的高分子薄膜。並經由精確的分子量 控制和去除乳酸片段,製作成具有孔洞的高分子材料。在鏈段共聚物的合 成部份,以poly(3-hexyl- thiophene) (P3HT)取代PS,並藉由末端修飾的格林 納 置 換 聚 合 反 應 和 配 位 插 入 開 環 聚 合 反 應 的 合 成 方 法 合 成 出 poly(3-hexylthiophene)- block-poly(l-lactide) (P3HT-PLLA)的鏈段聚合物,並 利用基質輔助激光解析飛行時間質譜(MALDI-TOF MS)做末端官能基的 分析並對照1 H NMR的圖譜來鑑定高分子。此外,針對P3HT及PLLA片段進 行不同分子量的合成。在太陽能電池元件製作部分,將此具有相分離特性 的薄膜應用至元件活化層,接著將PLLA片段以溫和的方式裂解後,可得到 具奈米孔洞結構的P3HT活化層。本研究期望能夠結合具吸光性質的太陽能 電池材料及特殊的奈米孔洞形態兩項特性,進而增加與電子接受體的接觸 面積,有效達到電荷分離效果。ii
第二部份為結合多面體矽氧烷寡聚物(POSS),將末端修飾雙鍵的聚噻 吩高分子與POSS連接製作成星狀結構之材料。由於矽氧烷具有相當好的熱 穩定性,本研究期望藉由此特性改善元件的熱穩定性。此外,由於分子量 的增加,可以有效改善高分子的成膜性,使得元件的效率得以提升。
Synthesis and Controlled Self-Assembly of End-Capped
Modified Poly(3-hexylthiophene) and Their Application in
Organic Solar Cell
Student: Yun-Chen Wu Advisers: Dr. Chain-Shu Hsu
Department of Applied Chemistry National Chiao Tung University
Abstract
Organic Photovoltaics (OPVs) have several advantages such as flexible, easy fabrication and low cost. The goal of this research is to study the synthesis and application of new poly(3-hexylthiophene) derivatives. The first part of this study is to incorporate poly(3-hexylthiophene) (P3HT) into copolymer structure with poly(l-lactide) (PLLA) blocks via a feasible Grignard metathesis method to control the end-capping group, and ring-opening polymerization method to combine the l-lactide monomer. Different volume fractions of P3HT-PLLA were prepared with different molecular weight of PLLA. The chemical structure of the copolymers were investigated by 1HNMR and MALDI-TOF MS. Well-defined and phase-separated microdomains of P3HT-PLLA thin film can be observed by appropriate solvents and post-thermal treatment. The block copolymer films reveal pit-like surface morphology. After degradation of PLLA block, the films with large amounts of porous texture were formed and used as templates for the fabrication of organic solar cells. The OPV devices with P3HT nanoporous film show higher power conversion efficiency than that with pristine P3HT thin film. The second part of this study, we combined vinyl terminated poly(3-hexylthiophene) with 1,3,5,7,9,11,13,15-Octakis(dimethylsilyloxy)- pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxane to synthesized new star-like P3HT
iv
polymer. This star-like polymer shows better film forming property and better device stability than the original vinyl-terminated P3HT.
謝誌 這是我打第二次的謝誌,事實上,我原來寫好的第一篇被我自己不小 心剪下然後忘記貼上,所以我要打第二次。一直以來,「不要去想,就不 會後悔」是我把持的信念,用我所有的勇氣與動力,渴望為自己爭取屬於 我自己的人生,也感謝許多一路上陪伴在我左右的人。 我很感謝我的指導老師 許千樹教授,謝謝他給我機會成為研究生,也 謝謝他提供一個這麼好的實驗環境,讓我不管是在合成或是製程都能夠隨 心所欲去嘗試,也謝謝他不厭其煩的指導我,讓我對於作研究有很深刻的 認識。此外,也感謝 何榮銘教授對於我研究內容的指教。 謝謝實驗室的大家們,謝謝黃軍浩博士對我的許多叮嚀,感謝他對於 我在合成及製程部分的教導,此外,謝謝他陪我玩超多類型的遊戲解悶。 謝謝陳奎百博士常常跟我討論許多實驗上的細節及給我很多實驗方向的意 見,他更是讓我了解到了˝登扔˝的厲害。謝謝霹靂毛謝朝翔學長對博愛作出 的重大貢獻,使得我論文能有快速的進展,謝謝他在我的實驗進入製程階 段與我彼此砥礪、互相切磋,讓我的元件製作有著相當全面的進步。謝謝 楊勝雄博士幫我把論文做最後的考核,更謝謝學長把實驗室管理的有條有 序,讓大家在安全及整齊的實驗環境下作實驗。謝謝廖永明學長常常在假 日陪我作實驗,更讓我對ptt有了更深入的認識,希望你之後能夠一見雙叼、 手到擒來。謝謝愛學人買相同鞋子的李智文學長及愛好運動的羅志楠學長 常常陪大家一起打球。謝謝宋建宏博士告訴我許多自組裝的作法,也謝謝 你常常找我打桌球讓我的球藝大進。謝謝愛亂捏人的龔亮仁博士對我實驗 技巧指點,讓我不管做什麼都很順利。謝謝何敏碩學長提供很多作實驗時 要注意的小細節,希望你之後的實驗也能夠很順利。謝謝張晉彥學長願意 花時間跟我討論旋光性及SEM的小細節。謝謝施宏旻學長陪我看情書,也 因為你,實驗室得到了許多歡樂的氣息,希望你趕快找夢中情人。謝謝汪
vi 佩瑄學姐從大一時期對我四年多來的照顧,從你那邊我知道了許許多多應 化系有趣的人事物。謝謝又加、怡碩、迪迪陪我度過我的碩一生活。謝謝 跟我同一屆的同學們,好麻吉吳彧群常常陪我買東西吃、開車出去玩,希 望你的洋基今年能有榮幸跟紅襪在世界大賽對決,然後被紅襪淘汰。謝謝 林芳常常跟我說啥時要考試以及提醒我許多事情。謝謝黃鈺婷跟我分享你 看過好看的電影。謝謝湖人隊的林承叡,謝謝女人很多的許光輝,謝謝可 愛的李天心,謝謝只有一開始裝禮貌的陳秋翔,謝謝半途插花的吳仁棋, 希望你們的實驗也能順順利利。還要特別感謝兩位助理小姐欣怡與小燕 姐,因為有了你們的幫助,作起實驗來才特別有勁。謝謝所有從大學就一 路相伴的同學們以及到了研究所才認識的同學學弟妹們,陳璟昆、黃偉杰, 謝謝你們常常提供儀器讓我使用, 陳宏哲、詹鎮瑋,謝謝你們常陪我出去 玩,王慧茹、馮蘭英、陳鈺君,謝謝你們常常陪我聊天。 最後我要謝謝我的家人,謝謝你們努力栽培我,謝謝你們不管我做什 麼都這麼的支持我,謝謝你們。
目 錄 中文摘要...i 英文摘要...iii 謝誌...v 目錄...vii 圖目錄...xi 表目錄...xiv 附圖目錄...xv 第一部份 聚3-己烷噻吩鏈段聚L型乳酸共聚物之合成、自組裝調控及其在太陽能電 池之應用...1 第一章 緒論...2 1-1. 太陽能電池簡介 ...2 1-1-1. 有機太陽能電池的起源 ... 3 1-1-2. 有機太陽能電池的原理 ... 4 1-2. 有機太陽能電池元件... 5 1-2-1. 雙層元件(BilayerDevices) ... 6
1-2-2. 異質接面元件(Bulk Heterojuntion Devices)... 6
1-3.有機太陽能電池的元件特性...7
1-3-1. 有機太陽能電池的元件特性(Open Circuit Voltage,Voc)... 8
1-3-2. 有機太陽能電池的元件特性(Short Circuit Current,Isc) ... 10
1-3-3. 有機太陽能電池的元件特性(Fill Factor,FF) ... 10 1-4.聚噻吩的簡介...11 1-5.各式聚噻吩鏈段共聚物...15 1-6.高分子太陽能電池整理...17 1-7.研究動機...19 第二章 實驗部份...20 2-1.試藥...20
viii
2-2.量測儀器...20
2-2-1. 核磁共振光譜儀(Nuclear Magnetic Resonance,NMR) ... 20
2-2-2.質譜儀(Matrix Assisted Laser Desorption/Ionization-Time of Flight Mass Spectroscopy,MALDI-TOF MS) ... 20
2-2-3.微差掃瞄卡計(Differential Scanning Calorimeter,DSC)...21
2-2-4.熱重分析儀(Thermal Gravimetric Analyzer,TGA) ...21
2-2-5. 凝膠滲透層析儀(Gel Permeation Chromatography,GPC) ... 21
2-2-6.紫外線與可見光光譜儀(UV-Vis Spectrophotometer) ...21
2-2-7.掃描式電子顯微鏡(Scanning Electron Microscope,SEM) ...21
2-2-8.原子力顯微鏡(Atomic Force Microscope,AFM) ...22
2-2-9.太陽光模擬元件量測系統(AM 1.5)...22 2-3.合成部份...22 2-3-1. 單體的合成 ... 22 2-3-2. 聚合物及其鏈段共聚物之合成 ... 24 2-4.合成路徑示意圖...27 2-5. 高分子有機太陽能電池元件製程...28 2-5-1.ITO pattern的製作 ... 28 2-5-2.高分子有機太陽能電池元件製作 ...29 第三章 結果與討論...31 3-1.單體結構鑑定...31 3-2.聚合物P1、P2及鏈段共聚物P3之合成與結構鑑定 ...31 3-3.分子量分析...38 3-4.熱性質分析...40 3-5.紫外光-可見光吸收光譜之光學性質 ...41 3-6.薄膜XPS之量測 ...43 3-7.元件薄膜形態之觀測...46 3-8.元件光電性質之量測...48 3-9.孔洞活化層之熱處理在元件光電性質的效應...50 3-10.碳六十層之熱處理在元件光電性質的效應...54
3-11.掃描式電子顯微鏡(SEM)分析 ... 58 第二部份 以多面體矽氧烷寡聚物為中心的星狀結構之合成及其在太陽能電池之應 用...59 第四章 緒論...60 4-1. 多面體矽氧烷寡聚物(POSS)材料的發展 ...60 4-2.研究動機...61 第五章 實驗部份...62 5-1.試藥...62 5-2.量測儀器...62
5-2-1. 傅立葉轉換式遠紅外光光譜儀 (Fourier Transform Infrared Spectroscopy,FTIR)... 62
5-2-2.廣角度X射線繞射儀 (Wide-Angle X-ray Diffraction,WAXD)20 5-3.合成部份...62 5-3-1.聚合物及其鏈段共聚物之合成 ...62 5-4.合成路徑示意圖...64 5-5. 高分子有機太陽能電池元件製程...65 5-5-1.ITO pattern的製作 ... 65 5-5-2.高分子有機太陽能電池元件製作 ...65 第六章 結果與討論...66 6-1. 星狀聚合物P4之合成與結構鑑定 ...66 6-2. 分子量分析...69 6-3. 熱性質分析...69 6-4. 紫外光-可見光吸收光譜之光學性質 ...70 6-5. 元件光電性質之量測...72 6-6. 溶劑揮發效應在元件光電性質之影響...77 第七章 結論...81 第八章 參考文獻...82
x 圖 目 錄
圖1-1. Charge transfer through potential gradient ...5
圖1-2. The device structure of (a) bilayer photovoltaics (b) BHJ photovoltaics...5
圖1-3. I-V curve of solar cell device...7
圖1-4. The equation of (a) efficiency (b) fill factor ...8
圖1-5. Relation between donor and different acceptors...9
圖1-6. Linear correlation between Voc and the different oxidation potential energy ... 9
圖1-7. Four regioisomers in the P3AT polymer chain...12
圖1-8. The mechanism of oxidative polymerization...12
圖1-9. The mechanism of oxidative addition to C-Br bond primarily at the 5-poision..13
圖1-10. The polymerization of 97-98% HT-HT poly(3-alkylthiophene) ...13
圖 1-11. The polymerization of HT-HT poly(3-alkylthiophene) by using LDA/MgBr2OEt2... 14
圖1-12. The Grignard metathesis polymerization...14
圖1-13. The end-capped Grignard metathesis polymerization ...15
圖1-14. The ideal structure of BHJ solar cell ...18
圖3-1. Mechanism of hydroboration reaction...32
圖3-2. Mechanism of hydroxylation ...32
圖3-3. Mechanism of Ring-Opening polymerization ...34
圖3-4. MALDI-TOF MS Spectra (a) P1 and (b) P2 ...35
圖3-5. 1 H NMR Spectra of P1 ... 36
圖3-6. 1 H NMR Spectra of (a) P2 and (b) P3... 37
圖3-7. UV-Vis absorption spectra of C1 ~ C3...42 圖3-8. XPS spectra of P3HT-PLLA thin film (a) Full scale image for C, O and S atom of the thin films before and after degradation (b) Carefully scanning for C atom of
the thin films before and after degradation (c) Carefully scanning for O atom of
the thin films before and after degradation ... 45
圖3-9. Tapping mode AFM height images of (a) P3HT-OH thin film (b) P3HT-PLLA thin film prior to etching (c) P3HT-PLLA thin film after etching. ... 47
圖3-10. Structure of Solar Cell Device ...48
圖3-11. I-V curve of device I and device II ...49
圖3-12. Tapping mode AFM height images of P3HT-PLLA thin film (a) before annealing (b) after annealing ... 52
圖 3-13. The short-circuit current and power conversion efficiency as function of annealing temperature (b) The open-circuit voltage and fill factor as function of annealing temperature ... 53
圖3-14. Tapping mode AFM 3D height images of C60 layer (a) before annealing (b) after annealing at 130°C (c) after annealing at 150°C (d) after annealing at 170°C... 56
圖3-15. (a) The short-circuit current and power conversion efficiency as function of annealing temperature (b) The open-circuit voltage and fill factor as function of annealing temperature ... 57
圖3-16. SEM image of P3HT-PLLA ...58
圖4-1. The structure of T8... 60
圖6-1. The mechanism of hydrosilylation (Chalk-Harrod’s)...66
圖6-2. FTIR spectrum of (a) POSS (b) reaction solution(after 1 day) (c) POSS-P3HT ... 68
圖6-3. UV-Vis absorption spectrum ...71
圖6-4. Structure of Solar Cell Device ...72
圖6-5. UV-Vis spectrum of device III under different temperature treatment ...75
圖6-6. UV-Vis spectrum of device IV under different temperature treatment ...75
圖6-7. I-V curve of device III... 76
圖6-8. I-V curve of device IV ...76
圖6-9. I-V curve of device under different solvent annealing time ...78
xii
圖6-11. WAXD spectrum of POSS-P3HT:PCBM layer...79
圖6-12. WAXD spectrum of POSS-P3HT:PCBM layer (part)...80
圖S1. Synthesis of poly(3-hexylthiophene)-block-poly(l-lactide)...27
表 目 錄
表 1-1 Different types of polythiophene block copolymers ... 16
表 1-2 Different donor-type solar cell materials and their efficiency ...17
表 2-1 Preparation of polymer solution...29
表 3-1 Characterization of P3HT-b-PLLA...39
表 3-2 Thermal analysis of block copolymers...40
表 3-3 UV-Vis absorption ...42
表 3-4 XPS Binding Energy Position ...43
表 3-5 The performance of polymer solar cells made from different nanomorphology under AM 1.5G illumination (100 mW/cm2) ... 49
表 3-6 The performance of device II made from different annealing temperature on P3HT-PLLA thin film under AM 1.5G illumination (100 mW/cm2) ... 52
表 3-7 The performance of device II made from different annealing temperature on C60 layer under AM 1.5G illumination (100 mW/cm2) ... 57
表 6-1 Characterization of P3HT-Vinyl and POSS-P3HT...69
表 6-2 Thermal analysis of star-like polythiophene polymers ...70
表 6-3 UV-Vis absorption peaks ...71
表 6-4 The performance of polymer solar cells made from different thermal treatment under AM 1.5G illumination (100 mW/cm2) ... 73
xiv 附 圖 目 錄 附圖 1. 3-Hexylthiophene,化合物1的1 H-NMR光譜圖 ... 89 附圖 2. 3-Hexylthiophene,化合物1的13 C-NMR光譜圖 ... 90 附圖 3. 3-Hexylthiophene,化合物1的質譜圖 ... 91 附圖 4. 2,5-Dibromo-3-hexylthiophene,化合物2的1 H-NMR光譜圖 ... 92 附圖 5. 2,5-Dibromo-3-hexylthiophene,化合物2的13 C-NMR光譜圖 ... 93 附圖 6. 2,5-Dibromo-3-hexylthiophene,化合物2的質譜圖... 94 附圖 7. Vinyl-terminated poly(3-hexylthiophene),高分子P1的1 H-NMR光譜圖... 95 附圖 8. Hydroxy-terminated poly(3-hexylthiophene),高分子P2的1 H-NMR光譜圖 .. 96 附圖 9. Poly(3-hexylthiophene)-block-poly(l-lactide),高分子P3的1 H-NMR光譜圖.. 97
第一部份
聚 3-己烷噻吩鏈段聚L型乳酸共聚物之合成、自組裝調控及其在太陽能電池
之應用
Synthesis and Controlled Self-assembly of Poly(3-hexylthiophene)-
第一章 緒論 1.1 太陽能電池簡介 由於近年來能源危機及環保意識抬頭,開發太陽光能源的太陽能電池 (Solar cell,又可稱為「太陽能電池」、「光感電池」),被譽為21世紀最具有 發展潛力的光電技術之ㄧ。1973年發生了石油危機,讓世界各國察覺到能 源開發的重要性,而今溫室效應及全球暖化現象日漸嚴重,更讓世界各國 紛紛投入在綠色能源的研究。由於太陽光是取之不盡,用之不竭的天然能 源,除了沒有能源耗盡的疑慮之外,也可以避免繼續產生二氧化碳的溫室 問題,因此加速發展太陽能源的應用科技為必然的趨勢,並期望藉由增加 太陽能源的利用來減低對石化能源的依賴性及對地球環境的傷害。目前應 用在太陽能電池的主要材料是矽,其主要可分為單晶矽(single crystal
silicon)、多晶矽(polycrystalline silicon)、非晶矽(amorphous silicon)、微晶矽 (microcrystalline silicon)共四種;合物半導體(compound semiconductor), 主要的材料有:CuInSe2 (CIS)、CuInGaSe2 (CIGS)、GaAs、GaInP、InGaAs、 CdTe等,這幾類無機材料的研究已有相當長久的歷史。以矽及化合物半導 體等無機材料製成的太陽能電池,通稱無機太陽能電池,效率高且性能穩 定,目前已有量產並被廣泛應用於太空及陸地上,但有製作過程過於複雜、 製作成本高等缺點。
另一大類為有機太陽能電池,包括有染料敏化太陽能電池(Dye- Sensitized Solar Cell)、有機小分子太陽能電池(Organic Solar Cell)、高分子太 陽能電池 (Polymer Solar Cell)等,這類材料則較為新穎,目前尚屬於研究階 段。相較於價格昂貴的無機太陽能電池,價格上則相對低廉且製作較為簡 單,使得以開發有機化合物作為太陽能電池材料的研究極具潛力。 1.1.1 有機太陽能電池的起源 光電效應是太陽能電池將光能轉換成電能的機制,此效應在1887年由 Heinrich Hertz 實驗發現的,而在1905年由愛因斯坦使用光子的概念在理論 上予以成功的解釋。第一個具功率性的固態太陽能電池在1883年由Charles Fritts 所做出,以硒及金薄膜製作成具有1%轉換效率的接面。1953年貝爾 研究實驗室(Bell Laboratories,USA)製作出轉換效率6%的第一個矽太陽能 電池。在自然界中,太陽所發出的光為太陽系內各生物體維繫生命之泉源, 地球上的植物須經陽光照射才能進行光合作用,其中葉綠素扮演著進行光 合作用不可或缺的角色,也因此引起了科學家的重視。1975年四月C. W. Tang 和A. C. Albrecht 兩位教授在 Nature 期刊上共同發表一篇論文,以 chlorophyll-a(即葉綠素A)作為太陽能電池的材料,為有機太陽能電池跨出了
一大步,但其轉換效率仍相當低 [1]。早期有機太陽能電池的研究多以
chlorophyll及phthalocyanine等衍生物為主[2,3],元件製作方法則相當簡易,
僅將有機材料以兩種不同電極夾在中間的單層太陽能電池[4-6],又稱作三明
治電池。1986年美國Kodak研究實驗室 C. W. Tang 博士以CuPc(copper phthalocyanine)及PV(perylene tetracarboxylic derivative)兩種有機化合物作為
材料,製作出雙層的有機太陽能電池,將其光電轉換效率提升至1%[7]。有
1.1.2 有機太陽能電池的原理 一般來說,大部分有機太陽能電池的材料結構多具備π鍵結的電子系 統,主要功能在於可吸收光線並且利用共軛π電子傳遞電荷。由兩個sp2混成 軌域頭對頭(head-to-head)形成的碳-碳單鍵結稱作σ鍵結,σ鍵結為平行平面 的方向,而由兩個pz軌域在側邊混成(side-to-side)形成的碳-碳雙鍵結稱作π 鍵結,π鍵結為垂直平面的方向,共軛π鍵結的電子系統構成填滿電子的最 高電子軌域(the highest occupied molecular level,HOMO)及未填滿電子的最 低電子軌域(the lowest unoccupied molecular level,LUMO),HOMO及LUMO 用於定義材料的光學能隙值差。適用於有機太陽能電池的材料必須具備低 能隙值的條件,能隙值 1.1 eV(1100 nm)的材料根據計算可以有效吸收 77% 的太陽光[11],大多數的材料的能隙值約為1.4 eV至 3 eV之間(絕緣體的能隙 值大於 3 eV),如此高的能隙值可能限制材料對於太陽光的吸收,且在室溫 無 光 線 環 境 下 , 僅 僅 只 有 微 乎 其 微 的 電 荷 密 度 , 為 了 提 高 電 荷 密 度,通常以摻入一些其他物質的方式使有機材料的導電效應提升[12]。 初期的太陽能電池為簡易的單層結構,利用不同電極兩端功函數的差 異,達到分離電子跟電洞的效果,而電池的效率往往受限於電極與吸光材 料之間接面的接合程度,若電極與吸光材料之間出現缺陷,則容易導致電 子或電洞的再結合或是不必要的漏電流[1-6]。 有機材料主要以光誘導的方式形成以庫倫作用力相互吸引的激子,並 非直接形成自由電荷,但此激子卻僅有10 %被分離成自由電荷[14],電荷分 離的區域主要在於接面處,原因主要來自於接面處位能差異性所形成的內 建電場(E = -grad U),吸光產生的激子必須在生命週期內擴散至接面處,否 則將返回至基態並以輻射或非輻射形式散失能量,因此,在接面處相分離 的長度必須受到限制。電子-電洞對在有機材料的擴散距離約為 10 至 15 nm。產生的自由電荷必須經由傳輸電荷的材料至電極,此過程藉由位能遞
差的方式來傳遞電荷,位能遞差是由施體及受體之間HOMO及LUMO的差 異來決定,類似階梯的原理達到傳遞電荷的效果。除藉由位能遞差及漂移 電荷之外,利用不對稱電極(一端為低工作函數金屬作為收集電子用途,另 一端為高工作函數金屬作為收集電洞用途)在短路下可產生外部電壓。此 外,電荷濃度差異造成的擴散電流亦可作為電荷傳遞的驅動力。事實上, 電荷傳遞的過程必定伴隨著電子與電洞的再結合,特別是當傳遞電子與電 洞的介質為相同物質。
Figure 1-1. Charge transfer through potential gradient
1.2 有機太陽能電池元件
有機太陽能電池的元件製作方式常見有兩類:雙層元件(bilayer photovoltaic) 、異質接面元件(bulk heterojunction photovoltaic)。
1.2.1 雙層元件 在文獻中有相當多種材料及製程方式用於有機太陽能電池雙層元件的 製作[15-19],一般說來,雙層元件的結構為一層施體層接著在其上層製作一 層受體層(圖 1-2a),此類有機太陽能電池藉由兩種不同材料特性來達到電荷 分離的效果,例如:phthalocyanine為提供電子的材料,亦稱p型(即傳導電 洞)的材料,perylene為接受電子的材料,亦稱n型(即傳導電子)的材料[7],電 子由一種材料傳至另一種材料即為施體-受體(Donor-Acceptor)型,其功能相 當類似半導體材料的p-n接面,施體材料吸收光子之後轉換成較高能量的激 發態,即未分離具有高束縛能量的電子-電洞對(electron-hole pair)(亦稱激 子),此庫倫作用力造成的束縛能量約為 0.4 eV,激子藉由擴散的形式到達 可以進行電荷分離的區域,即p型材料與n型材料的接面,接著電子與電 洞再藉由各自的傳導材料到達電極。 此種元件製作的方式有厚度上的限制,其原因來自於激子的生命週期 及擴散距離,與實際吸收光子所產生的激子數目進行比較,僅有少數在擴 散距離內所產生的激子可以移動至接面進行電荷分離,如此一來,容易造 成元件在效率上相當大的損失。 1.2.2 異質接面元件 異質接面元件的做法與雙層元件不同之處在於將施體及受體兩者均勻 混合製作成單層活化層(圖 1-2b) ,此類型作法常見於以碳六十衍生物為主 要接受電子材料的高分子太陽能電池上,其特色在於施體材料與受體材料 並沒有明顯的接面,均勻混合的活化層吸收光子之後產生的激子在分子與 分子之間相當微小的相分離處進行電荷分離,因為產生的激子能夠在擴散 距離內的接面進行電荷分離,因此能夠有效提升轉換效率[13]。 雙層元件的的施體與受體可分別與兩端電極相接,在異質接面元件僅
有一均勻混合的活化層,施體與受體無法分別與兩端電極相接。活化層中 由於施體與受體的均勻混合,造成無特定方向性的內部電場,因此,所產 生的電子與電洞也沒有特定的方向性,自由電荷利用擴散的方式自活化層 移動至兩端電極,電荷移動的過程需藉助傳電子與傳電洞的傳導材料。異 質接面元件的活化層可利用共蒸鍍或溶液塗佈的方式製作,共蒸鍍的製作 方式是將施體與受體的小分子同時蒸鍍製作,溶液塗佈的製作方式則是將 施體與受體溶解於溶劑中塗佈製作,可用於高分子與高分子、高分子與小 分子的混合作法。 1.3 有機太陽能電池的元件特性 有機太陽能電池需量測光照射下及黑暗中的電流-電壓曲線(current- voltage curve)或稱I-V曲線(圖 1-3),虛線表示為無光源下的暗電流,實線則 表示為照光源下的光電流,實線與x軸相交點表示為開路電壓(open circuit voltage,Voc),理論上此數值被定義為施體HOMO與受體LUMO的差值,實 際上仍由接面產生的內部電場所決定(圖 1-1)。與y軸相交點表示為短路電流 (short circuit current,Isc),將矩形面積(白色部分)除以總面積 (白色部分+黑 色部分)可得填充係數(fill factor,FF),此填充係數可做為衡量太陽能電池是 否 能 夠 作 最 大 轉 換 程 度 , 最 大 值 為 1 , 即 將 光 完 全 轉 換 成 電 流 。
矩形與y軸相交點表示為填充比例區域的最大電流值(maximum power output current,Imax),矩形與x軸相交點表示為填充比例區域的最大電壓值 (maximum power output voltage,Vmax)。一般來說,在黑暗中幾乎沒有電流 的產生,直到正向偏壓大於開路電壓,電流才開始產生。 太陽能電池光轉換成電流的轉換效率可以下列公式定義(a):Voc表示為 開路電壓,Isc表示為短路電流,FF表示為填充比例,Pin表示為注入光子數。 測 量 用 的 標 準 光 源 為 採 用 距 地 球 表 面 仰 角 4 8 . 2 ° 的 太 陽 光 光 譜 分佈,其強度標準值為 1000 W/m2,稱作AM 1.5 光譜。 (a) (b)
Figure 1-4. The equation of (a) efficiency (b) fill factor
1.3.1 開路電壓(Open Circuit Voltage,Voc)
根據傳統太陽能電池概念,在MIM元件(metal-insulator-metal,MIM, 即兩金屬中夾入太陽能電池材料)的開路電壓值是由兩不同金屬電極的功函 數差異所決定。在p-n接面的系統中,開路電壓值則需引入p型與n型材料之 間的準費米能階的概念來決定,尤以碳60 的還原電位對異質接面元件的影 響甚大’。對於太陽能電池來說,開路電壓是與施體的HOMO與受體的LUMO 呈現線性的關係[20]。Brabec et al. 藉由改變不同受體(不同碳 60 的衍生物) 製作成太陽能電池並測量Vo c的值來表示此種線性關係(圖 1-5),隨著 受體第一個還原電位的改變,元件測得Voc值亦有明顯的不同[20]。
Figure 1-5. Relation between donor and different acceptors[20] 當施體的第一個氧化電位改變時,Mulliaras et al. 測量到Voc值亦伴隨產 生不同的值[21]。Scharber et al. 利用 26 種不同施體材料製作成太陽能電池, 討論施體氧化電位與Voc的線性關係(圖 1-6),斜線表示線性關係(斜率為 1)。此外,開路電壓的值受到活化層形態的影響,利用non-aromatic及aromatic 兩類不同的溶劑製作活化層,所測得元件Voc有明顯的不同[22]。金屬與有機 材料之間存在的介面效應(Interfacial Effect),介面效應可能來自於金屬電 極 表 面 氧 化 物 的 產 生 , 此 效 應 會 影 響 金 屬 電 極 的 功 函 數 , 而 導 致 Voc的改變。[23]
1.3.2 短路電流(Short Circuit Current,Isc) 理想的元件,在各接面無任何缺陷情形下,Isc受到光誘導產生的電荷 載子濃度及電荷載子移動速率的影響。Isc可以下列公式定義: Isc = neμE n表示為電荷載子的濃度,e表示為單位電荷(elementary charge),μ表示為 移動速率(mobility),E表示為電場強度,假設元件效率能夠達到 100 %,n 即為每單位體積吸收的光子數。事實上,Isc的值並非受限於材料本身,而取 決於元件的製作過程,其中以活化層的形態影響最大[24-28],事實上,活化 層的形態是經由製作的過程來控制,可以影響的因素如溶劑的選擇、揮發 時間的控制、試片熱處理溫度的控制及蒸鍍的方式皆會造成不一樣的活 化層形態[29-32]。
外部量子效率(external quantum efficiency,EQE)或稱入射光轉換電效率 (incident photon to current efficiency,IPCE)可用來計算個別波長時轉換效 率,以下列公式定義: IPCE = 1240 Isc / λPin λ表示為入射光波長(單位為nm),Isc表示為元件測得電流大小(單位為 μA/cm2),Pin表示為使用的入射功率。 1.3.3 填充係數(Fill Factor,FF) 填充係數被定義為電流及電壓同時達到最大值時實際功率的比例值, 在 I-V 圖的表示上為總面積內的矩形面積(圖 1-3),填充係數容易受到串聯
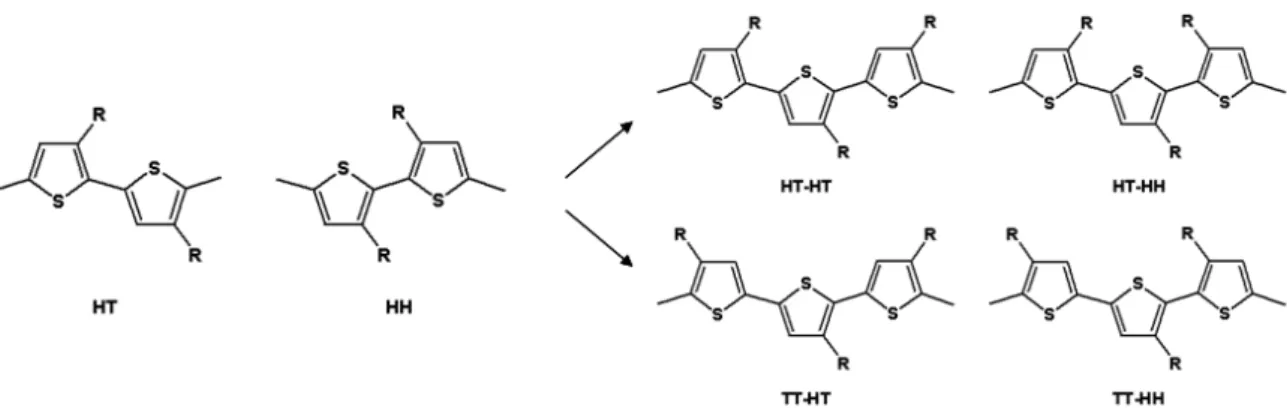
電阻(Series Resistance)的影響,串聯電阻主要來自於材料本身的特性、金屬 接面或是內部的接合作用力。 1.4 聚噻吩的簡介 聚噻吩(polythiophene,PTh)的研究始於 80 年代初期,其單體結構由於 相當類似吡咯(pyrrole)使得其製備方式亦相當類似[54],1980 年,Yamamoto et al. 發表一篇利用化學聚合方法合成聚噻吩高分子[55],但由於其溶解度不佳 的問題,在聚合的過程中無法得到高分子量的聚合物,為改善此問題, Daoust et al. 在噻吩的三號位置上引入不同的烷基合成聚(3-烷基)噻吩 (poly(3-alkylthiophene),P3AT)可以有效地改善其溶解度的問題[56-58]。引入 烷基側鏈對於聚噻吩高分子的物理性質及化學性質有相當顯著的影響,隨 著烷基的鏈長增加,其薄膜的氧化還原電位逐漸增加,而使得導電性會下 降。引入具分支結構的烷基側鏈可以改善規則度,但由於分支彼此會造成 立體阻礙,而使得薄膜的缺陷增加。事實上,較長的側鏈會改變高分子鏈 的對稱性、主鏈上環與環之間的共平面性而造成熱穩定性的下降。 聚噻吩的製備方式可分為三大類:電化學聚合方法[59]、使用氧化劑FeCl3 的 氧 化 聚 合 方 法 [ 6 0 , 6 1 ]及 去 鹵 基 縮 合 聚 合 法( d e h a l o g e n a t i o n p o l y - condensation)[62,63]。對於合成聚(3-烷基)噻吩而言,(3-烷基)噻吩在形成二聚 物後,由於三號位置取代基的關係會產生兩種規則度:頭對尾(head-to-tail) 及頭對頭(head-to-head),接著衍生出四種高分子主鏈上的立體規則結構: HT-HT、HT-HH、TT-HT及TT-HH[64-67] (圖 1-7),立體規則度高的聚(3-烷基) 噻吩高分子有較佳的導電性、光學性質及磁性,無規則的聚(3-烷基)噻吩高 分子由於主鏈上的HH結構導致相當大的立體阻礙,導致缺陷增加而破壞 材料本身的特性。
Figure 1-7. Four regioisomers in the P3AT polymer chain
Figure 1-8. The mechanism of oxidative polymerization[68]
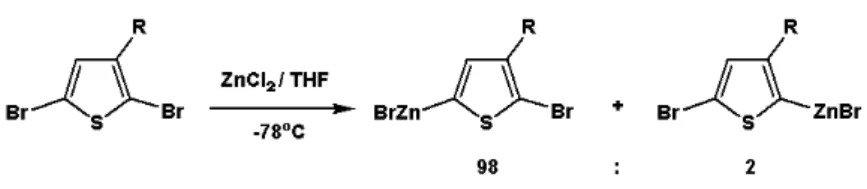
為了要得到立體規則度高的聚(3-烷基)噻吩高分子,並不能使用電化學 及氧化聚合方法(圖 1-8)[68],因此,必須利用去雙鹵基縮合聚合的方式,使 噻吩單體在聚合過程中先形成不對稱的中間產物再進行聚合。文獻上常見 的合成方式大多先利用有機金屬起始劑形成不對稱的中間產物,如:Rieke Zinc [63,69,70]、MgBr2OEt2[71]及CH3MgBr[33],接著加入有機金屬催化劑來進 行聚合反應,如Ni(dppe)Cl2、Ni(dppp)Cl2[72,73]等。 金屬起始劑Rieke Zinc先經過氧化加成至C-Br鍵上(圖 1-9),將反應控制 在-78oC時,由於在五號位置上的C-Br鍵有較佳的反應性,因此中間產物大 多數(98%)在五號位置的Br上形成C-ZnBr,而二號位置的Br僅有相當少部分 (2%)形成C-ZnBr,接著加入有機金屬催化劑Ni(dppe)Cl2 ([1,3-Bis(di- phenylphosphino)ethane]-dichloronickel (II))進行聚合反應,即可得到立體規 則度相當高的聚(3-烷基)噻吩高分子(圖 1-10)。
Figure 1-9. The mechanism of oxidative addition to C-Br bond primarily at the 5-poision
Figure 1-10. The polymerization of 97-98% HT-HT poly(3-alkylthiophene)
另一類作為起始劑的金屬為鎂,常見為MgBr2OEt2、CH3MgBr及
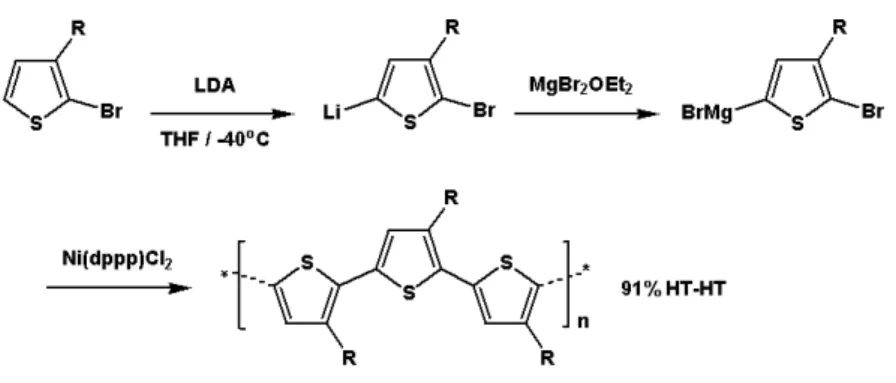
t-ButylMgBr。以MgBr2OEt2為例,先將二號位置作單邊溴化的噻吩單體與
LDA(lithium diisopropylamide)反應,接著將MgBr2OEt2加入可得到不對稱的 中間產物,接著加入有機金屬催化劑Ni(dppp)Cl2 ([1,3-Bis(diphenylphos- phino)propane]-dichloronickel (II))進行聚合反應,即可得到立體規則度相當 高的聚(3-烷基)噻吩高分子(圖 1-11)。目前來說,最廣為使用的方法是以 CH3MgBr及t-ButylMgBr作為起始劑的格林納置換聚合反應(Grignard metathesis polymerization,GRIM),此方法在 1999 年由McCullough et al. 所 發表,此方法的步驟簡單且可以有效合成出相當高立體規則性的聚噻吩高 分子[33] (圖 1-12) 。
此外,利用Suzuki Coupling及Stille Coupling先合成不對稱的噻吩單體, 接著使用有機金屬催化劑進行聚合反應也可得到立體規則性的聚 噻 吩
Figure 1-11. The polymerization of HT-HT poly(3-alkylthiophene) by using LDA/MgBr2OEt2
1.5 各式聚噻吩鏈段共聚物
格林納置換聚合反應經改良被用來合成末端具官能基(end-functionized)
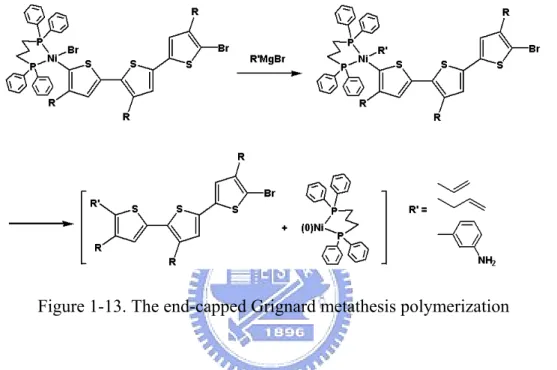
的不對稱聚噻吩高分子[34,35]。此反應機制是在進行中止反應前,加入末端修
飾的格林納試劑(R’MgBr)與高分子的一端相接(圖 1-13)。
Figure 1-13. The end-capped Grignard metathesis polymerization
末端具官能基的聚噻吩高分子可利用二次聚合的方式,採用的方式如 原子轉移自由基聚合反應(atom transfer radical polymerization,ATRP) [36,37]、 陰離子聚合反應[38]及開環聚合反應(ring-opening polymerization,ROP)[39]合 成出不同的聚噻吩鏈段共聚物,鏈段共聚物本身由於兩鏈段不同的分子特 性,在製作成薄膜後在表面產生奈米尺度的相分離現象。此外,隨著製作 薄膜的技術進步及有效控制相分離程度,使得有著相分離現象的薄膜能被 進一步發展出更多應用價值[40,41],目前已被合成的各式聚噻吩鏈段共聚物如 表 1-1。
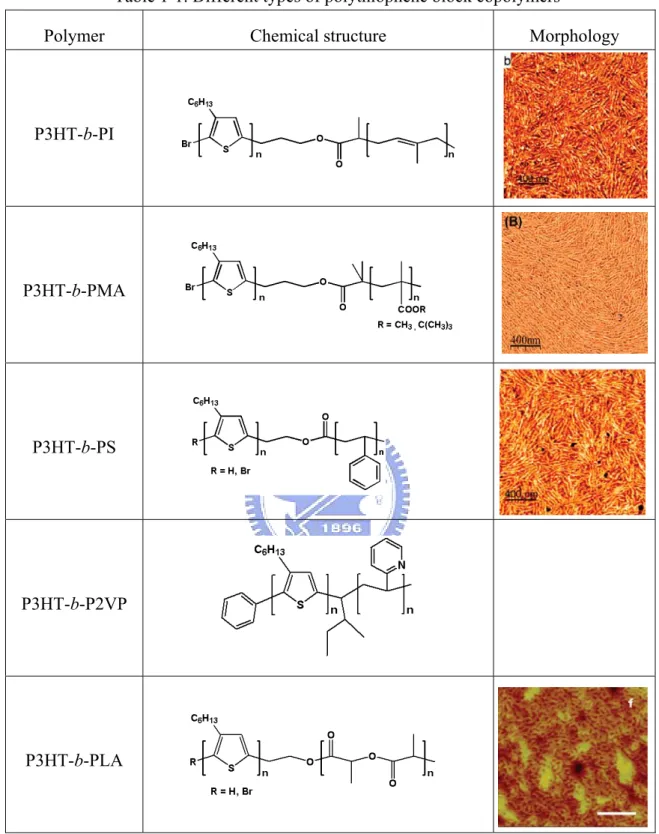
Table 1-1. Different types of polythiophene block copolymers
Polymer Chemical structure Morphology
P3HT-b-PI
P3HT-b-PMA
P3HT-b-PS
P3HT-b-P2VP
1.6 高分子太陽能電池整理
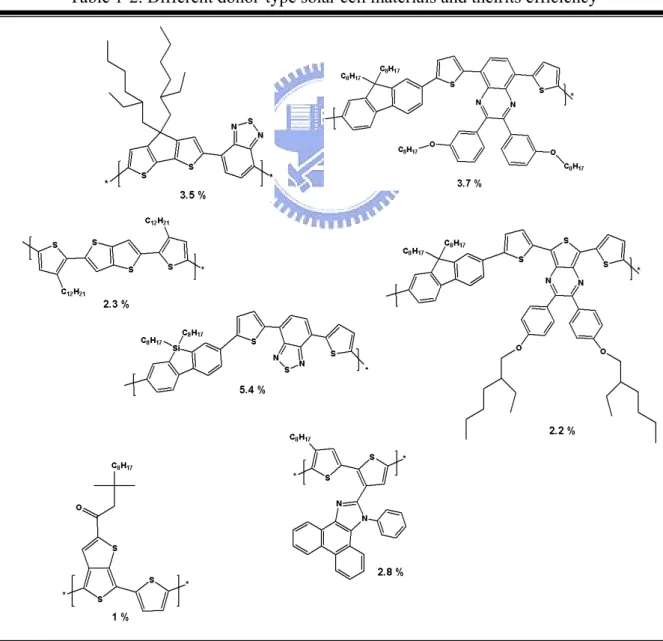
以P3HT/PCBM ([6,6]-phenyl-C61-butyric acid methyl ester)作為有機太 陽能電池的材料仍為目前研究的主流,利用在製程上的改良使元件的效率 能夠獲得改善,如: Yang et al. 用控制溶劑揮發的時間及加熱溫度這兩項重 要因素將效率提升至4.4%[76],Heeger et al. 在活化層與陰極金屬之間引入 TiOx作為入射光重分配層,此元件效率可達5%[9],Chen et al. 在PSS:PEDOT 中加入mannitol,使電洞的傳導效果提升並將效率提升至5.2%[10],而新施體 材料(表1-2)的設計也不斷推陳出新。
經由新材料的開發,降低施體材料的LUMO能階可以提高Voc值,提高 混合材料中傳遞電子及電洞的速率,Scharber et al.預估未來有機太陽能電池 的效率可能達到10%[11]。未來的發展可著眼於以下: 1. 合成新式低能隙高分子 2. 合成新式受體材料 3. 利用不同能隙高分子製作堆疊型電池 4. 減少散射影響 5. 結合無機物的混成材料 理想的異質接面元件結構如下:施體材料及受體材料之間形成垂直的 電子及電洞通道,由於傳電子及傳電洞彼此交錯,可以減少電子與電洞再 結合的機會,且介面面積增加,激子的電荷分離效率因此提高。
1.7 研究動機 目前太陽能電池是相當熱門的產業之ㄧ,由於國際油價的不斷上漲, 應用次世代環保新能源更造成太陽能產業的蓬勃發展,使其極具潛力成為 明星產品。雖然目前主要作為商業用途的太陽能電池仍以矽做為材料,不 過,由於上游供料取得不易且價格昂貴,開發便宜且製程簡易的新式材料 即成為此產業的新興目標,有機太陽能電池為以有機化合物作為太陽能電 池的材料,符合低價格且製程簡易的優點,使有機太陽能電池在未來有機 會成為新式的商業太陽能電池。 目前較常見於文獻的元件製作有雙層元件及異質接面元件等兩種作 法,雙層元件的限制在於可用於激子作電荷分離的介面太少,以致於產生 的激子無法做有效的分離,而異質接面元件雖能夠有效改善介面問題,但 兩種材料彼此並無法作最均勻的分佈,也因此元件的效率受限於活化層的 形態[22,24-28]。目前多數的研究多著重於合成新式低能階共軛高分子及製程方 式的改良,然而,本研究之目的在於希望將現有材料做進一步的改良,並 且結合薄膜製作的技術,改善雙層元件介面的問題。
本研究之高分子的構思源自於 Ho, R. M. et al. 將polyisoprene-b- poly(l-lactide) (PI-PLLA)的鏈段共聚物製作成具有自組裝現象的高分子薄 膜,並經由精確的分子量控制和去除乳酸片段,製作成具有孔洞的高分子 材料。在鏈段共聚物的合成部份,以poly(3-hexylthiophene) (P3HT)取代PI, 並藉由不對稱的合成方法合成出poly(3-hexylthiophene)-b-poly(l-lactide) (P3HT-PLLA),此外,針對P3HT及PLLA片段進行不同分子量的合成。在太 陽能電池元件製作部分,將此具有奈米孔洞結構薄膜應用至元件活化層, 接著將PLLA片段以溫和的方式裂解而製作成具孔洞的聚噻吩薄膜,希望能 夠藉由高分子本身作為吸光材料之外,此奈米孔洞形態能夠增加與電子接
第二章 實驗部份
2.1 試藥
實驗中所使用之藥品均分別採購自 Aldrich、Merck、Janssen、Lancaster、 TCI 與聯工公司。所有溶劑皆購自 Merck 及 Fischer 公司。無水 tetrahydrofuran (THF)及無水 ether 皆以鈉金屬除水,並加入 benzophenone 為指示劑,在氮 氣條件下迴流二日後蒸餾出使用。無水 toluene 以氫化鈣除水,在氮氣條件 下迴流二日後蒸餾出使用。 2.2 量測儀器 為了鑑定或測試所得的中間產物、前驅物單體或鏈段共聚物之化學結 構及物理特性,採用下列測試儀器:
2.2.1 核磁共振光譜儀(Nuclear Magnetic Resonance,NMR)
使用Varian-300 MHz核磁共振光譜儀。其中以CDCl3為溶劑,氫譜以 tetramethylsilane作為δ= 0.00 ppm為內部標準,碳譜則以 77.24ppm作為內部 標準,化學位移單位為ppm。光譜資料中:符號s表示單峰(singlet),d表示 二重峰(doublet),t表示三重峰(tirplet),q表示四重峰(quartet),m表示 多重峰(multiplet)。
2.2.2 質譜儀(Matrix Assisted Laser Desorption/Ionization-Time of Flight Mass Spectroscopy,MALDI-TOF MS)
使用MALDI-TOF質譜儀型號Bruker Daltonics Briflex III (Leipzig, Germany)。首先,將基質 2,5-dihydroxybenzonic acid溶於acetonitrile共溶液 (ACN / H2O = 2:1)配製基質溶液,接著將待測高分子溶於氯仿,先將基質 溶液滴在承載鋼板上,待溶劑自然揮發後,再滴待測物溶液,接著將鋼板 置入質譜儀,以雷射游離待測高分子並藉由飛行時間儀分析各不同分子量
大小的高分子。
2.2.3 微差掃瞄卡計(Differential Scanning Calorimeter,DSC)
使用 Perkin Elmer Pyris Diamond DSC 及冷卻系統。溫度以 indium 及 tin 做校正,實驗時秤取樣品 5 ~ 10 mg,加熱及冷卻速率分別為 10℃/min 及 50℃/min,用以量測樣品之玻璃轉移溫度,而玻璃轉移溫度則取相變化曲線 現之反曲點。
2.2.4 熱重分析儀(Thermal Gravimetric Analyzer,TGA)
使用 Perkin Elmer Pyris 熱重分析儀。實驗時秤取樣品 5 ~ 10 mg,樣品 之加熱速率為 10 °C/min,並在氮氣流量 100 mL/min 下測量其熱裂解情形, 熱裂解溫度取兩趨勢切線之相交點。
2.2.5 凝膠滲透層析儀(Gel Permeation Chromatography,GPC)
使用Viscotek VE2001GPC高壓幫浦系統,偵測器為LR125 Laser refractometer Refractive Index。儀器使用三支一組之American Polymer Column,所填充之Gel尺寸大小各為 105、104和 103Å,並使用polystyrene 標 準樣品製作分子量校正曲線。測試時以THF為沖提液,並保持於 35℃的恆 溫槽中。樣品溶液之配製方式為將秤取好的2.0 mg聚合物溶於 1 mL THF中 並加一滴Toluene作為內標準品,將配置溶液超音波震盪 15 分鐘後,以 0.45 μm 的 Nylon filter 過濾後使用。 2.2.6 紫外線與可見光光譜儀(UV-Vis Spectrophotometer) 使用HP 8453 型UV-Visible光譜儀。用以偵測樣品之吸收光譜,量測時 樣品以溶劑溶解後,旋轉塗佈成膜於ITO玻璃表面量測。Film的製備:配置 樣品濃度在個別溶液中的濃度為 1.7 wt%,以 3×3×0.15 cm3的ITO玻璃 當作基材,個別試片以不同轉速旋轉 30 秒塗佈於 ITO 玻璃上。
2.2.7 掃描式電子顯微鏡(Scanning Electron Microscope,SEM)
解後,以 0.45 μm的Telfon過濾篩進行過濾,接著將水逐滴滴入配置好的溶 液中,直到少許沉澱物析出後再靜置一小時,以塑膠滴管取澄清溶液滴於 玻 璃 上 , 蓋 上 培 養 皿 讓 溶 劑 慢 慢 揮 發 , 接 著 抽 真 空 一 小 時 讓 剩 餘 溶 劑完全揮發,接著置入機台觀測。
2.2.8 原子力顯微鏡(Atomic Force Microscope,AFM)
使用VEECO Bioscope型號SZ 004-994-000 原子力顯微鏡,首先,將聚噻 吩鏈段共聚物溶於CHCl3中,濃度為17 mg / mL,製作成薄膜後觀測。
2.2.9 太陽光模擬元件量測系統(AM 1.5)
使用 YAMASHITA DENSO 型號 YSS-50A 太陽光模擬元件量測系統測
量元件的電流及電壓值。 2.3 合成部份 2.3.1 單體的合成 單體之合成流程圖見 2.4 之圖 S1。 Grignard 試劑之製備 於 250 mL 雙頸瓶內置入鎂粉 (6.56 g,0.269 ml),裝置迴流管及血清 塞,真空下以火焰除水後通以氮氣,以乾燥的針筒取除過水 ether 100 mL 注入,冰浴下以針筒取 1-bromohexane (29.7 g,0.18 ml)緩緩加入,劇烈反 應並有氣泡產生,冰浴下反應2 小時至室溫,溶液由透明無色變為鐵灰色, 確認無大量鎂粉殘存即為反應完全。 3-Hexylthiophene(1)之合成 於 250 mL 雙頸瓶內置入 3-bromothiophene (25 g,0.15 ml)及Ni(dppp)Cl2 (0.48 g,0.88 mmol),裝置迴流管及血清塞,真空下以火焰除水後通以氮氣,
以乾燥的針筒取除過水的ether 100 mL注入,將製備好的Grignard試劑以乾 燥的針筒取至加液漏斗內並緩緩加入,劇烈反應並有氣泡產生,室溫下反 應 24 小時。以去離子水中止反應,用ether/H2O作萃取,收集有機層,再用 MgSO4乾燥後濃縮。以hexane為沖提液做管柱層析純化,得黃色液體狀混合 物 , 最 終 於 9 0 o C 作 減 壓 蒸 餾 得 透 明 無 色 液 體 2 2 g , 產 率 86%。 1H-NMR (300 Mz,CDCl 3,TMS,ppm) δ:0.96 (t,3H,-CH3)、1.38 (m, 6H,-CH2-)、1.72 (m,2H,-CH2-)、2.8 (t,2H,-CH2-)、6.94 (m,2H,aromatic protons)、7.25 (m,1H,aromatic protons)。13C-NMR (300 Mz,CDCl3,TMS, ppm) δ:13.66、22.17、28.57、29.83、30.08、31.24、119.30、124.58、127.85、 142.83。MS(EI,m/z):168.10。Element Anal.:calculated for C10H16S:C, 71.37;H, 9.58;S, 19.05;found:C, 70.98;H, 9.62;S, 19.4。 2,5-Dibromo-3-hexylthiophene(2)之合成 於 250 mL雙頸瓶內置入 3-hexylthiophene (10 g,59.48 mmol)及NBS (23.2 g,65.4 mmol),裝置迴流管及血清塞,以針筒取CHCl3 60 mL及acetic acid 60 mL注入至雙頸瓶內,室溫下反應 12 小時,溶液顏色由透明黃色變 為深褐色,並且有固體沉澱析出。以等體積去離子水稀釋,接著將水層移 除,配製稀釋KOH水溶液再次洗CHCl3層,接著將水層移除,再以等體積去 離子水洗CHCl3層,收集有機層並用MgSO4乾燥後濃縮。以hexane為沖提液 做管柱層析純化,得黃色液體狀混合物。最終於 110 oC作減壓蒸餾得 透明微黃色液體 15 g,產率 79%。 1H-NMR (300 Mz,CDCl 3,TMS,ppm) δ:0.96 (t,3H,-CH3)、1.38 (m, 6H,-CH2-)、1.72 (m,2H,-CH2-)、2.8 (t,2H,-CH2-)、6.8 (s,1H,aromatic
29.46、29.53、31.54、107.90、110.27、130.94、143.99。MS(EI,m/z):326。 Element Anal.:calculated for C10H14SBr2:C, 36.83;H, 4.33;S, 9.83;Br, 49.01;found:C, 36.75;H,4.21;S, 9.24;Br, 49.8。
2.3.2 聚合物及其鏈段共聚物之合成
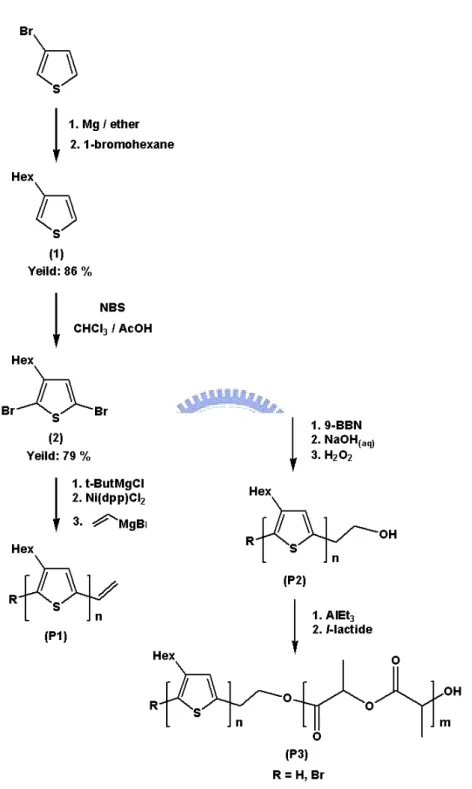
聚合物及鏈段共聚物之合成路徑如 Scheme 1 所示。其所合成之聚合物 及鏈段共聚物命名如下。
Synthesis of P1(Vinyl-terminated poly(3-hexylthiophene))
於 250 mL三頸瓶內置入 2,5-dibromo-3-hexylthiophene (10 g,61.3 mmol),固體加料瓶內置入金屬催化劑Ni(dppp)Cl2 (0.065 g,0.6 mmol),裝 置迴流管及血清塞,真空下以火焰除水後通以氮氣,以乾燥的針筒取除過 水的THF 100 mL注入,待單體溶解均勻後,以乾燥的針筒取t-butyl magnesium chloride 30.67 mL (1M溶於THF)至加液漏斗中並緩慢滴入,水浴 下加熱至 60oC反應 90 分鐘,溶液由透明無色變為黃色,待反應降至室溫, 將Ni(dppp)Cl2加入,室溫下反應15 分鐘後,溶液瞬間由黃色變為深紅色,
以乾燥的針筒取vinyl magnesium bromide 7.67 mL (1M溶於THF)加入,室溫
下反應 10 分鐘後,將溶液逐滴滴入methanol中作再沉澱,抽氣過濾收集沉
澱固體,置入 60oC烘箱烘乾後,將固體收集置入thimble filter作Soxhlet Extraction (methanol → hexane → CHCl3) 以浸洗聚合物,收集chloroform濃
縮,得金屬光澤顆粒狀固體聚合物 2 g,產率 0.54%。
1H-NMR (300 Mz,CDCl
3,TMS,ppm) δ:0.96 (t,-CH3)、1.38 (m,-CH2-)、 1.72 (m,-CH2-)、2.8 (t,-CH2-)、5.1 (d,=CH2)、5.5 (d,=CH2)、6.85 (m, -CH=)、6.94(m,aromatic protons)。
Synthesis of P2(Hydroxy-terminated poly(3-hexylthiophene)) 於 250 雙頸瓶內置入P1 (1g),裝置cork及血清塞,真空下以火焰除水 後通以氮氣,以乾燥的針筒取除過水的THF 100 mL注入,待P1 溶解均勻 後,溶液呈現橘紅色,以乾燥的針筒取9-borabicyclononane 2 mL (0.5 M溶 於THF),油浴下加熱至 45oC,反應 24 小時後,以針筒取NaOH水溶液 1 mL (6M) 注入,油浴下維持 45oC繼續反應 15 分鐘,待反應降至室溫,以針筒 取 33% 過氧化氫水溶液 1 mL緩慢滴入,白色固體析出,油浴下維持 45oC 再反應 24 小時,溶液由混濁變為澄清橘紅色。將溶液逐滴滴入methanol-H2O 共溶液(v/v = 25:1)中作再沉澱,抽氣過濾收集沉澱固體,用methanol-H2O 共溶液及acetone潤洗數次,置入 60oC烘箱烘乾後,將固體收集置入thimble filter作Soxhlet Extraction (acetone) 浸洗聚合物一天,收集thimble filter內
固體,得黑色粉末狀固體聚合物 0.92 g,產率 92%。 1H-NMR (300 Mz,CDCl 3,TMS,ppm) δ:0.96 (t,-CH3)、1.38 (m,-CH2-)、 1.72 (m,-CH2-)、2.8 (t,-CH2-)、3.02 (t,-CH2-)、3.86 (t,-CH2-)、6.94 (m, aromatic protons)。 Synthesis of P3(poly(3-hexylthiophene)-b-poly(l-lactide)) 將 25 mL高壓反應試管、P2、除過水的toluene、(3S)-cis-3,6-dimethyl- 1,4-dioxan-2,5-dion (l-lactide)及金屬催化劑triethyl aluminum (1M溶於toluene) 置入手套箱內,將P2 (0.1g)置入高壓反應瓶內並以乾燥的針筒取toluene 1.2 mL注入,加熱至 40oC至P2 完全溶解,以乾燥的針筒取triethyl aluminium (0.017 mL,0.017 mmol) 注入,將高壓反應試管封閉,加熱至 70oC反應 4 小時後,將l-lactide (0.2 g,1.38 mmol)置入高壓反應試管,將高壓反應試管
烘箱烘乾後,將固體收集置入thimble filter作Soxhlet Extraction (acetone) 浸 洗 鏈 段 聚 物 一 天 , 收 集t h i m b l e f i l t e r 內 固 體 , 將 固 體 收 集 得 黑 色粉末狀固體鏈段共聚物 87 mg。 1H-NMR(300 Mz,CDCl 3,TMS,ppm) δ:0.96(t,-CH3)、1.38(m,-CH2-)、 1.72(m,-CH2-)、2.8(t,-CH2-)、3.08(t,-CH2-)、4.36(t,-CH2-)、5.2 (q,-CH)、 6.94(m,aromatic protons)。
2.4 合成路徑示意圖
2.5 高分子有機太陽能電池元件製程 2.5.1 ITO pattern 的製作
本實驗所使用的玻璃基板為 Merck Display Technology 公司所生產的 indium-tin oxide(ITO)玻璃,其規格為 30 cm × 30 cm 大小,電阻值為 20 Ω / square,使用時切割為 6 cm × 6 cm 的正方形試片。接著製作元件的圖形 (pattern),參照以下所列步驟: (1) 上光阻: 使用長春人造樹酯股份有限公司 AF5040 之乾式光阻,將 6 cm × 6 cm 的正 方形試片置於加熱器上加熱至 150 ℃十分鐘後,將光阻貼上。 (2) 曝 光: 將貼好光阻的試片置於圖形(pattern)上方,在 300 ~ 400 nm 波長紫外光曝 光 55 秒。 (3) 顯 影: 以 1 % ~ 2 % 重量百分率濃度之碳酸鈉水溶液顯影。 (4) 蝕 刻: 將顯影過後的 ITO 玻璃基板浸入 50℃的濃鹽酸水溶液蝕刻約 30 秒。 (5) 去光阻: 以 1 % ~ 3 % 重量百分率濃度之氫氧化鈉水溶液剝除光阻。 (6) 清 理: a. Detergent 超音波震盪 10 min b. H2O超音波震盪 10 min c. NaOH(aq) 超音波震盪10 min d. D.I. water 超音波震盪 10 min e. Acetone 超音波震盪 10 min
f. IPA 超音波震盪 10 min,氮氣下吹乾 g. Oven 150˚C 12 hr 2.5.2 高分子有機太陽能電池元件製作 本研究合成了一系列不同比例的鏈段共聚物,利用其具有相分離的現 象來製作具孔洞的的吸光材料,接著將此薄膜應用至元件,為了了解此孔 洞材料的光電特性,因此製作太陽能電池多層元件。其元件結構為ITO /
PEDOT:PSS / Electron Donor Layer / Electron Acceptor Layer / Ca / Al,其中 poly(3,4-ethylene dioxythiophene)(PEDOT:PSS Baytron® P)作為電洞傳輸 層,其具有高導電度以及良好的熱穩定性,在製作多層元件時溶解於水中, 在塗佈時不會與有機層產生互溶的問題。在元件的製作上,將圖形化的ITO 玻璃切割成適合製作元件的大小 30 mm × 30 mm,在經過清洗後使用。在 溶液配置的部份,將聚噻吩高分子溶於氯仿(20 mg / mL) (R1),聚噻吩鏈段 共聚物溶於氯仿(17 mg / mL) (R2),再以超音波震盪使其完全溶解後,以 0.45 μm 的 Telfon 過濾篩進行過濾。
Table 2-1. Preparation of polymer solution
Sample Weight Solvent
R1 P3HT 20 mg CHCl3
R2 P3HT-PLLA (A3) 17 mg CHCl3
元件中每一層高分子材料均以旋轉塗佈的方式製作,首先將電洞傳輸 材料PEDOT:PSS以 3000 rpm旋轉塗佈 30 秒後,於 140°C下烘烤一小時。將 塗佈好PEDOT:PSS之元件置入手套箱內,將配置好的R1 溶液以 3000 rpm旋
轉塗佈,將未乾的R1 層置入培養皿中讓溶劑慢慢揮發,接著以 60°C烤 5 分 鐘讓剩餘溶劑完全揮發,接著滴入 1,2-propylene作為緩衝層以 6000 rpm旋 轉塗佈,以避免製備R2 層時與R1 層產生互溶的情形,接著將配置好的R2 溶液以 3000 rpm旋轉塗佈,接著在真空下以 190°C加熱 5 分鐘讓剩餘有機 溶劑完全揮發,並將元件靜置冷卻。將製作好的元件置於0.2 N NaOH 溶液 中(methanol / H2O = 20 / 80)進行裂解聚乳酸片段。裂解過程完成後,以 乾淨甲醇潤洗,並將元件置於真空中抽去有機溶劑。接著將元件送入金屬 蒸鍍機之腔體內進行蒸鍍,先將碳六十(20 nm)蒸鍍至高分子孔洞材 料上,接著蒸鍍陰極金屬鈣(30 nm)及鋁(100 nm)。
第三章 結果與討論 3.1 單體結構鑑定 單體(化合物 2)之合成步驟見Scheme 1,結構鑑定如下說明。化合物 2 的1H-NMR圖譜,其結構特徵為噻吩環上長碳側鏈旁有一組單重峰訊號(δ = 6.94 ppm),單體的長碳鏈上碳上的氫分為四群,分別為三重峰訊號(δ = 0.96 ppm)、多重峰訊號(δ = 1.38 & 1.72 ppm)、三重峰訊號(δ = 2.8 ppm),因此, 所有測量得到的氫訊號皆符合預期。對照其MASS的圖譜,其所顯示的分子 量位置於 326 亦與理論分子量 326.091 相同,由此可知化合物 2 被合成出 來。 3.2 聚合物 P1、P2 及鏈段共聚物 P3 之合成與結構鑑定 聚合物P1 是利用格林納置換法進行聚合[33,37]。將單體(化合物 2)一當 量,加入等當量莫耳數之起始劑t-ButMgCl及 0.02 莫耳比的金屬催化劑 Ni(dppp)Cl2進行聚合反應,利用改變聚合的時間來控制聚合條件,因此精 準控制起始劑及催化劑的反應時間是必要的條件。為了使起始劑及催化劑 在聚合過程能夠保持活性,必須將反應溶液盡量除水氣及氧氣,且起始反 應在 45oC下加熱進行一至二小時,以確保單體皆能夠與起始劑完全反應, 接著加入金屬催化劑Ni(dppp)Cl2進行聚合反應,待反應進行五分鐘後即可 加入終止反應的格林納試劑,根據加入的格林納試劑可以得到不同的末端 雙鍵的不對稱高分子[34,35]。 聚合物 P2 首先是利用氫硼化反應(hydroboration reaction)將氫化硼的硼 -氫鍵加成至烯烴之碳-碳雙鍵上,氫硼化反應的機制如圖 3-1 所示,硼元 素有一親電性的 2p 空軌域可以讓烯烴雙鍵的 π 電子對進入其空軌域形成 π
的鍵結,最後硼上的一個氫轉移至雙鍵另一碳上形成四中心的中間態。
Figure 3-1. Mechanism of hydroboration reaction
接著反應會加入雙氧水及氫氧化鈉,目的是將硼氫官能基轉換成氫氧 官能基,如圖 3-2 所示。雙氧水及氫氧化鈉會先反應產生過氧化氫陰離子 (step 1),此陰離子會攻擊與高分子形成不穩定中間體的硼鍵結(step 2), 進而和硼鍵結的碳會轉移攻擊氧,轉化成較穩定的中間態,此中間態再與 水反應形成含氫氧官能基的高分子(step 3)。 Step 1: Step 2:
Step 3:
Figure 3-2. Mechanism of hydroxylation
鏈段共聚物P3 是利用配位插入開環聚合反應進行聚合。將聚合物P2 一 當量,加入等當量莫耳數之催化劑triethyl aluminum進行聚合反應,利用改 變聚合的時間來控制聚合條件,因此精準控制起始劑的起始反應時間及乳 酸的開環反應時間是必要的條件。為了使起始劑在聚合過程能夠保持活 性,必須將反應溶液盡量除水氣及氧氣,少數的水氣可能造成反應的高分 子鏈引發終止而使得反應難以得到有效控制和聚合,所以整個反應過程在 手套箱內進行以利控制水氣及氧氣的含量。起始反應使用等當量莫耳數的 triethyl aluminum在 70oC下加熱進行,反應過程產生乙烷,反應需進行四小 時以確保P2 與起始劑完全反應成為高分子起始劑(macroinitiator),催化劑的 量須謹慎控制,使用量過少則活性點數目少,不足以引發所有單體的聚合, 使用量過多則活性點太多,能夠增長的單體數目變少。乳酸的開環聚合反 應需加熱至 110oC,以確保與高分子起始劑能夠進行配位插入開環反應,此 外,聚合時間對聚乳酸的分子量也有一定影響,聚合時間短,聚合反應沒 有進行完全,所得到的聚乳酸分子量較低,聚合時間過長,所得到聚乳酸 又 受 到 一 定 程 度 的 熱 降 解 , 且 此 狀 況 在 聚 合 反 應 溫 度 較 高 時 更 為 嚴
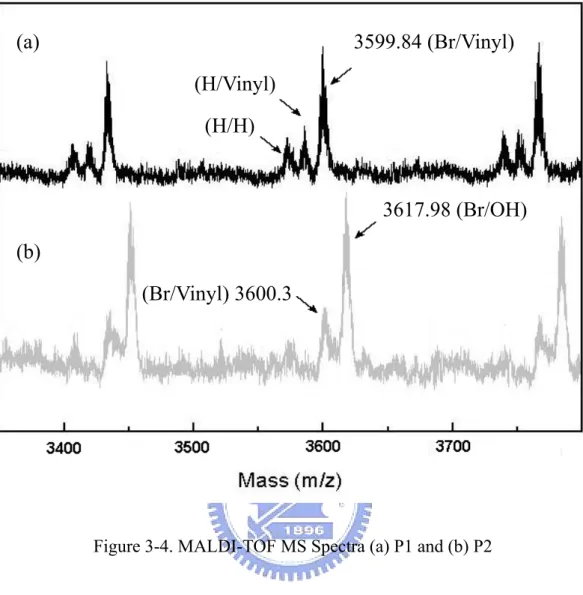
Figure 3-3. Mechanism of Ring-Opening polymerization 有關配位插入開環聚合反應的反應機制,如圖 3-3 所示,催化劑一般為 有機金屬化合物,如烷基金屬和烷氧基金屬化合物。這類反應一般認為單 體上氧原子與催化劑金屬的空軌道配位絡合,乳酸單體在金屬-碳鏈或 金屬-烷氧鏈上進行插入、增長。 將合成得到的不對稱的聚噻吩高分子P1 及P2 用基質輔助激光解析飛行 時間質譜(MALDI-TOF MS)做末端官能基的分析[35,43],如圖 3-4 所示。經由 MALDI-TOF MS可得知P1 及P2 的分子量分佈及精確的分子量,每一峰值間 的差值約為 166,即聚噻吩高分子單體 3-己烷噻吩的分子量,每一根鋒 值可由下列計算式得到末端官能基的分子量。 Mw高分子 = Mw單體 × n + Mw末端官能基 *n 表示相連接的單體數目 以鋒值 3599.8 為例,3599.8 = 166 × 21 + 113.8,113.8 為Br / vinyl末端 官能基的分子量總和,由此可推知不對稱的聚噻吩高分子P1 被合成出來, 此高分子的兩邊末端一邊為Br一邊為vinyl官能基,此外,在圖 3-4a中,另 兩支峰值分別來自於H / H及H / vinyl末端官能基的高分子。利用圖 3-4b
(a) 3599.84 (Br/Vinyl) (H/Vinyl) (H/H) 3617.98 (Br/OH) (b) (Br/Vinyl) 3600.3
Figure 3-4. MALDI-TOF MS Spectra (a) P1 and (b) P2
可以比較P1 經氧化反應前後分子量的差異,峰值 3617.98 為接上 -OH官能 基的聚噻吩高分子,3617.98 - 3600.3 ~ 18 為兩個H一個O的分子量總和,即 雙鍵經過氧化接上 -H及 -OH,此高分子的兩邊末端一邊為Br一邊為 OH官能基,由此可推知不對稱的聚噻吩高分子P2 被合成出來。 將合成得到的P1 ~ P3 用核磁共振儀測高分子上氫的訊號來分析末端的 官能基,如圖 3-5 所示,在δ = 5.109、5.146 及 5.484、5.541 ppm兩處分別 為末端vinyl官能基的其中一組雙重峰氫訊號(i),另一組氫訊號為δ = 6.85 ppm的多重峰氫訊號(h),此外,聚噻吩高分子P1 的氫訊號與單體(化合物 2) 相似,其結構特徵為 噻 吩環上長碳側鏈旁有一組單重峰訊號(δ = 6.94
ppm)、多重峰訊號(δ = 1.38、1.72ppm)、三重峰訊號(δ = 2.8 ppm),由此可 推知具有vinyl官能基的聚噻吩高分子(P1)被合成出來。 Figure 3-5. 1H NMR Spectra of P1 利用相同方式測量P2 及P3 聚噻吩高分子,如圖 3-6a所示,在δ = 3.86ppm處為末端 -OH官能基相鄰 -CH2- 上的三重鋒氫訊號(k),在δ = 3.02 處為末端 -CH2-OH官能基相鄰 -CH2- 上的三重鋒氫訊號(j),此外,在圖 4-6 中兩處δ = 5.109、5.146 及 5.484、5.541 ppm並無vinyl官能基上氫的訊號, 由此可推知末端的雙鍵皆轉變成OH官能基,此外,聚噻吩高分子P2 的氫訊 號與P1 相似,其結構特徵為噻吩環上長碳側鏈旁有一組單重峰訊號(δ = 6.94 ppm),單體的長碳鏈上碳上的氫分為四群,分別為三重峰訊號(δ = 0.96 p p m ) 、 多 重 峰 訊 號 ( δ = 1 . 3 8 、 1 . 7 2 p p m ) 、 三 重 峰 訊 號 ( δ = 2 . 8
ppm)。由噻吩環結構上的氫訊號皆無產生變化來得知,經由硼氫化反應再 經由過氧化氫與氫氧化鈉的氫氧化的反應,並不會破壞噻吩環上的雙鍵結 構。如圖 3-6b為鏈段共聚物(P3)的圖譜,在δ = 5.2 ppm處為聚乳酸 -CH官 能基上四重峰氫訊號(o、q),在圖 3-6a 中δ = 3.86 ppm處的 -CH2- 上的三 重峰氫訊號(k),由於進行配位插入開環聚合,高分子末端的醇基反應形 成酯基官能基,在圖 3-6b 中的化學位移改變至 δ = 4.36 ppm(m)。 在圖 3-6a 中δ = 3.02 ppm處的 -CH2- 上的三重峰氫訊號(j),由於化學 位移環境並無太大改變,在圖 3-6b 中δ = 4.36 ppm處可以偵測到三重鋒的 氫訊號(l),最後,其它來自於聚噻吩的氫訊號與P1 及P2 相似,其結構特徵 為噻吩環上長碳側鏈旁有一組單重峰訊號(δ = 6.94 ppm),單體的長碳鏈上 碳上的氫分為四群,分別為三重峰訊號(δ = 0.96 ppm)、多重峰訊號(δ = 1.38、1.72 ppm)、三重峰訊號(δ = 2.8 ppm)。藉由比較P2 及P3 的1H NMR 訊號,可得知聚噻吩鏈段共聚物被合成出來。 3.3 分子量分析 本研究目的是為了合成出具有自組裝相分離現象之有機太陽能電池材 料,聚噻吩衍生物為目前最被廣泛利用作為研究有機太陽能電池的材料, 聚噻吩高分子可作為吸收太陽光的材料產生激子,因此本研究選擇聚噻吩 作為鏈段共聚物的主體,利用合成方法結合聚乳酸高分子。在許多文獻上, 可以利用不同實驗的方式來調控鏈段共聚物製作成薄膜後的型態,鏈段共 聚物可利用自組裝的方式形成各種型態規則排列的奈米結構。 由於聚噻吩鏈段共聚物之比例會影響自組裝的型態,且必須考量製作 成的薄膜需為聚噻吩作為基體(matrix)亦稱模板(template),在聚乳酸的片段 需為分子量較小的部份,而聚噻吩的片段為分子量較大的部份。聚合物A1 及B1 為聚噻吩高分子,其末端經由合成方法修飾 -OH官能基(結構參考圖
S1 之P2)來作為配位插入開環聚合反應中乳酸單體的開環起始劑。高分子 (A1 ~ A3 及B1 ~ B2)的分子量是利用凝膠滲透層析儀來測量,如表 3-1 所 示,A1 分子量為 6662,PDI值為 1.15,B1 分子量為 9530,PDI值為 1.42。 經由配位插入開環聚合反應可以成功合成出聚噻吩鏈段共聚物(A2、A3 及 B2),而聚噻吩高分子與聚乳酸高分子之相對應分子量比例(number average molecular weight ratio)可以經由計算得聚乳酸的重量比(wPLLA)來得知,分別 為 0.17 及 0.25,換算比例為一比五及一比三。
Table 3-1. Characterization of P3HT-b-PLLA
Sample Mn(g/mol) P3HT Mn(g/mol) PLLA Mn(g/mol) total Mw/ Mn wPLLA A1 P3HT-OH 6662 6662 1.15 A2 P3HT-PLLA(1) 6662 1385 8047 1.23 0.17 A3 P3HT-PLLA(2) 6662 2277 8939 1.37 0.25 B1 P3HT-OH 9530 9530 1.41 B2 P3HT-PLLA(1) 9530 1415 10946 1.85 0.13
wPLLA = Mn(PLLA) / [Mn(PLLA) + Mn(P3HT)]
聚噻吩片段及聚乳酸片段之分子量控制是經由合成時間控制,較長的 反應時間可得到較大分子量的高分子,利用格林納置換法聚合反應合成的
聚噻吩僅需要五分鐘時間即可得到分子量 6662 的高分子,反應時間相當快
速,而合成聚乳酸片段則需相對較長的反應時間,才能合成出適合的分子 量大小。
3.4 熱性質分析 太陽能電池是藉由吸收太陽光而得到自由電子,然而在長時間照射 下,溫度會逐漸提高而破壞元件的特性,因此材料本身的穩定性相當重要, 穩定性可分為化學性及物理性的穩定。化學性的穩定及材料本身的化學結 構是否產生化學鍵的斷裂而影響材料本質,進而造成元件效率降低。物理 性的穩定即材料在製作過程可能產生的缺陷,缺陷容易產生局部漏電流或 是電荷的再結合,這些現象皆會導致元件效率的降低。為了得到高分子材 料對於熱的穩定性,藉由TGA及DSC來獲得這方面資訊。聚合物A1 ~ A3 及 B1 ~ B2 之TGA及DSC得到的裂解溫度、Td及Tg值列於下表 3-2 中。
Table 3-2. Thermal analysis of block copolymers
TGA DSC Sample Td(˚C) Tg(˚C) A1 P3HT-OH 350 5.1 A2 P3HT-PLLA(1) 255 5, 43.4 A3 P3HT-PLLA(2) 243 5.1,47.5 B1 P3HT-OH 352 5.9 B2 P3HT-PLLA(1) 258 5.4,44.2 由 TGA 的溫度來判斷,含有乳酸片段的共聚物裂解溫度較低,約為 243 ~ 258 ˚C,且含有較大分子量乳酸片段裂解溫度更低,其原因可能來自於聚 乳酸片段較易裂解。 經由DSC的測量得知含有poly(l-lactide)的共聚物P2、P3 及P5 的Tg約為 43 ~ 47 ˚C,此玻璃轉移溫度來自於poly(l-lactide),而另一個玻璃轉移溫度
來自於poly(3-hexylthiophene)約為 5.1 ~ 5.9 ˚C,不含有poly(l-lactide) 的聚噻吩高分子無另外一個玻璃轉移溫度。 3.5 紫外光-可見光吸收光譜之光學性質 本研究中所合成之鏈段共聚物主要是以聚噻吩作為比例較多的部份, 其紫外光-可見光之吸收光譜主要在可見光範圍(350 ~ 650 nm),如圖 3-7 所 示,測量試片C1 ~ C3 之紫外光-可見光光譜最大特徵峰列於下表 3-3。在薄 膜製作上首先須配製待測物溶液,將高分子P3HT-OH(A1)及P3HT-PLLA(2) (A3)溶於氯仿中,濃度為 17 mg / mL,再以超音波震盪使其完全溶解後, 加熱至 45°C約十五分鐘,以 0.45 μm的Telfon過濾篩進行過濾,以 3000 rpm 旋 轉 塗 佈 於 石 英 基 板 上 , 接 著 在 1 9 0 ° C 烤 1 0 ~ 1 5 分 鐘 讓剩餘有機溶劑完全揮發。 C1 為聚噻吩高分子製作成的薄膜,所測得之吸收位置較溶液態有紅位 移的現象,最大吸收的特徵峰由 450 nm移至 510 nm,此紅位移現象的產生, 是由於分子在薄膜狀態時,其分子鏈與分子鏈之間的距離較溶液態來的靠 近,其中彼此的堆疊造成高分子鏈的能階形成簡併態(Degenerency),因此 薄膜時的最大吸收鋒位置會產生紅位移的現象。薄膜態的紫外光-可見光之 吸收範圍相當寬廣,從 350 ~ 650 nm 皆有吸收訊號,在 599 nm位置為聚噻 吩高分子鏈共振的吸收,在溶液態則無此峰值被偵測到。C2 使用聚噻吩鏈 段共聚物,由於聚乳酸片段在可見光範圍並無吸收訊號,所以吸收光譜的 分佈相當相似於C1,在吸收強度大小則較C1 小,由於C2 為聚噻吩鏈段共 聚物所製作的薄膜,在試片的吸收面積分佈上含有聚乳酸高分子,因而影 響到薄膜的吸收強度。C3 利用與C2 相同的方式來製作薄膜,接著經由裂解 方式除去聚乳酸片段,其吸收光譜的分佈與吸收強度大小皆與C2 相似,未
Table 3-3. UV-Vis absorption Sample UV-Vis (nm) Film UV-Vis (nm) Solution C1 P3HT-OH 510,599 450 C2 P3HT-PLLA(2) 510,595 450
C3 P3HT-PLLA(2) after degradation 510,596
*C2 及 C3 為利用 P3HT-PLLA(2) (A3)來製作薄膜 裂解去除聚乳酸片段之後,薄膜表面因而產生許多孔洞,這些孔洞的產生 來自於原先聚乳酸片段在薄膜上所佔據的體積,由於孔洞無任何的吸收訊 號,因此,對於吸收光譜的分佈與吸收強度無任何顯著影響。 由於聚噻吩鏈段共聚物所含聚噻吩比例較聚噻吩高分子少,因此在製 作 成 薄 膜 後 的 吸 收 強 度 降 低 , 可 能 導 致 在 製 作 元 件 後 由 於 吸 收 強 渡變弱而影響的光電流產生。