行政院國家科學委員會專題研究計畫 成果報告
以抗氧化物減低腎臟缺血再灌流誘發腎小管細胞凋亡的效
應(I):細胞凋亡基因的研究(3/3)
計畫類別: 個別型計畫 計畫編號: NSC92-2314-B-002-163- 執行期間: 92 年 08 月 01 日至 93 年 07 月 31 日 執行單位: 國立臺灣大學醫學院醫學系 計畫主持人: 賴明坤 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 93 年 11 月 2 日
行政院國家科學委員會補助專題研究計畫
■ 成 果 報 告 □期中進度報告(計畫名稱)
以抗氧化物減低腎臟缺血再灌流誘發腎小管細胞凋亡的效應(I):細胞
凋亡基因的研究(3/3)
計畫類別:■
個別型計畫 □
整合型計畫
計畫編號:NSC 92-2314- B -002 -163
執行期間:92 年 8 月 1 日至 93 年 7 月 31 日
計畫主持人:賴明坤
共同主持人:
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整
報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究
計畫、列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:台大醫學院醫學系
中 華 民 國 93 年 10 月 30 日
行政院國家科學委員會專題研究計畫執行進度報
告
計畫題目:以抗氧化物減低腎臟缺血再灌流誘發腎小管細胞凋亡的效
應(I):細胞凋亡基因的研究(1/3)
計畫編號:NSC 90-2314-B-002-445
執行期限:90 年 08 月 01 日至 91 年 07 月 31 日
主持人:賴明坤
執行機構及單位名稱:台大醫學院醫學系
一、中文摘要 腎臟移植或缺血灌流造成自由基的釋放可能是造成腎細胞死亡之主因。為驗 證此一假說我們已利用不同時間之腎缺血觀察腎自由基與細胞凋亡的產生。我們 也以超冷光儀偵測活體腎臟及離體腎小管與全血之自由基表現,TUNEL 免疫染 色, DNA 階梯生化證據及西方墨點法探討致死基因 bax/bcl-2, CPP32, and PARP 的細胞凋亡的分子機轉。結果發現增加缺血再灌流引發自由基的釋放量增加, 增 加 TUNEL 正染色細胞(apoptotic cell), 與 DNA 階梯之表現。此機轉可能是因活化 bax/bcl-2, CPP32, and PARP 的細胞凋亡的分子機轉有關。我們亦發現 Mn SOD, CuZn SOD 與 catalase 會隨著缺血與再灌流時間的增加而逐漸降低。為進一步探究 自由基的增加是造成腎細胞凋亡的產生主因。我們曾以添加抗氧化物, SOD, C60(FC4S)來評估腎細胞凋亡的反應。結果發現抗氧化物可降低腎細胞凋亡數目, DNA 階梯現象與 Bax/Bcl-2, CPP32 , PARP 的反應。此結果證明抗氧化物的添加可 以改善腎細胞凋亡與自由基的清除是正相關。為強化抗氧能力我們以目前的缺氧 預處理發現抗氧化保護蛋白 MnSOD、CuZnSOD、Catalase 的表現在 10%缺氧後之第 二天,此三種蛋白顯著增加。以 DNA laddering 數據發現腎臟遭受缺血/再灌流產 生細胞凋亡之時機主要是一小時的缺血與四小時再灌流。重要發現是自由基種類 與數量的測定發現缺氧前處理能減低這些自由基的數量。Bax, Bcl-2, CPP32, PARP 之表現之結果亦發現缺氧預處理有助於減低缺血再灌流後受損腎臟的腎功 能與細胞凋亡之表現。缺氧預處理實驗的結果則能讓我們了解氧化壓力(自由基) 的減低是造成肝腎胞凋亡減少的主因。藉由腎細胞產生抗氧化之防禦機轉,藉以 減低腎缺血/再灌流傷害所引發腎細胞凋亡與腎功能失調的病徵;此種有意義的研 究結果將有助於減清臨床上因腎臟移除後之移植腎之延遲作用或是經腎缺血後 再灌流的傷害。 關鍵詞:自由基;細胞凋亡; 缺血再灌流; 抗氧化物 ABSTRACTShort periods of hypoxia termed "hypoxic preconditioning" are known to increase protection against sustained ischemia/reperfusion injury. Hypoxia-inducible factor-1α (HIF-1α) is essential for the activation of hypoxia-inducible genes and its activation can promote cell survival in hypoxic tissues. We explored the effect of hypoxic preconditioning on HIF-1 expression and ischemic tolerance in the kidney subjected to ischemia/reperfusion damage. To support the role of HIF-1α in protective preconditioning, we also studied the effect of HIF-1 inducer, cobalt chloride (CoCl2)
and HIF-1 inhibitor, 3-(5'-hydroxymethyl-2'-furyl)-1-benzylindazole (YC-1), on HIF-1 expression and protection in the kidney. Rats for hypoxic induction were placed in a hypobaric chamber (10% O2, 380 Torr, 5,500 m) 15 h/day. After hypoxic
preconditioning, these kidneys subjected to 45-min ischemia followed by reperfusion were conducted to analyze apoptosis-related mechanism and to determine reactive oxygen species (ROS) amount in vivo by a chemiluminescence method. Results showed hypoxic predonditioning upregulated HIF-1α, heat shock protein 70 (HSP 70),
Bcl-2 and Bcl-xL expression in preconditioning kidneys. The upregulation in HSP 70
and Bcl-2 by hypoxia and CoCl2 can be depressed by YC-1, supporting a
HIF-1α-dependent protection. In response to ischemia/reperfusion insult, potentiated apoptotic mechanisms including increases in Bax/Bcl-2 ratio, caspase 3 expression, and poly-(ADP-ribose)-polymerase fragments, and enhanced oxidative stress by increases in ROS release from kidney surface and renal venous blood were found. The increased oxidative injury led to the increased DNA fragmentation and apoptotic cell number in the ischemia/reperfusion kidney. However, all these oxidative insults were significantly reduced by a mechanism of Bcl-2 and Bcl-xL upregulation during hypoxic
preconditioning. In proximal tubule cultures from preconditioning kidneys, antisense oligodeoxyribonucleotides of HSP 70 and Bcl-2 treatment abrogated the partial protection against hypoxia/reoxygenation injury. Our results suggest that HIF-1α activation could contribute to protective kidney preconditioning by upregulation of HSP 70 and Bcl-2, which are important in the acquisition of renal protection against ischemia/reperfusion induced oxidative stress.
INTRODUCTION
Cellular hypoxia is an important component of several pathophysiological conditions, including tumerogenesis and ischemia-related disorders. In these and other hypoxic situations mammalian cells alter gene expression to counter the effects of limited O2. Hypoxia induces the transcriptional activation of a variety of genes,
including erythropoietin, vascular endothelial growth factor, and transferring [Bunn and Poyton, 1996]. Increased expression of these genes is mediated primarily by the heterodimeric transcription factor hypoxia-inducible factor 1 (HIF-1), which is composed of HIF-1α and HIF-1β subunits [Bunn and Poyton, 1996; Wang et al., 1995]. Decreased O2 tension stimulates HIF-1α protein accumulation by decreasing its
proteasomal degradation [Kallio et al., 1997]. Hypoxic stabilization of HIF-1α is blocked in the presence of H2O2, suggesting that hypoxia-induced changes in the level
of reactive oxygen species (ROS) may be involved in HIF-1 activation [Huang et al., 1997]. Hypoxic preconditioning (8% O2 for 3 hours), a treatment known to protect the
newborn rat brain against hypoxic-ischemic injury, markedly increased HIF-1α and HIF-1β expression [Bergeron et al., 2000]. HIF-1α and HIF-1β protein levels were markedly increased after intraperitoneal injection of CoCl2 (60 mg/kg).
Preconditioning with CoCl2 24 hours before hypoxia-ischemia afforded 75% brain
protection, compared with that in vehicle-injected littermate controls. Thus activation of HIF-1 by hypoxia or CoCl2 could be efficient in increases of ischemic tolerance.
Lack of oxygen, i.e. hypoxia, plays a fundamental role in many pathologic processes. In ischemic diseases, including stroke,myocardial infarction, and acute renal failure, hypoxia leadsto cell death and determines tissue pathology (Cotran et al., 1998). Interestingly, cells can be protected when a non-injurious hypoxic stress is performed several hours or days before a lethalhypoxic-ischemic stress (preconditioning). This phenomenon is called tolerance. Ischemic tolerance can be achieved in organs by several preconditioning sublethal stresses such as hypoxia(Bernaudin et al., 2002), ischemia itself (4), and hyperthermia (6).
As hypoxic preconditioning is non-invasive and reproducible, this model has been used to study the mechanisms protecting the kidney against hypoxia-ischemia particularly in rats. These studies suggested that hypoxia-inducible factor-1 (HIF-1) could be an important mediator of hypoxia-induced tolerance toischemia (13, 14, 16). Indeed, hypoxic preconditioning inducesexpression of HIF-1 and its target genes in neonatal (14) andadult brain (16). In addition, cobalt chloride, one agent that activates
HIF-1 (17), also induces tolerance againsthypoxia-ischemia in neonatal rat brain (13). HIF-1 is an importanttranscription factor regulating gene expression in response to hypoxia. Moreover, HIF-1 target genes such as erythropoietin (EPO) and vascular endothelial growth factor (VEGF) protect the brainagainst ischemia (11, 18-20). This suggests that HIF-1 mightbe involved in the establishment of ischemic tolerance in brain.However, it is possible that there are other adaptive mechanismsunderlying this protection. However, sufficient evidence indicates a lack of correlation between stress-inducible expression (HSP 70) and early preconditioning, which is short-lived and lasts between 1 and 3 h, depending on the model and species (Heads et al., 1995). Heads RJ, Latchman DS, Yellon DM. Differential stress protein mRNA expression during early ischemic preconditioning in the rabbit heart and its relationship to adenosine receptor function. J Mol Cell Cardiol 27: 2133-2148, 1995.
Understanding changes in gene expression in kidney following exposure to hypoxia could reveal new mechanisms of ischemic tolerance. Accordingly, we exploited DNA microarray technology to investigatethe brain genomic response of neonatal rat to hypoxia (3 h, 8% O2). In an effort to analyze the mechanisms of
oxygen sensing at the molecular level, HIF-1 was identified as a key regulator of oxygen-dependent gene expression. The HIF-1 transcription factor is a heterodimer consisting of two basic helix-loop-helix PAS proteins, HIF-1 and HIF-1β, also known as ARNT (aryl hydrocarbon receptor nuclear translocator) (reviewed in ref 12). While the HIF-1β subunit is constitutively expressed, degradation of HIF-1α by the ubiquitin-proteasome pathway is regulated in an oxygen-dependent manner involving hydroxylation of two proline residues in the oxygen-dependent degradation domain (ODD) of the molecule (13, 14). Prolyl hydroxylation requires 2-oxoglutarate and iron as cofactors, which explains the hypoxia-mimetic effect of transition metal ions such as cobalt (reviewed in ref 15).
Complete or partial cessation (ischemia) followed by restoration of blood flow (reperfusion) is a serious event that affects several organs, including brain, heart and kidney [1-7]. The mechanisms of acute tissue damage following ischemia/reperfusion (I/R) injury are thought to involve a complex interaction of immediate cellular damage caused by reactive oxygen species (ROS) [1-3,7]. Many studies indicate that ROS formation during reperfusion may evoke an abnormal signal transduction or cellular dysfunction [8,9] and initiate the cascade of apoptosis/necrosis, and subsequent inflammatory infiltration [6,7,10]. The consequences of I/R induced ROS-oxidative stress including lipid peroxidation, as well as damage to proteins and nucleic acids, may contribute to activation and expression of the genes/proteins responsible for apoptosis [17]. For example, increases in the Bax/Bcl-2 ratio [18], expression of caspase and its activity [19,20], and caspase-mediated cleavage of poly-(ADP-ribose)-polymerase (PARP) [21] have been found in organs subjected to I/R injury or in cells after a cytotoxic insult.
Apoptosis or programmed cell death is a genetically controlled response for cells to commit suicide [11-13] and is a crucial event that can initiate inflammation and subsequent tissue injury [14-16]. Studies in heart and kidney tissues, both in vitro and in vivo, suggest that cell death upon reperfusion is largely apoptotic in nature [6,22]. However, because of tissue-specific pattern of stress kinase activation and different sources of ROS in I/R heart and kidney [22-24], the time course for onset of apoptosis during I/R injury is different between heart (within 30 min of reperfusion) and kidney (2-4 h after reperfusion) [22].
The activation of EPO, VEGF, and ENO1 gene transcription in response to decreased O2 availability is mediated by HIF-1, abasic helix-loop-helix transcription
Semenza, 1995 ). Expression of HIF-1 increases exponentially as cellular O2
concentrationsare decreased (Jiang et al., 1996 ). Expression of the limitingHIF-1 subunit is precisely regulated by O2 concentration anddetermines the level of HIF-1
DNA-binding activity and transcriptionalactivity within the cell (Huang et al., 1996 ; Jiang et al., 1996 ,1997b ; Semenza et al., 1996 ; Pugh et al., 1997 ). In addition to hypoxia, divalent metals (such as CoCl2) and iron chelators (such as DFO) induce
HIF-1 expression, HIF-1 DNA-binding activity, and trans-activation of genes containing HIF-1 binding sites (Wangand Semenza, 1993a ,b ; Wang et al., 1995a ; Jiang et al., 1997b ;Pugh et al., 1997 ). The mechanisms of action of these compounds have not been determined but seem to be distinct from the hypoxiasignal-transduction pathway (Gleadle et al., 1995b ; Ehleben etal., 1997 ; Fandrey et al., 1997 ).
Recently, exposure of cardiomyocytes [25-27] or isolated perfused heart [28] to brief hypoxia termed "hypoxia preconditioning" (HP) may render the heart more tolerant to a subsequent I/R injury. The cardioprotection afforded by HP from in vitro data could be ascribed to mitochondrial ROS release [27], intracellular ionic alterations and upregulation of endogenous catalase and heat shock protein-70 (HSP-70) gene expression [29]. Adaptation to chronic hypoxia, or a long-lasting HP may provide a more enhanced and prolonged cardioprotection [30], however, despite of extensive in vitro and in vivo data of the heart, the in vivo relevance to the kidney is not yet determined.
The overall objective of this study was designed to address whether a long-lasting HP could efficiently alleviate I/R induced oxidative stress and apoptosis formation by an enhanced protective mechanism. We also determined whether a long-lasting HP enhances and/or elongates the renal protection ability. We adapted a long-lasting HP method (chronically intermittent hypoxia-10% O2) to address apoptosis
cell death, expression in proto-oncogenes and proteinase, and ROS release during I/R injury. Our results had in detail addressed the protective mechanism induced by long-lasting HP and provided a new strategy to increase tolerance against I/R injury.
MATERIALS AND METHODS Hypoxic preconditioning (HP)
Female Wistar rats (200-250 g) were housed at the Experimental Animal Center, National Taiwan University at a constant temperature and with a consistent light cycle (light from 07:00 to 18:00). The animal care and experimental protocol are in accordance with the guidelines of the National Science Council of Republic of China (NSC 1997). The method for HP induction was described previously [Chien et al., 1997]. In brief, rats were placed in a high altitude chamber (HP rats) 15 h/day for different periods, whereas control age-matched animals were maintained at sea level (SL) at the same temperature and light cycle. A level of 380 torr (5,500 m) was selected, because it represents the maximal altitude to which most rats can successfully adapt. The animals were exposed to hypoxia from 17:00 to 08:00 and then returned to room air. The body weight of the animals was measured once a week. Food and water were provided ad libitum.
Surgery
Four groups of rats, sham-operated SL (sham SL) and HP (sham HP) rats and SL (I/RSL) and HP (I/RHP) rats with I/R injury, were used. On the experimental day all rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and were tracheotomized. Catheters were placed in the left carotid artery and left renal vein for blood sampling and in the left femoral vein for drug and blood supplement. The rat was then placed on its right side, and the left kidney was exposed via a flank incision and dissection from the surrounding tissue.
For induction of unilateral ischemia in the left kidney, the left renal artery was clamped by using small vascular clamps for a 45-min period. Sham-operated animals received similar operative procedures without occlusion of the renal artery. Reperfusion was initiated by removing the clip. All rats were randomly subjected to different time frames of reperfusion.
Experimental protocols
Three experiments were performed. We first explored HIF-1α expression in nuclear protein of rat kidney subjected to different time frames of 10% hypoxia. In the second part of experiment, we determined renal expression of HIF-1α and from HP rats subjected to 2 day-, 1 week-, 2 week-, and 4 week-HP. These samples were quickly frozen in liquid nitrogen, and stored at –70°C for determination of HIF-1α, Bcl-2, Bcl-xL, and HSP-70 expression. In the third part of study for examination of HP induced renal protection, we evaluated I/R induced oxidative injury, including ROS generation, apoptosis formation and apoptosis-related protein expression, in the kidneys of HP and SL rats.
The rats were sacrificed with overdose of anesthetics at the end of ischemia (n=3 in each group), 1 h (n=4 in each group), 4 h (n=4), 10 h (n=3), and 24 h (n=3) of reperfusion. The kidney was resected and divided into three parts. One was stored in 10% neutral buffered formalin for routine histology and in situ assay for DNA fragmentation, one was prepared for DNA fragmentation electrophoresis, and the other was quickly frozen in liquid nitrogen, and stored at –70°C for protein and RNA isolation.
RNA isolation and real-time quantitative PCR anslysis.
Total RNA preparations from rat kidney samples were performed by using trizol reagent (GIBCO/BRL). Real-time quantitative PCR to measure the HIF-1α, bax, bcl-xl, and bcl-xs transcripts of treated and normal kidney tissue were individually used with two primer and one oligonucleotide probe by using an Applied Biosystems PRISM 7700 Sequence Detector (Perkin-Elmer) [35]. The primers and probes sequences are: HIF-1α (forward primer: 5’-CAGCAGACCCAGTTACAGAA-3’, reverse primer:
5’-TCAGTTAACTTGATCCAAAGCTCT-3, probe: 5’-TTCTCGTTCTCGCCGCCGG-3’), bax (forward primer:
5’-AGACACCTGAGCTGACCTTGGA-3’, reverse primer:
5’-CCTGAGACACTCGCTCAGCTT-3’, probe:
5’-CGCCCCAGGACGCATCCAC-3’), bcl-xs (forward primer:
5’-CACCCGAGAGCCGGAAA-3’, reverse primer:
5’-CAGAACTACACCAGCCACAGTCAT-3’, probe: 5’-TTTCAACCGCTGGTTCCTGACGG-3’), bcl-xl (forward primer:
5’-GCCACAGCAGCAGTTTGGAT-3’, reverse primer:
5’-AAACTCATCGCCAGCCTCTCT-3’, probe: 5’-CTCCCCATGGCAGCAGTGAAGC-3’), and GAPDH (forward primer:
5’-TGCCTTCTCTTGTGACAAAGTG-3’, reverse primer:
5’-TGCCGTGGGTAGAGTCATACT-3’, probe: CATCAACGACCCCTTCATTGACCTC-3’).
The thermal cycling conditions included 2 min at 50°C, 30 min at 60°C and 5 min at 95°C. Thermal cycling proceeded with 40 cycles of 94°C for 15 sec and 60°C for 1 min. All reactions were performed in the Model 7700 Sequence Detector (PE Applied Biosystems), which contains a Gene-Amp PCR system 9600. Reaction conditions were programmed on a Power Macintosh 7100 (Apple Computer, Santa Clara, CA) linked directly to the Model 7700 Sequence Detector. Analysis of data was also performed on the Macintosh computer. Collection and analysis software was
developed at PE Applied Biosystems.
HIF-1α on HSP 70 and Bcl-2 expression
The rats were exposed, in parallel, to room air (20% O2) or hypoxia (10% O2)
for 15 h and then the nuclear extract of kidneys were isolated for HIF-1α analysis by using western blot. YC-1, 3-(5'-hydroxymethyl-2'-furyl)-1-benzylindazole, an agent developed for circulatory disorders that inhibits platelet aggregation and vascular contraction and inhibits HIF-1 activity in vitro and in vivo (Yeo et al., 2003). We examined whether YC-1 inhibits HIF-1 and depressed hypoxia-, and cobalt-enhanced Bcl-2 and HSP 70 protein levels. The rats were subjected to room air, 10% hypoxia, intraperitoneal CoCl2 (60 mg/kg, Sigma), hypoxia in the presence of 0.1 mg YC-1
(HIF-1α inhibitor), and CoCl2 in the presence of 0.1 mg YC-1 for 15 h followd by
western blot analysis.
Preparation of protein for HIF-1α
The expression of HIF-1α protein was determined in the kidney using immunoblotting methods. Kidney tissue was homogenized in five volume of lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM KCl, 2mM EDTA, 0.5 mM dithiothreitol (DTT), 0.4 mM phenylmethyl-sulfonyl fluoride, and 2% nonidet P-40 containing a protease inhibitor cocktail (Sigma) in a Potter-Elvehjem homogenizer. The homogenate was centrifuged at 12,000 ×g for 10 min 4°C, and the resulting supernatant was analyzed for HIF-1α (Kim et al., 2002). Protein concentrations were determined using the bicinchoninic acid method (Bio-Rad) with bovine serum albumin as a standard. The lysates (150 µg protein) were incubated with 5 µL of a purified mouse anti-HIF-1α antibody (Novus Biological, Inc., Littleton, CO, USA) for 12 h, followed by incubation with 10 µL of protein A-Sepharose beads (Amersham Pharmacia Biotech) for 3 hrs. After washing, the immunocomplexes were eluted by boiling for 3 minutes in 20 µL of the SDS sample buffer containing 10 mM DTT. The elute was electrophoresed on 7% SDS-polyacrylamide gels, and then the proteins were transferred to Immobilon-P membranes (Millipore), which were then incubated with 5% skim milk in Tris-buffered saline for 2 h to block nonspecific binding. The membranes were incubated overnight at 4°C with a mouse anti-HIF-1α antibody (1:2000), and re-incubated with a goat anti-rat IgG antiserum conjugated with horseradish peroxidase (1:5000) for 2 h. Immunoreactive protein bands were visualized using the enhanced chemiluminescence plus detection system (ECLplus, Amersham-Pharmacia).
Immunoblot analysis for Bcl-2, Bcl-xL, HSP-70, Bax, CPP32, and PARP.
Kidney samples from HP and SL rats with or without I/R injury were used to measure the amounts of HSP-70, Bcl-xL, Bax/Bcl-2, caspase 3 (CPP32), and PARP
[Chien et al., 2001]. For protein analysis, kidney samples were homogenized with a prechilled mortar and pestle in extraction buffer, which consisted of 10 mM Tris-HCl (pH=7.6), 140 mM NaCl, 1 mM PMSF, 1% NP-40, 0.5% deoxycholate, 2% β-mercaptoethanol, 10 µg/ml pepstain A, and 10 µg/ml aprotinin. The mixtures were homogenized completely by vortexing and kept at 4°C for 30 min. The homogenate was centrifuged at 12,000 ×g for 12 min at 4°C, the supernatant was collected, and the protein concentrations were determined by BioRad Protein Assay (BioRad Laboratories, Hercules, CA, USA). Antibodies that react with Bax (Chemicon, Temecula, CA, USA), Bcl-2, Bcl-xL, HSP-70, the activation fragment of caspase 3 (CPP32/Yama/Apopain)
(Transduction, Bluegrass-Lexington, KY, USA), and PARP (Biomol, Plymouth Meeting, PA, USA) were used.
SDS-PAGE was performed on 12.5% separation gels in the absence of urea and stained with Coomassie brilliant blue. Proteins on the SDS-PAGE gels, each lane
containing 30 µg of total protein, were transferred to nitrocellulose filters. The immunoreactive bands were detected by incubation with the tested antibody, followed by secondary antibody-alkaline phosphatase, and finally with NBT and 5-Bromo-4chloro-3-indolyl phosphate, toluidine salt (Roche Diagnostic GmbH, Mannheim, Germany) stock solution for 30 min at room temperature.
In vivo and in vitro CL-ROS recording
In vivo. The method for detection of CL-ROS from the renal surface after intrarenal arterial MCLA injection was adapted for demonstration of ROS production in the I/R kidney [Chien et al., 2001]. Briefly, a continuous infusion of MCLA (1 mM in 0.1 ml) (Sigma, St. Louis, MO, USA) was administered into the control or I/R kidney via an intrarenal arterial catheter. The MCLA-enhanced CL-ROS signal from the kidney surface was measured continuously using a CL Analyzing System (CLD-110, Tohoku Electronic Industrial Co., Sendai, Japan).
In vitro. A series of blood samples (0.2 ml) from the renal vein was obtained immediately after reperfusion for various times. The blood samples were immediately wrapped with aluminum foil and kept on ice until CL-ROS measurement, usually done within 2 h [Chien et al., 2001]. The CL-ROS was measured in a completely dark chamber of the Chemiluminescence Analyzing System. After 100-sec background level determination, 1.0 ml of 0.1 mM lucigenin in PBS (pH 7.4) was injected into the sample. The CL-ROS was continuously monitored for an additional 600 sec. The total amount of CL-ROS was calculated by integrating of the area under the curve and subtracting it from the background level. The assay was performed in duplicate for each sample and was expressed as CL-ROS counts/10 sec. The mean standard error (SE) of the CL-ROS level of each sample was calculated.
In situ demonstration of superoxide formation by NBT
An NBT perfusion method was used for localizing de novo ROS generation in the I/R kidney [Chien et al., 2001]. Three rats of I/RSL and I/RHP were sacrificed after 4 h of reperfusion. An 18-gauge needle connected to an infusion pump (Infors AG, CH-4103, Bottmingen, Switzerland) was inserted in the upper abdominal aorta just above both kidneys, whereas the lower abdominal aorta below both kidneys was ligated. After the bilateral renal veins were cut, the kidneys were perfused with 37°C Hanks' balanced salt solution (flow rate, 10 ml/min; pH 7.4). Once blood had been removed, NBT (1 mg/ml) was added to the Hanks' balanced salt solution, and the kidneys were perfused for an additional 10 min at a flow rate of 5 ml/min. All unreacted NBT was removed from the kidneys by postperfusion with Hanks’ solution. The NBT-perfused kidneys were cut and fixed in zinc/formalin for histologic examination for formazan deposits, and some samples were prepared for localization of apoptotic cells. In separate experiments, SOD was included in the perfusate (500 µg/ml) for confirmation of the specificity of the NBT deposition as the product of ROS (Chien et al., 2001).
In situ apoptotic assay (ISAA)
The method for ISAA (i.e., terminal deoxynucleotidyl transferase-mediated nick-end labeling method, TUNEL) was performed according to the method of Gavrieli et al. [1992] with minor modifications [Chien et al., 2001]. Briefly, 4-6 µm thick sections of the kidney were prepared, deparaffinized, and stained by the TUNEL-ABC method. Twenty high-power (×400) fields of the outer medulla were randomly selected in each section to count the number of apoptotic cells. Proximal tubules were distinguished from other segments by their characteristic brush border and their relatively large epithelial cell bodies. The distal tubules were recognized by their smaller (the nuclei are close together) and more regular cells [Chien et al., 2001]. The number of apoptotic cells was expressed per 100 of the proximal or distal tubular cells in each section.
DNA Extraction and Electrophoresis
The kidney samples were homogenized and lysed with a 20 ml of buffer containing 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, and 0.1 M NaCl, 1 ml of 10% sodium dodecyl sulfate. Then they were incubated with 200 µl of 10 mg/ml proteinase K at 50°C overnight. DNA were extracted twice with phenol and twice with phenol/chloroform (1/1); the upper layer were collected, to which 5 M NaCl was added at a 1:50 (v/v) ratio and then participated overnight at –20°C in absolute ethanol. DNA was pelleted by centrifugation at 3,000 rpm at 4°C for 10 minutes, washed with 70% ethanol, and dried. DNA obtained was resuspended in TE (10 mM Tris-HCl, 1 mM EDTA) at 1 µg/ml and incubated with 0.1 mg/ml DNAse-free RNAse at 37°C for 30 minutes. Four µl of 0.25% bromophenol blue and 0.25% xylene cyanol in 40% sucrose were added to DNA samples at 1:5 (v/v) ratio. Electrophoresis was performed at 50 V in 1.6% agarose gels. DNA was visualized with ethidium bromide.
Isolation of proximal and distal tubules
Proximal and distal renal tubules were isolated as previously described [Chien et al., 2001]. To determine the role of Bcl-2 and HSP70 in protecting renal cells against hypoxia/reoxygenation injury, HSP 70 and Bcl-2 oligodeoxynucleotide (ODN) were added into the cell medium. ODN of HSP 70 (sense 5’-ATGGCCAAGAAAACA-’3 and antisense 5’-TGTTTTCTTGGCCAT-3’) and Bcl-2 (sense 5’-ATGGCGCAAGCCGGGA-3’ and antisense 5’-TCCCGGCTTGCGCCAT-3’) treatment was applied to the isolated PT cultures. The efficacy of antisense ODN uptake by the PT cultures was quantified by measuring the levels of targeted HSP 70 and Bcl-2 protein in the cultures. Western blot analyses were performed on the PT cultures (n=4 in each group) after 24 h.
Induction of hypoxia/reoxygenation of the renal tubule cells was performed as described previously [Chien et al., 2001]. The cultures were first placed in an atmosphere of 95% O2/5% CO2 at 37°C for 30 min. Hypoxia was achieved by gassing
with 95% N2/5% CO2 for 15 min, whereas reoxygenation was performed by
reintroduction of 95% O2/5% CO2 for 30 min. For determination of the number of
apoptotic cells in culture, apoptotic cell death in damaged cells were analyzed by using fluorescent dye and examined by fluorescence microscopy to visualize and count cells with condensed chromatin organization. The PT and DT cells were stained with annexin V-FITC and propidium (PI) to identify apoptotic cells. The apoptotic cells were determined as the cells with annexin V-FITC positive and PI-negative staining by a Couter flowcytometer (EPICS XL-MCL). The amounts of ROS in PT cell cultures (106 cells/ml) were detected by the lucigenin-enhanced CL-ROS test described above.
Statistical analysis.
All values are expressed as mean ± SE. For comparisons of group data, one-way analysis of variance and then the Student’s unpaired t-test were conducted. P < 0.05 was considered to indicate statistical significance.
RESULTS
Effect of hypoxic preconditioning on kidney HIF-1α expression
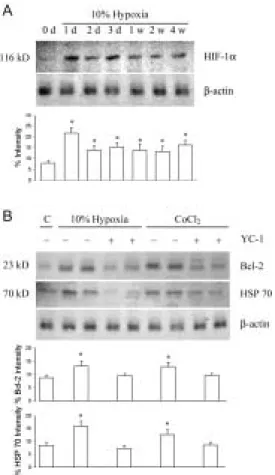
HIF-1α is a heterodimer made of two protein subunits, HIF-1α and HIF-1β (Wang et al., 1995). Whereas HIF-1β is constitutively expressed,HIF-1α expression is tightly regulated by cellular oxygen concentration(Huang et al., 1996). Thus HIF-1α determines HIF-1 DNA binding activity andtranscriptional activity during hypoxia. As shown in Fig. 1A, exposure of rats to hypoxia (10% O2, from 1 d to 4 weeks) induced
an increase in HIF-1α expression in nuclear extracts of the whole kidney, when compared with control animals. The increased HIF-1α expression of the
preconditioning kidney was 2.8, 1.8, 2.0, 1.8, 1.7, and 2.1 folds of control kidney after 1 day, 2 day, 3 day, 1 week, 2 weeks, and 4 weeks of hypoxic exposure, respectively. Histologic detection of apoptotic cells in reperfused renal tissue
The apoptotic cells were distinguished by their dark stained nuclei. Apoptotic cells were not detected, or only rarely present, in sections from sham-operated SL and HP rat kidney and from all kidneys subjected to ischemia without reperfusion.
In the SL kidneys with I/R injury, the apoptotic cells were detected in both PT and DT at 1-4 h of reperfusion (Fig. 1A). The numbers of apoptotic cells reached a plateau when the kidneys were reperfused for 4 to 6 h. No apparent apoptotic cells were detected in the kidneys subjected to 10-24 h of reperfusion, probably due to the removal of apoptotic cells by tissue repair mechanism.
In the HP kidneys with I/R injury, the number of apoptotic cells was also appeared in both PT and DT at 4 h of reperfusion (Fig. 1B). However, the number of apoptotic cells was significantly reduced in HP kidneys when compared to the SL kidneys.
In the SL kidneys with I/R injury, light microscopic examination showed severe tubular obstruction and necrotic cell death at 24 h of reperfusion (Fig. 1C). However, the severely tubular obstruction and necrotic cell death was not found in the HP kidneys with I/R injury (Fig. 1D).
Analysis of DNA fragmentation
By gel electrophoresis, no apparent DNA fragmentation was detected from sham-operated kidney or ischemic kidneys without reperfusion (Figure 2). An apoptotic DNA laddering pattern was clearly evident when ischemic kidneys were subjected to 1-4 h of reperfusion (Figure 2). However, the appearance of DNA fragmentation was significantly depressed in the HP rats when compared to SL rats. In these two ischemic groups of animals, there was an increasing "smear" DNA pattern after prolonged reperfusion (i.e., >10-24 h) (Figure 2).
Expression of Bcl-2, Bcl-xL and HSP-70 protein during HP
We examined the protein expression of Bcl-xL, Bcl-2, and HSP-70 in cortex and medulla of SL and HP kidneys using specific antibodies to probe Bcl-xL, Bcl-2 and HSP-70 homologs by Western blot analysis. As shown in Fig. 3, Bcl-xL expression appeared to be induced on 2 days of HP and was markedly increased by 1-4 weeks of HP. Bcl-2 expression as well as HSP-70 expression was increased after 1 week of HP and was persisted or enhanced by the prolonged period of HP (Fig. 3).
Expression of proto-oncogene mRNA in the I/R kidney
We addressed the apoptosis-related gene expression responding to I/R injury by real time QT-PCR analysis. A typical data of baseline level of bax and bcl-xL of SL and HP rats was shown in Fig. 4. The averaged expression of bax, bcl-xS and bcl-xL mRNA was shown in Fig. 5. bax mRNA rapidly increased, peaking at 1 h of reperfusion and then gradually declining by 4 h of reperfusion in I/R kidneys of HP and SL rats. bax mRNA was less expressed in HP kidney, although this difference is not significant. Expression of bcl-xS and bcl-xL mRNA
Expression of anti-apoptotic and pro-apoptotic proteins in the I/R kidney
The expression of Bcl-2 and Bcl-xL proteins in the I/R kidneys of HP and SL rats is shown in Fig. 6. Before I/R injury, the baseline level of Bcl-2 and Bcl-xL protein expression was increased in the HP kidney when compared to SL rats. In response to ischemia or 1-4 h of reperfusion, the expression of Bcl-2 and Bcl-xL was markedly enhanced in the HP kidneys.
The expression of Bax, CPP32, and PARP in the kidney samples after I/R was displayed in Fig. 7. Expression of Bax, but not of CPP32 and PARP, was detected in sham-operated SL kidneys. The expression of all three of proteins was increased
during ischemic periods and after 1-4 h of reperfusion. However, the expression of all three of proteins was not increased in the HP kidneys. By densitometry analysis, the Bax/Bcl-2 ratio was significantly reduced in HP kidneys with I/R injury (Fig. 8). ROS release after I/R
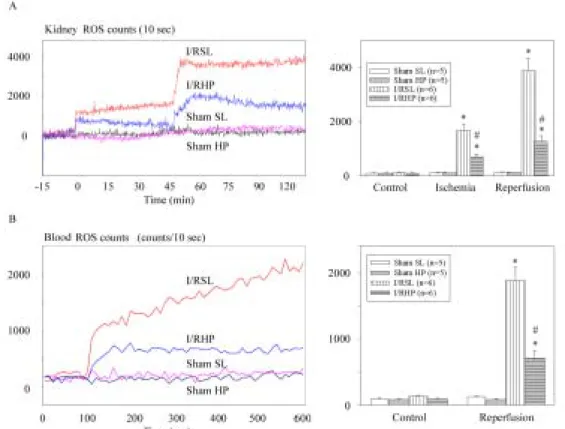
Kidney CL in vivo. Intrarenal arterial infusion of MCLA into the I/R kidney led to a rapid rise in CL counts to a peak at 2 min postinjection (Fig. 9A). The peak value of MCLAlucigenin-enhanced CL from the I/R kidney surface was then rapidly decreased to a basal level as renal circulation resumed (usually took 4 min). The basal CL level detected from the normal kidney surface was about 1,800-3,500 counts/10 sec. The sham-operated kidneys and the ischemic kidney without reperfusion did not show increased CL after administration of lucigenin (1,850±105 counts/10 sec in 5 sham-operated SL; 1,680±110 counts/10 sec in 5 sham-operated HP; 1,950±146 counts/10 sec in 5 ischemic SL; 2,190±155 counts/10 sec in 5 ischemic HP), suggesting that the enhanced CL resulted from reperfusion injury. To further confirm this hypothesis, in I/R SL rats treated with SOD prior to lucigenin injection at 1-4 h of reperfusion, the peak value of CL from the kidneys was depressed significantly (not presented), indicating that the increased CL counts were derived from ROS after I/R insult.
In Fig. 9A, lucigenin enhanced CL on the kidney surface was increased ~4-fold in 5 SL rats (8,702 ± 1,544 counts/10 sec), whereas increased ~2-fold (4,866 ± 1,344 counts/10 sec) in 5 HP rats at 5 min of reperfusion. Lucigenin-enhanced CL was not demonstrated in kidneys after 10-24 h of reperfusion in both SL and HP groups. It is uncertain whether these increases in CL following I/R derived from renal tubular cells, from endothelial cells, or from an increased number of phagocytes that generate ROS event in the lucigenin amplified system. However, increased CL counts from the kidney surface may represent the direct renal ROS generation and renal damage degree after I/R insults.
Effect of I/R on renal venous blood CL. We also adapted the CL method to examine the amounts of ROS in the renal venous samples. The CL levels of whole blood samples, wrapped with aluminum foil and stored in an iced box, were quickly determined within 2 h. The pH value in the control blood is around 7.38-7.45, whereas the pH value after ischemia is around 7.33-7.36. After 45 min-ischemia, the renal venous CL was increased 6-fold (from 1,173 to 8,511 counts/10 sec) in HP rats within 5 min of reperfusion, whereas the renal venous CL was increased 15-fold (1,098 to 15,120 counts/10 sec) in the SL rats. The increases in renal venous CL persisted for 4 h after reperfusion and returned nearly to their basal levels after 10-24 h of reperfusion.
DISCUSSION
Our previous studies, which found that HP kidneys adjust to their situation, becoming more tolerant to ischemic insults [Chen, 1993; Chien and Chen, 1994], suggest an important protective mechanism developed during HP. In the present study, we have demonstrated that HP led to a reduction in apoptosis formation and ROS generation in the kidney subjected to I/R injury. Upregulation in anti-death genes, Bcl-2, Bcl-xL, and HSP-70, in the kidney after HP provides direct protection against I/R induced oxidative stress.
A recent report demonstrated that adaptation to chronic hypoxia, or long-lasting hypoxia preconditioning (HP) increased tolerance to ischemia in a manner dissimilar to short-lasting HP [Tajima et al., 1994] and showed that preconditioning and adaptation were additively protective [26]. Furthermore, another important feature of long-lasting HP is that the protective effect still persists for a certain period after the removal of hypoxic environment [30].
During ischemia the cellular internal milieu resulting from hypoxia alone changes profoundly with the intracellular accumulation of protons [Holloway et al., 1986] and hypoxanthine [Paller et al., 1984]. These changes are complicated by the ROS stress and the marked increase in intracellular calcium during the stage of reperfusion [Opie, 1989]. Under these circumstances, the tertiary structure of proteins may change sufficiently to alter function. Because of the adverse effect of ischemia and reperfusion, it has been suggested that reduced damage during I/R can be improved in two major ways: (i) by limiting ischemic injury during surgery or graft preservation; and (ii) by protecting organ from the aggression of the initial reperfusion [Bonventre, 1992].
Renal protection follows HP [Chen, 1993; Chien and Chen, 1994; Chien et al., 1995] can The response of apoptotic cell death to I/R injury primarily found in the renal cortex and outer medulla was diminished in the HP group. In this investigation, apoptotic cell death, as a damaged marker determined by nuclear fragmentation with The association of lesser tubular damage with less apoptotic cell death in HP rats suggests that HP could attenuate I/R induced apoptosis by three possible mechanisms: a) an enhanced cellular protection against oxidant injury, b) an increase in free radical scavenging activity, and c) a reduction in ROS generation.
Cellular protection against oxidant injury by HSP 70, Bcl-2, Bcl-xL.
In this investigation synergetic upregulation of HSP 70, Bcl-2 and Bcl-xL during HP may provide more renal protection against ischemia/reperfusion damage. HSP 70 can function as chaperones [Yellon et al., 1992], improve transportation of repair proteins across cellular and subcellular membranes, prevent intracellular calcium overload, increase catalase activity [Karmazyn et al., 1990; Van Why et al., 1992], and reduce the ischemia/reperfusion induced oxidative injury [Kukreja et al., 1992; Mocanu, 1993]. Although HSPs can be induced in ischemia/reperfusion injury [Schober et al., 1997] by ATP depletion [Van Why et al., 1994], superoxide [Schoeniger et al., 1994] or increased cell calcium concentrations [Kiang et al., 1994], the accumulated amount of HSPs may not surmount the severe ischemia/reperfusion injury completely. It is therefore possible that an overabundance of HSP 70 before ischemia/reperfusion insult is able to attenuate the consequences of ischemia/reperfusion at the protein level. Kume and coworkers reported that heat shock pretreatment of rats, as another type of preconditioning method, induces more HSP 72 in the liver, reduces the succeeding ischemia/reperfusion injury, and results in a related improvement in the survival rate after reperfusion in an in vivo model [Kume et al., 1996]. A similar explanation by Murry et al. [1986] is that ischemic preconditioning can delay lethal cell injury in ischemic myocardium. Further evidence in support of this hypothesis is that over-expression of the rat inducible HSP 70 in a transgenic mouse increases the resistance of the heart to ischemic injury [Marber et al., 1995]. Taken together, these results and our data consistently implicate that the enhanced expression of HSP 70 provides a protective function against oxygen distress.
In contrast to the pattern of expression for HSP 70 during HP, Bcl-2 and a Bcl-2 related-gene, Bcl-xL are also activated in the HP kidney. The Bcl-2 family of proteins constitutes a critical checkpoint in cell death [Oltvai et al., 1994] and contains agonists and antagonists of apoptosis, and alterations in their ratio determine the life or death of a cell [Hanada et al., 1995]. These two opposite effects are the consequence of changes in the amount of proteins that promote cell survival, such as Bcl-2, or facilitate the activation of the endogenous cell death pathway, such as Bax [Oltvai et al., 1993]. Bax forms heterodimers with Bcl-2, depressing its protective influence on cell viability; if Bax homodimers predominate, cells are more disposed to undergo apoptosis [Miyashita et al., 1994]. The protective mechanisms by which Bcl-2 decreases the
sensitivity of cells to die are multi-factorial, including the regulation of calcium ion [Marin et al., 1996], attenuation in superoxide anion generation [Hockenbery et al., 1993], interference with the stimulation of proteases of the interleukin-1β-converting enzyme family [Shimizu et al., 1996], and stabilization of mitochondrial membrane permeability [Krajewski et al., 1993]. Another possible mechanism underlying the protective effect of Bcl-2 is its anti-proteolytic action []. Bcl-xL is similar in size and predicted structure to Bcl-2, and prevents apoptotic cell death. Over-expression of Bcl-xL regulates survival decisions within susceptible cells by functioning downstream of oxidant production [Fang et al., 1995] and is more efficient than Bcl-2 in preventing cell death induced by hypoxia [Shimizu et al., 1995], like the ischemic condition. Shimizu et al (1995) further suggested that Bcl-2 and Bcl-xL exert an anti-cell death function by a mechanism other than regulation of ROS activity.
In response to I/R damage, the expression of Bcl-2 and Bcl-xL is highly enhanced, whereas the expression and activity of caspase 3-like protease is significantly depressed in the HP group as compared to that in non-HP rats. In Caenorhabditis dlegans CED-3/ICE-mediated cell death is inhibited by gain-of-function mutations in the ced-9 gene. Ced-9 encodes a protein, which shows significant homology to the gene product of the mammalian bcl-2 gene [Martin, 1995]. Over-expression of Bcl-2 and its homolog Bcl-xL have recently been reported to inhibit activation of caspase 3 (CPP32) protease and subsequent cell death following various apoptotic stimuli [Chinnaiyan et al., 1996; Monney et al., 1995]. The ability to over-express HSP 70, Bcl-2 and Bcl-xL within the HP kidney may contribute to the decrease in caspase 3-like proteolytic activity, ROS generation and subsequent renal tubular cell death.
HP reduces free radical release during reperfusion
In conclusion, I/R injury is clinically relevant during renal surgical procedures such as anatrophic nephrolithotomy and kidney transplantation. The therapeutic approach from the current result shows that HP protects the kidney from subsequent I/R injury by reducing oxidative stress induced apoptosis cell death. The protective role of HP my be mediated by the up-regulation of HSP70, Bcl-2 and Bcl-xL.
Within the intact heart, ROS release from the cardiomyocytes, endothelial cells, and neutrophils [25,26Ferrari et al., 1993; Hess and Manson, 1984], whereas in the kidney, ROS come from the neutrophils, endothelial cells, and epithelial cells [23,24].
REFERENCES
1. Emerson MR. Nelson SR. Samson FE. Pazdernik TL. Hypoxia preconditioning attenuates brain edema associated with kainic acid-induced status epilepticus in rats. Brain Research. 825:189-93, 1999
2. Emerson MR. Nelson SR. Samson FE. Pazdernik TL. A global hypoxia preconditioning model: neuroprotection against seizure-induced specific gravity changes (edema) and brain damage in rats. Brain Research. Brain Research Protocols. 4:360-6, 1999.
3. Yamaoka Y, Shimahara Y, Nakatani K. Clinical role of blood ketone body ratio as an indicator evaluating hepatic tolerance for portal triad cross-clamping in cirrhotic liver section. Surg Res Comm 3: 87-93, 1988.
4. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74: 1124-1136, 1986.
5. Shiki K, Hearse DJ. Preconditioning of ischemic myocardium: reperfusion-induced arrhythmias. Am J Physiol 253: H1470-H1476, 1987.
6. Mocanu MM, Steare SE, Evans MCW, Nugent JH, Yellon DM. Heat stress attenuates free radical release in the isolated perfused rat heart. Free Radic. Biol Med. 15: 459-463, 1993.
7. ZN, and Korsmeyer SJ. (1994) Checkpoints of dueling dimers foil death wishes. Cell 79:189-192.
8. Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348: 384-387, 1992.
9. Bissonnette RP, Echeverri F, Mahoubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature 359: 552-554, 1992.
10. Fanidi A, Harrington EA, Evan GI. Co-operative interaction betwen c-myc and bcl-2 proto-oncogenes. Nature 359: 554-556, 1992.
11. Ansari R, Coates PJ, Greenstein BD, Hall PA. In situ end-labeling detects DNA strand breaks in apoptosis and other physiological and pathological states. J Pathol 170: 1-8, 1993.
12. Hockenbery DM, Otlavi ZN, Yin XM, Milliman CL, Korsmeyer SL. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75: 241-251, 1993. 13. Jacobson M, Burne JF, King MP, et al. Bcl-2 blocks apoptosis in cells lacking
mitochondrial DNA. Nature 362: 365-369, 1993.
14. Oltavi ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accerelerates programmed cell death. Cell 74: 609-619, 1993.
15. Boise LH, Gonzalez-Garcia M, Postema CE et al. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74: 597-608, 1993. 16. Shimizu S, Eguchi Y, Hosaka H, Kamiike W, Matsuda H, Tsujimoto Y. Prevention
of hypoxia-induced cell death by Bcl-2 and Bcl-xL. Nature 374: 811-813,1995. 17. Karmazyn M, Mailer K, Courrie RW. Acquisition and decay of heat sock enhanced
postischemic ventricular recovery. Am J Physiol 258: H424-H431, 1990.
18. Van Why SK, Hildebrandt F, Ardito T, Mann AS, Siegel NJ, Kashgarian M. Induction and intracellular location of HSP 72 after renal ischemia. Am J Physiol 263: F769-F775, 1992.
19. Van Why SK, Mann AS, Thulin G, Zuh H, Kashgarian M, Siegel NJ. (1994) Activation of heat-shock transcription factor by graded reductions in renal ATP, in vivo, in the rat. J. Clin. Invest. 94: 1518-1523.
20. Kiang JG, Carr FE, Burns MR, McClain DE. (1994) HSP-72 synthesis is promoted by increase in [Ca2+]i or activation of G proteins but not pHi or cAMP. Am J Physiol
267:C104-C114.
21. Nakajima T., Miyaji T., Kato A., Ikegaya N., Yamamoto T., and Hishida A. Uniphrectomy reduces apoptotic cell death and enhances renal tubular cell regeneration in ischemic ARF in rats.
22. Yellon, D.M., Latchman D.S. Stress proteins and myocardial protection. J Mol Cell Cardiol 24: 113-124, 1992.
23. Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J. Clin. Invest. 1995 95: 1446-1456.
24. Schoeniger LO, Andreoni KA, Ott GR, Risby TH, Bulkley GB, Udelsman R, Burdick JF, Buchman TG. Induction of heat-shock gene expression in postischemic pig liver depends on superoxide generation. Gastroenterology 1994; 106:117-184. 25. Opie LH. Reperfusion injury and its pharmacological modification. Circulation,
1989, 80:1049-1062.
26. Holloway JC, Phifer T, Henderson R, Welbourne TC. (1986) Renal acid-base metabolism after ischemia. Kidney Int 29: 989-994.
27. Paller MS, Hoidal JR, Ferris TF. Oxygen free radicals in ischemic acute renal failure. J Clin Invest 74: 1156-1164, 1984.
28. Fang W, Rivard JJ, Ganser JA, LeBien TW, Nath KA, Mueller DL, Behrens TW. Bcl-xL rescues WEHI 231 B lymphocytes from oxidant-mediated death following diverse apoptotic stimuli. J Immunol 155: 66-75, 1995.
29. Chen R. H., Su Y.H., Chuang R.L.C., Chang T.Y. Suppression of transforming growth factor-β-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene, 17: 1959-1968, 1998.
30. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labelling of nuclear DNA fragmentation. J Cell Biol 119: 493-501, 1992.
31. Chinnaiyan AM, Orth K, O'Rorke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway. J. Biol. Chem. 271: 4573-4596, 1996.
32. Monny L, Otter I, Oliver R, Ravn U, Mirzasaleh H, Fellay I, Poirier GG, Borner C. Bcl-2 overexpression blocks activation of the death protease CPP32/Yama/Apopain. Biochem. Biophys. Res. Commun. 221, 340-345, 1996.
33. Shimizu S, Eguchi Y, Kamiike W, Matsuda H, Tsujimoto Y. Bcl-2 expression prevents activation of the ICE cascade. Oncogene 12: 2251-2257, 1996.
34. Schott RJ, Rohmann S, Braun ER, Schaper W. Ischemic preconditioning reduces infarct size in swine myocardium. Circ Res 66: 1133-1142, 1990.
35. Li GC, Vasques JA, Gallagher KP, Lucchesi BR. Myocardial protection with preconditioning. Circulation 82: 609-619, 1990.
36. Liu X, Engelman RM, Moraru II, et al. Heat shock: a new approach for myocardial preservation in cardiac surgery. Circulation 86: II-358-363, 1992.
37. Maulik N, Engelman RM, Wei Z et al. Interleukin-1a-preconditioning reduces myocardial ischemia reperfusion injury. Circulation 88:II387-394, 1993.
38. Gross GJ, Auchampach JA. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res 70: 223-233, 1992.
39. Liu GS Thornton J, Van Winkle DM, Stanley AWH, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 84: 350-356, 1991.
40. Banerjee A, Locke-Winter C, Rogers KB et al. Preconditioning against myocardial dysfunction after ischemia and reperfusion by an alpha 1-adrenergic mechanism. Circ Res 73: 656-670, 1993.
41. Yang ZZ, and Zou AP. Transcriptional regulation of heme oxygenases by hypoxia-inducible factor-1α in renal medullary interstitial cells. Am J Physiol Renal Physiol. 281:F900-F908, 2001.
42. Semenza GL, and Wang GL: A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 12:5447-5454, 1992.
43. Kim CH, Cho YS, Chun YS, Park JW, Kim MS. Early expression of myocardial HIF-1α in response to mechanical stresses regulation by stretch-activated channels and the phosphatidylinositol 3-kinase signaling pathway. Circ Res 90:e25-e33, 2002.
44. Bunn HF and Poyton RO. Physiol Rev 76: 839-885, 1996.
45. Wang G, Jiang B, Rue E, and Semenza G. Proc Natl Acad Sci USA. 92, 5510-5514, 1995.
46. Kallio P, Pongratz I, Gradin K, McGuire J, and Poellinger L. Proc Natl Acad Sci USA, 94, 5667-5672, 1997.
47. Hanson ES, Foot LM, Leibold EA. Hypoxia post-transcriptionally activates iron-regulatory protein 2. J Biol Chem 19: 5047-5052.
48. Yeo EJ, Chun YS, Cho YS, Kim J, Lee JC, Kim MS, Park JW. YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J Natl Cancer Inst. 2003 95:516-25.
Fig. 1 Hypoxia effect on HIF-1α, Bcl-2, and HSP 70 protein expression. A. 10%
intermittent hypoxia exposure displayed the highest kidney HIF-1α expression (2.8 folds) after 1 day (1 d) of exposure. After two days (2 d) of exposure, the enhanced HIF-1α expression was partly decreased but still maintained at levels around 1.7-2.1 folds of control (0 d). B. After 2 days of 10% hypoxic exposure and intravenous CoCl2
administration (60 mg/kg), the kidney Bcl-2 and HSP 70 expression were enhanced. YC-1 significantly inhibited hypoxia and CoCl2-enhanced Bcl-2 and HSP 70 expression,
indicating that activation in HIF-1α can upregulate Bcl-2 and HSP 70 protein expression.
Fig. 2 Prolonged HP induced Bcl-xL, Bcl-2, and HSP 70 expression in the homogenates
of renal cortex (C) and medulla (M). After 2 d of hypoxic exposure, the expression in Bcl-xL, Bcl-2, and HSP 70 is significantly increased Note the increased expresion of Bcl-xL, Bcl-2, and HSP 70 after 1 w of HP.
Fig. 3 ROS detection from rat kidney surface in vivo and from renal venous blood. A:
Typical recordings of ROS from the kidney surface at 5 min of reperfusion in one sham-operated SL (sham SL) and HP (sham HP) rat and ischemia/reperfusion SL (I/RSL) and HP rat (I/RHP). The basal ROS levels before ischemia/reperfusion are around 1,00-220 counts/10 s. Lucigenin (1.0 mM) was injected at the 100 s after basal CL determination and elicited an increasing CL in the kidney subjected to I/R injury. The increased CLs return to control value within 5 min. B: After 45-min ischemia, the renal venous ROS increased at the time immediately after reperfusion (within 5 min). Note that a decrease of kidney surface and renal venous CLs was found in the HP rats.
Fig. 4 De novo formation of ROS and apoptotic cells in the I/R kidney. A: TUNEL
positive cells (brown stain) occur in both proximal (PT) and distal tubules (DT) in SL kidney subjected to 45-min ischemia and 4 h of reperfusion. ROS production (blue deposits) appears mainly in the PT, not DT. B: Significantly decreased TUNEL positive cells and ROS deposits display in HP kidney subjected to 45-min ischemia and 4-h reperfusion. C: The enhanced bax expression (brown stain) is concomitant with the ROS products in the I/R kidney of SL rat. However, a decreased Bax and ROS formation is observed in the I/RHP kidney.
Fig. 5 Western blot analysis using specific antibodies to Bax, Bcl-2, CPP32, and PARP
of homogenates of rat kidney subjected to I/R injury. Note the increased expression of Bax, CPP32, PARP after reperfusion. The expression of Bcl-2 appeared decreased after ischemia insult, and returned to its pre-ischemic level after reperfusion. A 15-min ischemia plus reperfusion. Western blot analysis using specific antibodies to Bax, Bcl-2, CPP32, and PARP of homogenates of rat kidney subjected to ischemia/reperfusion injury. Note the increased expresion of Bax, CPP32, PARP after reperfusion. The expression of Bcl-2 appeared decreased after ischemia insult, and returned to its pre-ischemic level after reperfusion. A 15-min ischemia plus reperfusion was sufficient to cause the increased expression of Bax/Bcl-2 ratio.
Fig. 6 Agarose gel electrophoresis of DNA from kidney with ischemia alone (0 h) and
ischemic/reperfusion (1, 4, 10, and 24 h) in SL (N) and HP (H) rats. DNA "ladder" pattern was visible during early reperfusion (1 h), grew progressively for 4 h, and diminished slowly thereafter. Note that DNA “ladder” pattern was significantly depressed in the HP rats within 1-4 h of reperfusion.
Fig. 7 Effect of hypoxia/reoxygenation (H/R) on renal proximal (PT) and distal tubular
(DT) cells. Isolated PT and DT cells were subjected to 15-min hypoxia (95% N2/5%
CO2) followed by 30-min reoxygenation (95% O2/5% CO2). Cell viability and tubular
apoptosis were measured by trypan blue exclusion and flow cytometry. Results are the mean ± SE of four cell preparations. H/R to PT and DT cells produced a statistically significant (P<0.05) decrease in cell viability and an increase in tubular apoptosis. Addition of 50 µM FC4S to PT and DT cells decreased cellular injury in PT, but not in
DT cells. * P <0.05 when compared to their control stage (C). # P<0.05 when compared to H/R stage.