E
ffect of Roaming Transition States upon Product Branching in the
Thermal Decomposition of CH
3
NO
2
R. S. Zhu,
†P. Raghunath,
‡and M. C. Lin*
,†,‡†Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States
‡Center for Interdisciplinary Molecular Science, Department of Chemistry, National Chiao Tung University, Hsinchu, Taiwan 300
*
S Supporting InformationABSTRACT: The kinetics for the thermal unimolecular decomposition of CH3NO2 and its structural isomer CH3ONO have been investigated by
statistical theory calculations based on the potential energy surface calculated at the UCCSD(T)/CBS and CASPT3(8, 8)/6-311+G(3df,2p) levels. Our results show that for the decomposition of CH3NO2at pressures less than 2 Torr, isomerization to CH3ONO via the recently located roaming transition
state is dominant in the entire temperature range studied, 400−3000 K. However, at higher pressures, the formation of the commonly assumed products, CH3+ NO2, becomes competitive and at pressures higher than 200
Torr the production of CH3+ NO2is exclusive. The predicted rate constants for 760 Torr and the high-pressure limit with Ar as
diluent in the temperature range 500−3000 K, producing solely CH3+ NO2, can be expressed respectively by kd760(CH 3NO2) =
2.94× 1055T−12.6exp(−35500/T) s−1and kd∞(CH3NO2) = 5.88× 1024T−2.35exp(−31400/T) s−1. In the low pressure limit, the
decomposition reaction takes place exclusively via the roaming TS producing internally excited CH3ONO, giving rise to both CH3O + NO and CH2O + HNO with the second-order rate constant kd0(CH3NO2) = 1.17× 1031T−10.94 exp(−32400/T) cm3
molecule−1s−1. For CH3ONO decomposition, a new roaming transition state connecting to the CH2O + HNO products has been located, lying 6.8 kcal/mol below the well-known four-member ring tight transition state and 0.7 kcal/mol below CH3O +
NO. The rate constants predicted by similar calculations give rise to the following expressions for the thermal decomposition of CH3ONO in He: kd760(CH3ONO) = 8.75 × 1041T−8.97exp(−22600/T) s−1 and kd∞(CH3ONO) = 1.58 × 1023T−2.18
exp(−21100/T) s−1in the temperature range 300−3000 K. These results are in very good agreement with available experimental data obtained under practical pressure conditions. The much different branching ratios for the formation of CH3O + NO and
CH2O + HNO in the decomposition of both CH3NO2and CH3ONO are also given in this work.
1. INTRODUCTION
The thermal decomposition of CH3NO2, a prototype energetic
material that contains the key [C,H,O,N] ingredients of nitro and nitro-amine compounds, is relevant not only to the combustion of energetic materials but also to its related recombination reaction CH3 + NO2, which is relevant to the
pollution chemistry in the troposphere. There have been many studies on the kinetics and mechanism of the unimolecular decomposition reaction of CH3NO2 and the related CH3 +
NO2reaction.
1
The results of some of the key measurements will be cited for comparison with the predicted values later. To date, the thermal decomposition of CH3NO2has been nearly exclusively assumed to take place by CN splitting:
→ +
CH NO3 2 CH3 NO2 (1)
Other potential decomposition channels via the well-known nitro-nitrite isomerization and the H-atom migration and dehydration processes producing HCNO via CH2N(O)OH all occur by tighter and energetically less favorable transition states2−6 and thus cannot compete with the CN breaking process with 60 kcal/mol dissociation energy aided by its loose transition state without an intrinsic barrier.
In 1986, an interesting result on the detection of CH3O +
NO was reported by Wodtke, Hinsta, and Lee7in their IRMPD (infrared multiphoton decomposition) of CH3NO2 under a
collision-free molecular beam condition using a high-power CO2 laser. This observation could not be satisfactorily
accounted for theoretically for nearly a quarter of a century with reliable computational methods based on the nitro-nitrite isomerization mechanism because the process is both entropi-cally unfavorable and enthalpientropi-cally inaccessible in the IRMPD experiment.3−6
In 2004, a new mechanism for molecular decomposition, termed “roaming mechanism” was first discovered by a joint experimental−theoretical study on the photodissociation of H2CO.8On the basis of the systems studied in the last several
years,9−12roaming transition states (RTS) are typically found to locate at the radical−radical separations of about 3−4 Ǻ, where the radicals may sample orientations leading to
Special Issue: Joel M. Bowman Festschrift
Received: January 31, 2013
Revised: June 10, 2013
Published: June 13, 2013
intramolecular abstractions with barriers around 1−2 kcal/mol below the lowest bond cleavage paths. The implication of this finding to the initiation mechanisms of combustion and explosion of energetic materials is profound because the thermal decomposition of energetic materials typically occurs via the lowest energy paths with favorable entropic changes.
In 2009, motivated by the roaming mechanism as aforementioned, we attempted to search for and successfully located a very loose roaming type transition state for the isomerization of CH3NO2 to CH3ONO using different
computational methods.13 The existence of the RTS below the CH3 + NO2 dissociation limit allowed us to reasonably
explain the experimental observation cited above by Wodtke, Hinsta, and Lee7in their IRMPD of CH3NO2with as much as
40% CH3O yield.
On the grounds that the thermal decomposition process occurs energetically close to that of IRMPD, the initial formation of CH3O + NO and potentially CH2O + HNO in
the thermal unimolecular decomposition of CH3NO2may be competitive with the commonly assumed CH3+ NO2product
formation. In this work, we have accordingly investigated computationally the kinetics for the thermal decomposition of CH3NO2on the basis of the potential energy surface (PES) reported previously13employing the variational RRKM theory to predict the formation of the three key product pairs CH3+ NO2, CH3O + NO, and CH2O + HNO as functions of
temperature and pressure; these results may be helpful for simulations of the decomposition and combustion of CH3NO2,
CH3ONO and larger nitro- and nitramine compounds.
2. COMPUTATIONAL METHODS
The computational approach for the mapping of PES including our extensive search for the roaming transition state (RTS1) and the calculation of the microcanonical rate constants for the fragmentation of excited CH3NO2and CH3ONO as functions
of energy have been described in detail previously (Figure 1).13
For the decomposition of CH3ONO, another roaming
transition state (RTS2) for the production of CH2O + HNO has been located in this work; it is distinctively different from the well-known four-center tight transition state (TS3). In RTS2 the dissociating NO group roams around the CH3O
fragment and abstracts one of the H atoms from the CH3group to yield CH2O + HNO (see SI-3, Supporting Information).
The rate constants were computed with a variational RRKM code (Variflex14) by solving the master equation15,16involving
multistep vibrational energy transfers for the excited intermediate CH3NO2* or CH3ONO* based on the PES calculated at the UCCSD(T)/CBS//UB3LYP/6-311+G-(3df,2p) and CASPT3(8,8)/6-311+GUCCSD(T)/CBS//UB3LYP/6-311+G-(3df,2p) levels of theory, as shown in Figure 1.
The energies used in the rate constant calculations are mainly based on the UCCSD(T)/CBS//UB3LYP/6-311+G(3df,2p) level; among them, the energy of the loose RTS1 obtained at the CASPT3(8,8)/6-311+G(3df,2p) level was used, similar to those of the variational transition states (VTS’s) for bond breaking processes described below. Frequencies and rotational constants used in the calculations are taken from the UB3LYP/ 6-311+G(3df,2p) level. The Cartesian coordinates of the related species are listed in Supporting Information (SI-1). The variational dissociation curve for CH3NO2→ CH3+ NO2 was calculated at the CASPT2(8,8)/6-311+G(3df,2p)// UB3LYP/6-311+G(3df,2p) level to cover the C−N bond with separation from 1.499 to 4.699 Å, with an interval step size of 0.2 Å, other geometric parameters were fully optimized. The dissociation curve can befitted to the Morse potential function E(R) = De[1− exp(−β(R − Re))]2, which was employed to
approximate the minimum energy path for the variational transition state of CH3NO2 → CH3 + NO2. In the above
equation, R is the reaction coordinate (i.e., the distance between the two bonding atoms; the C−N in this work), Deis
the bond energy excluding zero-point energy, and Re is the
equilibrium value of R. The computed potential energies could be fitted reasonably to the Morse potential function with the parameterβ = 2.223 Å−1 (see SI-2, Supporting Information). The barrierless dissociation of CH3ONO to CH3O + NO was calculated similarly with β = 2.552 Å−1. For both barrierless processes, the number of a variational transition quantum states, N†EJ, was given by the variationally determined minimum
in NEJ(R), as a function of the bond length along the reaction
coordinate R, which was evaluated according to the variable reaction coordinate flexible transition state theory.17,18 The estimation of the transitional mode contribution to the transition state number of states for a given energy is evaluated via Monte Carlo integration with 10 000 configuration numbers. The numbers of states for RTS1 and RTS2 are evaluated according to the rigid-rotor harmonic-oscillator assumption, the smaller frequencies (less than 50 cm−1) were treated as a free or hindered rotor.
For the collisional energy transfer calculations, the L-J parameters required for the RRKM calculations for the quenching of CH3NO2/CH3ONO are taken from ref 19, ε/k = 290.4 K andσ = 4.347 Å, and those for He and Ar, ε/k = 10 and 114 K andσ = 2.55 and 3.47 Å, respectively, were taken from the literature.20 The energy−transfer process was computed on the basis of the exponential down model with a ⟨ΔE⟩down value (the mean energy transferred per collision) of
150 and 400 cm−1for He and Ar, respectively.
3. RESULTS AND DISCUSSION
3.1. Rate Constants for the Decomposition of CH3NO2. On the basis of the PES summarized in Figure 1,
the thermal decomposition of CH3NO2 may take place
preferentially by the following three low-energy paths: Figure 1.Energy diagrams (in kcal/mol. at 0 K) for the CH3NO2
decomposition, computed at the UCCSD(T)/CBS level. The values for CH3NO2 and RTS1 in the parentheses were calculated at the CASPT3(8,8)/6-311+G(3df,2p)//CASSCF(8,8)/6-311+G(d) level.
→ + → → + → + CH NO CH NO (1) CH NO CH ONO CH O NO (2) CH O HNO (3) 3 2 3 2 3 2 3 3 2
The bond-breaking processes in reactions 1 and 2 occur by variational transition states that were computed at the CASPT2(8,8)/6-311+G(3df,2p)//UB3LYP/6-311+G(3df,2p) level of theory as aforementioned, whereas the first step in reaction 2 is mainly controlled by RTS1 in Figure 1. For reaction 3, as aforementioned, we have located a new roaming transition state, RTS2, lying 0.7 kcal/mol below CH3O + NO
predicted at the CCSD(T)/CBS//UB3LYP/6-311+G(3df,2p) level. It should be noted that the possibility of existence of the loose TS similar to RTS1 in the nitro−nitrite isomerization reaction had also been reported by Mckee4 in 1989 for the CH3NO2isomerization process using the CAS (4,4)/6-31G(d)
method; however, the loose TS was found to lie 10.0 kcal/mol above the CH3+ NO2limit at the
MRCI/6-31G(d)//CAS/6-31G(d) level. Subsequently, Saxon and Yoshimine6 had also located a loose TS at the MRCI(7,7)/6-31G (d)//CAS(4,4)/4-31G level of theory; their TS was found to lie 56.7 kcal/mol above CH3NO2 and 0.4 kcal/mol below the CH3 + NO2 asymptote, energetically akin to our RTS1 value computed at the CASPT2 level of theory. The well-known nitro−nitrite isomerization path occurring via TS2, however, lies above the CH3 + NO2 dissociation limit by 9.6 kcal/mol, which is
consistent with the results of previous calculations.3−6 The contribution from this path should therefore be kinetically negligible.
Experimentally, most of the CH3NO2 decomposition rates
were measured in the pressure range 150−30400 Torr Ar,21−29 the reported main products are CH3 + NO2. Due to the
location of the RTS1 below the CH3+ NO2dissociation limit,
CH3NO2 can isomerize to cis-CH3ONO, which further decomposes to give CH3O + NO and CH2O + HNO, as
shown in Figure 1. Figure 2 shows the pressure dependent rate constants predicted at 1000 and 1500 K for the two competing reactions 1 and 2 in the pressure range 10−5to 760 Torr Ar. At both temperatures, the decomposition is found to be dominated by the production of CH3ONO via RTS1 below
about 2 Torr pressure, above which the formation of CH3 +
NO2 becomes competitive and dominant at P > 200 Torr. Figure 3 presents the branching ratios for CH3ONO formation
via RTS1 in the decomposition of CH3NO2at various pressures
in the temperature range 400−3000 K. The results most vividly illustrate the competition between the two decomposition reactions; at pressures lower than 2 Torr, isomerization of CH3NO2 to CH3ONO via RTS1 is dominant in the whole
temperature range, whereas at P > 200 Torr, more than 90% of the products are CH3+ NO2. Our results can thus explain why
most of the investigators did not observe the CH3O + NO products under practical, higher pressure conditions.
The calculated first-order rate constants for the decom-position of CH3NO2 are plotted in Figure 4 for comparison
with available literature data. Calculated values at 760 and 30400 Torr Ar and at the high-pressure limit are plotted as dotted, dashed, and solid lines in the figure, indicating that, under most experimental conditions, our predicted values lie within the scatter of experimental data.
The rate constants for 760 Torr and the high-pressure limit with Ar dilution in the temperature range 500−3000 K, Figure 2.Predicted rate constants for the formation of CH3+ NO2
and CH3ONO via RTS1 in the decomposition of CH3NO2at 1000 and 1500 K under different pressures.
Figure 3.Predicted branching ratios for the formation of CH3ONO via RTS1 at pressures between 10−5 and 760 Torr for the decomposition of CH3NO2in the temperature range 400−3000 K.
Figure 4.Comparison of the predicted first-order rate constant for CH3NO2 decomposition with experimental values in Ar. Dotted, dashed, and solid lines are the predicted values at 760 and 30400 Torr Ar and infinite pressure. Symbols in the legend are the experimental values.
representing the values for production of CH3 + NO2, can be expressed respectively by = × − − − kd760(CH NO )3 2 2.94 1055T 12.6exp( 35500/ ) sT 1 = × − ∞ − − kd (CH NO )3 2 5.88 1024T 2.35exp( 31400/ ) sT 1
The CH3NO2 isomerization rate constant in the
collision-controlled, second-order, low-pressure limit, which represents exclusively for formation of CH3ONO, can be given by
= × − − − − k (CH NO ) 1.17 10 T exp( 32379/T) cm molecule s d 0 3 2 31 10.94 3 1 1
As aforementioned and depicted in Figure 1, the barrier of RTS2 is 6.8 kcal/mol lower than that of TS3, lying below the CH3O + NO dissociation limit by 0.7 kcal/mol. On the basis of this new RTS2, the calculated rate constants under the conditions of 10−5 to 760 Torr Ar and 300−3000 K temperature for CH3NO2 → CH3ONO* → CH3O + NO
(k2) can be represented as
= × − − −
k2 8.91 1019T 1.84exp( 30600/ ) sT 1
and CH3NO2→ CH3ONO* → CH2O + HNO (k3) represents
as
= × − − −
k3 2.15 1017T 0.75exp( 30200/ ) sT 1
The branching ratios of these two channels are shown in Figure 5. Combining Figures 3 and 5, one can see that, at low
pressures, the formation of CH2O + HNO from the
fragmentation of internally excited CH3ONO formed by the isomerization via RTS1 is dominant in the whole temperature range.
3.2. Rate Constants for the Decomposition of CH3ONO. We have also taken the opportunity to calculate
the rate constants for the competitive decomposition of CH3ONO to CH3O + NO and CH2O + HNO. The predicted rate constants for the products of CH3O + NO at 100 and 760
Torr and the high-pressure limit are plotted in Figure 6 for comparison with available literature data. The predicted values are seen to be in good agreement with experimental data measured with different diluents,30−37 as shown in the figure,
including He for which the rate constants measured at 710 Torr pressure with the extrapolated high pressure limit have been reported.30The predicted results for 760 Torr He and the high-and low-pressure limits in the temperature range 300−3000 K can be expressed respectively by
= × − − − kd760(CH ONO)3 8.75 1041T 8.97exp( 22600/ ) sT 1 = × − ∞ − − kd (CH ONO)3 1.58 1023T 2.18exp( 21100/ ) sT 1 = × − − − − k T T (CH ONO;He) 8.91 10 exp( 22300/ ) cm molecule s d0 3 19 7.92 3 1 1
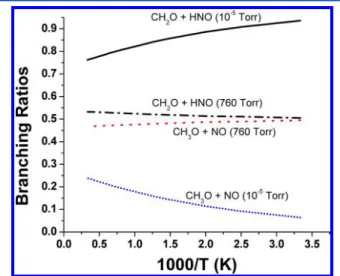
On the basis of RTS2, the branching ratios for the production of CH2O + HNO and CH3O + NO have been
calculated and shown in Figure 7 as a function of temperature at 10−5and 760 Torr Ar pressure. The results show that at low temperatures and low pressures, the main products are CH2O +
HNO; however, at P = 1 atm, two channels are very
Figure 5.Predicted branching ratios for the formation of CH3O + NO and CH2O + HNO in the thermal decomposition of CH3NO2 via CH3ONO at pressure 10−5to 760 Torr Ar.
Figure 6.Comparison of the predictedfirst-order rate constant for the decomposition of CH3ONO→ CH3O + NO with the experimental values in He. Dotted, dashed and solid lines are the predicted values at 100 and 760 Torr He and infinite pressure. Symbols in the legend are the experimental data.
Figure 7.Predicted branching ratios for the formation of CH3O + NO and CH2O + HNO in the thermal decomposition of CH3ONO at pressure 10−5and 760 Torr Ar.
competitive. It is worth noting that the branching ratios predicted for the formation of these two product channels in the thermal decomposition of CH3NO2(Figure 5) and those in the thermal decomposition of CH3ONO (Figure 7) are
distinctively different. The former values are pressure-independent between 10−5 and 760 Torr, reflecting the fragmentation of highly excited CH3ONO with 57.1 kcal/mol of internal energy via RTS1 (akin to a chemical activation process), whereas the latter values reflect the collisional activation of CH3ONO from the ground up as in a typical
thermal unimoleular decomposition reaction.
Due to the very flat potential energy surfaces around the roaming transition states RTS1 and RTS2, accurate determi-nation of relative contributions to the dissociation products from these roaming channels versus the conventional product channels needs extensive trajectory simulations based on a complete potential surface which is beyond the scope of this work.
4. CONCLUSION
In this study, the kinetics for the thermal unimolecular decomposition of CH3NO2 and its structural isomer
CH3ONO have been calculated by variational RRKM theory based on the PES recently computed by two of the present authors at the UCCSD(T)/CBS//UB3LYP/6-311+G(3df,2p) and CASPT3(8,8)/6-311+G(3df,2p) levels of theory.13 As alluded to in the Introduction, the location of a loose roaming type transition state for the isomerization of CH3NO2 to
CH3ONO allowed us to fully account for the production of CH3O with as much as 40% yield by IRMPD under the
collision-free molecular beam condition reported by Wodtke et al.7 The results of the present study based on the PES shows that under the thermal condition, which is similar to that of IRMPD, the isomerization production of CH3ONO is
dominant at pressures below 2 Torr in the temperature range studied, 400−3000 K, whereas the formation of the CH3 +
NO2products becomes competitive at pressures higher than 2 Torr. At 200 Torr pressure, for example, the production of the latter product pairs accounts for more than 90% of the decomposition reaction. Accordingly, the predicted high-pressure limit rate constant represents solely for the production of CH3 + NO2, whereas the predicted second-order,
low-pressure limit rate constant represents entirely for the formation of the CH3ONO, which further decomposes
competitively to CH3O + NO and CH2O + HNO via another roaming transition state.
■
ASSOCIATED CONTENT*
S Supporting InformationCartesian coordinates of the main species related to the decomposition of CH3NO2 (SI-1), calculated andfitted VTS curve for CH3NO2decomposition (SI-2), and the structure of
the roaming transition state RTS2 connecting to CH2O + HNO (SI-3). This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATIONCorresponding Author *E-mail: chemmcl@emory.edu. Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSThe authors acknowledge a partial support from the U.S. Office of Naval Research under contract No. N00014-08-1-0106. M.C.L. also thanks Taiwan’s National Science Council for the award of a distinguished visiting professorship at the National Chiao Tung University, Hsinchu, Taiwan, as well as the support from the Ministry of Education under its ATU Program.
■
REFERENCES(1) Manion, J. A.; Huie, R. E.; Levin, R. D.; Burgess, D. R., Jr.; Orkin, V. L.; Tsang, W.; McGivern, W. S.; Hudgens, J. W.; Knyazev, V. D.; Atkinson, D. B.; Chai, E.; Tereza, A. M.; Lin, C.-Y.; Allison, T. C. ; Mallard, W. G.; Westley, F. ; Herron, J. T.; Hampson, R. F.; Frizzell, D. H. NIST Chemical Kinetics Database, NIST Standard Reference Database 17, Data version 2008.12, Version 7.0 (Web Version), Release 1.4.3; National Institute of Standards and Technology, Gaithersburg, MD, 20899-8320; Web address http://kinetics.nist. gov/.
(2) Dewar, M. J. S.; Ritchie, J. P.; Alster, J. Ground States of Molecules. 65. Thermolysis of Molecules Containing NO2Groups. J. Org. Chem. 1985, 50, 1031−1036.
(3) Mckee, M. L. Ab Initio Study of Rearrangements on the Nitromethane Potential Energy Surface. J. Am. Chem. Soc. 1986, 108, 5784−5792.
(4) Mckee, M. L. MCSCF Study of the Rearrangement of Nitromethane to Methyl Nitrite. J. Phys. Chem. 1989, 93, 7365−7369. (5) Nguyen, M. T.; Le, H. T.; Hajgato, B.; Veszpremi, T.; Lin, M. C. Nitromethane − Methyl Nitrite Rearrangement: A Persistent Discrepancy between Theory and Experiment. J. Phys. Chem. A 2003, 107, 4286−4291.
(6) Saxon, R. P.; Yoshimine, M. Theoretical Study of Nitro-Nitrite Rearrangement of CH3NO2. Can. J. Chem. 1992, 70, 572−579.
(7) Wodtke, A. M.; Hintsa, E. J.; Lee, Y. T. Infrared Multiphoton Dissociation of Three Nitroalkanes. J. Phys. Chem. 1986, 90, 3549− 3558.
(8) Townsend, D.; Lahankar, S. A.; Lee, S. K.; Chambreau, S. D.; Suits, A. G.; Zhang, X.; Rheinecker, J. L.; Harding, L. B.; Bowman, J. M. The Roaming Atom: Straying from the Reaction Path in Formaldehyde Decomposition. Science 2004, 306, 1158−1161.
(9) Grubb, M. P.; Warter, M. L.; Suits, A. G.; North, S. W. Evidence of Roaming Dynamics and Multiple Channels for Molecular Elimination in NO3Photolysis. J. Phys. Chem. Lett. 2010, 1, 2455− 2458.
(10) Hause, Michael L.; Herath, Nuradhika; Rongshun, Zhu; Lin, M. C.; Arthur, G. Suits, Roaming-Mediated Isomerization in the Photodissociation of Nitrobenzene. Nat. Chem. 2011, 3, 932−937.
(11) Heazlewood, B. R.; Jordan, M. J. T.; Kable, S. H.; Selby, T. M.; Osborn, D. L.; Shepler, B. C.; Braams, B. J.; Bowman, Joel. M. Roaming is the Dominant Mechanism for Molecular Products in Acetaldehyde Photodissociation. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 12719−12727.
(12) Sivaramakrishnan, R.; Michael, J. V.; Wagner, A. F.; Dawes, R.; Jasper, A. W.; Harding, L. B.; Georgievskii, Y.; Klippensteina, S. J. Roaming Radicals in the Thermal Decomposition Of Dimethyl Ether: Experiment and Theory. Combust. Flame 2011, 158, 618−632.
(13) Zhu, R. S.; Lin, M. C. CH3NO2Decomposition/Isomerization Mechanism and Product Branching Ratios: An Ab Initio Chemical Kinetic Study. Chem. Phys. Lett. 2009, 478 (1−3), 11−16.
(14) Klippenstein, S. J.; Wagner, A. F.; Dunbar, R. C.; Wardlaw, D. M.; Robertson, S. H. VARIFLEX, version 1.00; 1999.
(15) Gilbert, R. G.; Smith, S. C. Theory of Unimolecular and Recombination Reactions; Blackwell Scientific: Carlton, Australia, 1990. (16) Holbrook, K. A.; Pilling, M. J.; Robertson, S. H. Unimolecular Reactions; Wiley: 1996.
(17) Wardlaw, D. M.; Marcus, R. A. RRKM Reaction Theory for Transition States of any Looseness. Chem. Phys. Lett. 1984, 110, 230− 234.
(18) Klippenstein, S. J.; Marcus, R. A. Unimolecular Reaction Rate Theory for Highly Flexible Transition States. J. Phys. Chem. 1988, 92, 5412−5417.
(19) Van Leeuwen, M. E. Derivation of Stockmayer Potential Parameter for Polar Fluids. Fluid Phase Equilib. 1994, 99, 1−18.
(20) Hippler, H.; Troe, J.; Wendelken, H. J. Collisional Deactivation of Vibrationally Highly Excited Polyatomic Molecules. J. Chem. Phys. 1983, 78, 6709−6715.
(21) Zaslonko, I. S.; Petrov, Yu. P.; Smirnov, V. N. Thermal Decomposition of Nitromethane in Shock Waves: The Effect of Pressure and Collision Partners. Kinet. Catal. (Engl. Transl.) 1997, 38, 321−324.
(22) Zhang, Y. X.; Bauer, S. H. Modeling The Decomposition Of Nitromethane, Induced by Shock Heating. J. Phys. Chem. B 1997, 101, 8717−8726.
(23) Zaslonko, I. S.; Kogarko, S. M.; Mozzhukin, E. B.; Petrov, Yu. P. Thermal Decomposition of Nitromethane in Shock Waves. Kinet. Catal. (Engl. Transl.) 1972, 13, 1001−1005.
(24) Glaenzer, K.; Troe, J. Thermische Zerfallsreaktionen von Nitroverbindungen I: Dissoziation von Nitromethan. Helv. Chim. Acta 1972, 55, 2884−2893.
(25) Dublikhin, V. V.; Nazin, G. M.; Manelis, G. B. Thermal Decomposition of Nitromethane. Bull. Acad. Sci. USSR Div. Chem. Sci. (Engl. Transl.) 1971, 20, 1247−1248.
(26) Crawforth, C. G.; Waddington, D. J. Reactions Of Nitroalkanes In The Gas Phase. Part 1. - Pyrolysis Of Nitromethane. Trans. Faraday Soc. 1969, 65, 1334−1349.
(27) Borisov, A. A.; Kogarko, S. M.; Skachkov, G. I. Thermal Decomposition of Nitromethane. Kinet. Catal. 1966, 1, 521−526.
(28) Makovky, A.; Gruenwald, T. B. The Thermal Decomposition of Nitromethane under High Pressure. Trans. Faraday Soc 1959, 55, 952−598.
(29) Cooper, S. C.; Lin, C. Y.; Argetsinger, A.; Lin, M. C. Relative Rate of CH3NO2, CH3ONO and CH3ONO2 Formation in the Thermal Reaction of NO2with Acetaldehyde and Di-tButyl Peroxide at Low Temperatures. J. Energ. Mater. 1989, 7, 55−75.
(30) He, Y.; Sanders, W. A.; Lin, M. C. Thermal Decomposition of Methyl Nitrite: Kinetic Modeling of Detailed Product Measurements by Gas-Liquid Chromatography and Fourier Transform Infrared Spectroscopy. J. Phys. Chem. 1988, 92, 5474−5481.
(31) Batt, L.; Milne, R. T.; McCulloch, R. D. The Gas-Phase Pyrolysis of Alkyl Nitrites. V. Methyl Nitrite. Int. J. Chem. Kinet. 1977, 9, 567−587.
(32) Batt, L.; McCulloch, R. D.; Milne, R. T. Thermochemical and Kinetic Studies of Alkyl Nitrites (RONO)-D(RO-NO), The Reactions between RO. and NO, and the Decomposition RO. Proc. Symp. Chem. Kinet. Data Upper Lower Atmos. 1975, 441−461.
(33) Zaslonko, I. S.; Kogarko, S. M.; Mozzhukhin, E. V.; Petrov, Yu. P.; Borisov, A. A. Thermal Decomposition of Methyl Nitrite in Shock Waves. 1. Initial Stage of Decomposition and Mechanism of Chemiluminescence of H2CO and HNO. Kinet. Catal. (Engl. Transl.) 1970, 11, 249−255.
(34) Phillips, L. The Pyrolysis of Methyl Nitrite. J. Chem. Soc. London 1961, 3082−3090.
(35) Shaw, R.; Trotman-Dickenson, A. F. The Reactions of Methoxyl Radicals with Alkanes. J. Chem. Soc. 1960, 3210−3215.
(36) Steacie, E. W. R.; Shaw, G. T. The Homogeneous Unimolecular Decomposition of Gaseous Methyl Nitrite. Proc. R. Soc. London A 1934, 146, 388−395.
(37) Hsu, D. S. Y.; Burks, G. L.; Beebe, M. D.; Lin, M. C. Thermal Decomposition of Methyl Nitrite in Shock Waves Studied by Laser Probing. Int. J. Chem. Kinet. 1984, 16, 1139−1150.