Chiral Phosphinophenyloxazolines Bearing Alkoxymethyl Substituents:
Synthesis and Application in the Palladium Catalyzed Allylic Substitution
Reactions
Rong-Jiunn Chen ( ) and Jim-Min Fang* ( )
Department of Chemistry, National Taiwan University, Taipei 106, Taiwan, R.O.C.
The chiral phosphine-oxazoline ligands 3 and 4 bearing 4-alkoxymethyl substituents on the oxazoline ring with (R)-configuration were prepared fromL-serine methyl ester in 66% and 33% yields, respectively. Along this synthetic pathway, theb-hydroxylamides derived fromL-serine methyl ester and 2-halobenzoyl chlorides were expediently converted to the corresponding oxazolines by using diethylaminosulfur trifluoride as the activation agent. Potassium diphenylphosphide was the reagent of choice for replacing the bromine atom on the phenyl ring, giving the desired oxazoline-phosphine ligands 3 and 4. Together with [Pd(h3 -allyl)Cl]2, ligands 3 and 4 induced an enantioselective allylic substitution reaction of 1,3-diphenyl-2-pro-penyl acetate by dimethyl malonate. Although ligands 3 and 4 exhibit the (R)-configuration, differing from the (S)-configuration of Pfaltz-Helmchen-Williams phosphine-oxazoline ligands, all these ligands led to the same enantiotopic preference in the allylic substitution reaction. To facilitate the recovery and reuse of the phosphine-oxazoline ligand, immobilization on Merrifield resin was attempted, albeit in low loading. Keywords: Phosphine; Oxazoline; Palladium; Chiral ligands; Allylic substitution.
INTRODUCTION
The chiral ligands prepared by connection of phosphine and oxazoline moieties have been successfully used to pro-mote a variety of metal catalyzed asymmetric organic reac-tions.1For example, Pfaltz,2Helmchen,3and Williams4groups have independently found the highly enantioselective substi-tution reaction of 1,3-diphenyl-2-propenyl acetate with di-methyl malonate by the catalysis of a palladium complex 2 that is prepared in situ from the phosphine-oxazoline ligand 1 and [Pd(h3-allyl)Cl]2.5The (S)-configuration of the chiral ligand 1 originates from (S)-valinol. We report herein the synthesis of the P,N-ligands 3 and 4 that bear 4-alkoxymethyl substituents on the oxazoline ring. In addition to the phospho-rus and nitrogen atoms, the 4-alkoxymethyl group may also serve as a coordination site for a metal ion to render a
stereo-control in organic transformation, such as the Pd-mediated allylic substitution reactions.5
RESULTS AND DISCUSSION
The condensation reaction ofL-serine methyl ester (as the hydrochloric salt 5) with 2-chlorobenzoyl chloride (6) in the presence of Et3N gave an amide 7 in 78% yield (Scheme I). Several attempts to form oxazoline by activation of the hydroxyl group in 7 failed to give the desired cyclization product. For example, treatment of 7 with p-TsCl or SOCl2 in the presence of Et3N gave only the elimination product 8.6 In another approach, the hydroxylamide 7 was heated with PPh3/CCl4in the presence of Et3N to afford a mixture of alkene 8 and the desired oxazoline 9 in 27% and 30% yields,
Dedicated to Professor Ching-Erh Lin on the Occasion of his 66thBirthday and his Retirement from National Taiwan University * Corresponding author. Fax: +886-2-23636359; E-mail: jmfang@ntu.edu.tw
P O N Ph Ph Pd H P O N Ph Ph H X P O N Ph Ph O PhCH2O H P O N Ph Ph MeO H S R R 1 2 3 4
respectively. Finally, diethylaminosulfur trifluoride (DAST) turned out to be the reagent of choice for the transformation of hydroxylamide 7 into oxazoline 9.7Thus, treatment of 7 with DAST at -78°C in the presence of K2CO3gave 9 exclu-sively in 99% yield, without complication of the side-product 8. Unlike the valinol derived oxazolines, e.g., compound 1, the oxazoline 9 containing an electron-withdrawing ester group renders the proton at C-4 susceptible to alkaline condi-tions. Indeed, oxazoline 9 was readily changed to alkene 8 upon treatment with a base, e.g., NaOH and NaH, presumably via the H-4 abstraction to cause a rupture of the oxazoline ring (Scheme I).
By a procedure similar to that for amide 7, the acyl chloride derived from 2-bromobenzoic acid (10) was reacted in situ with the serine derivative 5 to give the amide 11 in 85% yield (Scheme II). The direct amidation reaction of acid 10 with 5 was also carried out, albeit in modest yields (50-75%), by using dicyclohexylcarbodiimide (DCC) or 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide (EDCI) as the dehydrat-ing agents and Et3N and 4-dimethylaminopyridine (DMAP) as the reaction promoters. Hydroxylamide 11 was then treated with DAST in the presence of K2CO3to afford the desired oxazoline 12 in a quantitative yield.7
The reduction of ester 12 with LiAlH4at room tempera-ture afforded the alcohol 13, which was treated with NaH and MeI to give the ether 14 in a 92% overall yield (Scheme II). One should avoid using excessive amounts of LiAlH4; other-wise, the bromophenyl moiety in 12 would also be reduced.
The alkylation reaction of alcohol 13 with 1-benzyloxy-4-(3-bromopropyl)benzene was achieved in THF/DMF (5:1) solution by the assistance of Ag2CO3, giving ether 15 in 58% yield. Substitution of bromine atom with diphenylphosphine group was not trivial as one would expect. We have tried sev-eral methods using different combinations of reagents, e.g., BuLi (or t-BuLi)/Ph2PCl in tetramethylethylenediamine (TMEDA), Li (or Mg)/Ph2PCl (or PPh3) in various condi-tions, and even with catalysts of CuI or Pd(PPh3)2Cl2, but failed to procure the desired substitution product.8Finally, the bromophenyl compounds 14 and 15 were reacted with KPPh2to afford the desired oxazoline-phosphine ligands 3 and 4.9The1H NMR spectra indicated that the aromatic pro-tons (H3”) at the ortho-position of the PPh2group appeared at the relatively high fields ofd 6.49 (for 3) and 6.86 (for 4), pre-sumably due to the shielding effect of the phenyl group in a pseudo-equatorial orientation (Fig. 1, see below). The methy-lene protons (H1’) adjacent to the methoxy group in 3 also ap-peared at the relatively high fields ofd 2.44 and 2.66, presum-ably due to the shielding effect of the pseudo-axial phenyl group. The optimal yields of 3 (86%) and 4 (75%) were pro-cured by stirring 14 and 15, respectively, with 5 equiv of KPPh2in THF at room temperature for 24 h. Use of less NH3+Cl -HO O MeO Cl O Cl Cl O N MeO O H Et3N Cl O N O MeO H Cl O N HO O MeO H Cl O N MeO O H B B + 5 6 7 (78%) + 8 4 9 4 see the Text
Scheme I NH3+Cl -HO O MeO Br O N MeO O X O N O PhCH2O Br O HO Br O N HO Br O N HO O MeO H Br O N MeO PPh2 O N MeO 3 + 5 10 11 12 13 14 X = Br 15 i ii iii iv v vi X = PPh3 4 v 1' 1" 1' Scheme II
Reagents and conditions: (i) SOCl2, 25°C, 3 h; Et3N, CH2Cl2,
25°C, 4 h; 85%. (ii) DAST, CH2Cl2, -78°C, 1 h; K2CO3, -78
°C, 3 h; 99%. (iii) LiAlH4/Et2O, THF, 25°C, 4 h; 95%. (iv)
NaH, MeI, THF, 25°C, 3 h; 96%. (v) KPPh2, THF, 25°C, 24 h;
86% for 3 and 75% for 4. (vi) NaH, PhCH2OC6H4(CH2)3Br,
amounts (1-3 equiv) of KPPh2resulted in lower yields. When 15 was heated with KPPh2in refluxing THF (68°C), a com-plicated product mixture containing the alcohol 13 and 3-(4-benzyloxyphenyl)propanol, in addition to the desired phos-phine product 4, was obtained in considerable amounts. The cleavage of ether linkage in 15 might result from the nucleo-philic attack at the carbinyl carbons (C-1¢ and C-1²) by di-phenylphosphine or bromine anions.
Phosphine 4 were partially oxidized in the air by the ca-talysis of Pd/C under acidic conditions,10e.g., in AcOH/MeOH solution (1:10), to give the corresponding phosphine oxide 16. The characteristic trivalent phosphorus signal in 4 oc-curred atd -22.96 in the31P NMR spectrum, whereas the pentavalent phosphorus in 16 appeared at a much lower field ofd 32.76.
Interestingly, treatment of the chlorophenyl oxazoline 9 with t-BuLi/Ph2PCl in Et2O (-78 to 25°C, 18 h) gave a 32% yield of 17 with retention of the chlorine atom. By the chela-tion effect of the neighboring oxazoline moiety,11compound 9 might undergo the ortho-lithiation, and then react with Ph2PCl to afford the product 17.
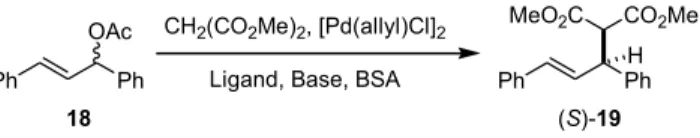
With compounds 3 and 4 in hand, a wide range of asym-metric organic reactions with catalysis of transition metals could be assessed by using these chiral oxazoline-phosphine ligands.1-5To demonstrate this possible application of com-pounds 3 and 4, we firstly investigated the substitution reac-tion of 1,3-diphenyl-2-propenyl acetate (18) with dimethyl malonate by the catalysis of [Pd(h3-allyl)Cl]2.5According to the previously reported reaction protocol,12 N,O-bis(trimeth-ylsilyl)acetamide (BSA) and a base (KOAc or CsOAc) were also used as the promoters in addition to the chiral ligands (3 or 4). The best result (entry 4, Table 1), in 96% chemical yield and 90% ee, was obtained from the reaction using 3 equiv of dimethyl malonate, 20 mol % of ligand 3, 5 mol % of
[Pd(h3-allyl)Cl]2, 1 mol % of CsOAc and 10 equiv of BSA in CH2Cl2solution. By comparison with the previous reports,13 the optically active product 19 with levorotation predomi-nated in the (S)-enantiomer. The ee value was determined by HPLC analysis on a Chiralcel OD column, where the (S)-en-antiomer was more polar than the (R)-en(S)-en-antiomer. Using ligand 4 for the allylic substitution reaction resulted in the same stereoselectivity, albeit in modest enantioselectivity (£ 65% ee).
In comparison, the (R)-configuration in ligands 3 and 4 (the alkoxymethyl substituents shown on thea-face) differs from the (S)-configuration in ligand 1 (the isopropyl substi-tutent shown on theb-face). However, either the (S)-ligand 1 or the (R)-ligands 3/4 induced the same enantiotopic prefer-ence in the allylic substitution reaction, giving (S)-19 as the dominant product. The rationale for the stereoselectivity us-ing ligand 1 has been proposed by Helmchen and cowork-ers.3,5Their proposed transition state (Fig. 1a) includes sev-eral important features: (i) the isopropyl substituents on the oxazoline ring forces the triarylphosphine scaffold to tilt to-ward thep-allyl moiety, (ii) the p-allyl moiety prefers the exo-orientation to minimize the steric effect against the pseudo-equatorial phenyl group on the phosphorus center, and (iii) the nucleophilic attack of dimethyl malonate occurs at C-1 that is trans to the phosphorus atom. On the other hand, the alkoxy group in ligand 3 (or 4) can coordinate to the palla-dium, and thus exposes the less hindered exo-face to accom-modate thep-allyl moiety (Fig. 1b). These two working mod-els interpret well how the allylic substitution reaction occurs in a highly stereoselective manner to afford the product
pre-P O N Ph Ph O PhCH2O P O N Ph Ph O PhCH2O O 4 16 air, Pd/C, H+ Cl O N O PhCH2O Ph2P Cl O N O PhCH2O 9 17 t-BuLi, Ph2PCl Et2O,-78oC to rt Ph Ph OAc Ph Ph MeO2C CO2Me H CH2(CO2Me)2, [Pd(allyl)Cl]2
Ligand, Base, BSA
18 (S)-19 N P Pd Ph H H O H Ph i-Pr N P Pd Ph H H O H O R Ph H3" Nu (a) Nu less hindered exo-face (b) pseudo-equatorial 1 3 pseudo-axial 1'
Fig. 1. Working models for the enantioselective allylic substitution reactions: (a) using (S)-ligand 1, and (b) using (R)-ligand 3 or 4.
dominating in the (S)-isomer. In comparison with the present study, somewhat higher enantioselectivity in the previously reports3,5may be accounted for by the slight orientation twist of the pseudo-equatorial phenyl groups in the two transition states.
Immobilization on polymer and other supports is a gen-eral method for facilitation of the recovery and reuse of the ligand and metal catalyst.14
In one approach, the benzyl pro-tecting group in 4 was removed by treatment with AlCl3and N,N-dimethylaniline,15and the resulting phenol product 20 was reacted with Merrifield resin (chloromethylated polysty-rene) in the presence of Cs2CO3in DMF to give the resin-supported oxazoline-phosphine ligand 21.16However, the loading of oxazoline-phosphine ligands was rather low, 0.33 mmol/g as estimated by elemental analysis of the nitrogen content.
CONCLUSION
In summary, we have devised an efficient method for the synthesis of chiral oxazoline-phosphine ligands bearing an alkoxymethyl group as the additional coordination site. The use of these chiral ligands has been demonstrated in a
palladium catalyzed asymmetric allylic substitution reaction. We are currently searching for an improved preparation of the immobilized oxazoline-phosphine ligands that may be applied to facilitate catalytic asymmetric reactions.
EXPERIMENTAL General Procedures
All reactions requiring anhydrous conditions were con-ducted in flame-dried apparatus under an atmosphere of ar-gon or nitrogen. Syringes and needles for the transfer of re-agents were dried at 100°C and allowed to cool in a desicca-tor over P2O5before use. Ethers were distilled from sodium benzophenone ketyl; (chlorinated) hydrocarbons, and amines from CaH2. Reactions were monitored by TLC using pre-coated with a 0.25 mm layer of silica gel containing a fluores-cent indicator. Column chromatography was carried out on Kieselgel 60 (40-63mm). Melting points are uncorrected. Op-tical rotations were measured on a digital polarimeter with a cuvette of 10 cm length. [a]DValues are given in 10-1deg cm2 g-1. Chemical shifts of1H,13C and31P NMR spectra are re-ported relative to CHCl3[dH7.24,dC(central line of t) 77.0] and H3PO4(dP= 0). Distortionless enhancement polarization transfer (DEPT) spectra were taken to determine the types of carbon signals.
Methyl (S)-2-(2-Chlorobenzamido)-3-hydroxypropanoate (7)
A mixture of 2-chlorobenzoic acid (11.6 g, 74 mmol) and SOCl2(52 g, 224 mmol) was stirred at room temperature for 3 h and then concentrated under reduced pressure to give
Table 1. Allyllic substitution reactions of 1,3-diphenyl-2-propenyl acetate (18) with dimethyl malonate using [Pd(h3 -allyl)Cl]2and chiral ligand (3 or 4), giving product 19a
Entry Ligand (mol %) [Pd(allyl)Cl]2 (mol %) Base (mol %) BSA (equiv) Solvent Reaction time (h) Yield (%) Ee (%) Config. b 1 3 (10) 5 KOAc (150) 0 CH2Cl2 72 00 - -2 3 (10) 5 KOAc (5) 3 CH2Cl2 48 21 03 NDc 3 3 (10) 5 KOAc (5) 3 CH2Cl2/DMF 48 70 49 S 4 3 (20) 5 CsOAc (1) 10 CH2Cl2 48 96 90 S 5 3 (20) 5 CsOAc (1) 10 CH3CN 48 98 79 S 6 4 (20) 10 KOAc (5) 1.5 CH2Cl2 40 96 53 S 7 4 (20) 10 KOAc (5) 1.5 THF 54 63 65 S 8 4 (20) 5 CsOAc (1) 10 CH2Cl2 06 97 42 S 9 4 (20) 5 CsOAc (1) 2 CH3CN 02 98 10 S
aThe reaction was conducted with 3 equiv of dimethyl malonate at 25°C. b
Configuration of major enantiomer. cNot determined. P O N Ph Ph O O H R 20 R = H R = Merrifield resin 21
the corresponding 2-chlorobenzoyl chloride. A solution of L-serine methyl ester hydrochloride (10 g, 67 mmol) in CH2Cl2(150 mL) and Et3N (18.9 g, 186 mmol) were added. The reaction mixture was stirred for 4 h at room temperature, and quenched by addition of water (100 mL). The aqueous phase was separated and then extracted with CH2Cl2(100 mL). The combined organic phase was dried (MgSO4), fil-tered, concentrated, and chromatographed on a silica gel col-umn with elution of EtOAc/hexane (3:7) to give amide 7 (14.8 g, 86%). TLC (EtOAc/hexane, 1:1) Rf= 0.50; [a]D23= +26.2 (c = 1.0, CH2Cl2); IR (KBr, cm-1) 3347, 2962, 1750, 1619, 1546, 1377, 1217, 1060;1H NMR (CDCl3, 400 MHz)d 2.75 (t, J = 5.9 Hz, OH), 3.78 (s, 3H), 4.00-4.10 (m, 2H), 4.83 (m, 1H), 7.15-7.40 (m, 4H), 7.62-7.65 (dd, J = 1.2, 8.0 Hz, 1H);13C NMR (CDCl3, 100 MHz)d 52.9, 55.3, 63.3, 127.1, 130.30, 130.38, 130.9, 131.7, 134.0, 166.5, 170.6; HR-FAB-MS calcd for C11H1335ClNO4(M++ H): 258.0455, found: m/z 258.0526.
Methyl 2-(2-Chlorobenzamido)-2-propenoate (8) and
(
S)-2-(2-Chlorophenyl)-4-methoxycarbonyl-4,5-dihydro-1,3-oxazole (9)
A mixture of hydroxylamide 7 (200 mg, 0.77 mmol), PPh3(300 mg, 1.2 mmol) and Et3N (120 mg, 1.2 mmol) in CCl4(30 mL) was heated under reflux for 24 h. The mixture was concentrated and chromatographed on a silica gel col-umn by elution with EtOAc/hexane (2:8) to give alkene 8 (50 mg, 27%) and oxazoline 9 (56 mg, 30%).
Hydroxylamide 7 was treated with DAST, by a proce-dure similar to that for 12, to give oxazoline 9 in a quantita-tive yield. Alkene 8: TLC (EtOAc/hexane, 3:7) Rf= 0.53; IR (KBr, cm-1) 3383, 2968, 1728, 1679, 1532, 1336, 1213, 1050;1H NMR (CDCl3, 400 MHz)d 3.85 (s, 3H), 6.00 (br s, 1H), 6.79 (br s, 1H), 7.33-7.44 (m, 3H), 7.69 (dd, J = 7.4, 1.8 Hz, 1H), 8.50 (br s, NH);13C NMR (CDCl3, 100 MHz)d 53.1, 109.7, 127.2, 130.2, 130.5, 130.7, 130.9, 131.8, 134.5, 164.4, 164.8; HR-FAB-MS calcd for C11H11ClNO3(M++ H): 240.0349, found: m/z 240.0428. Oxazoline 9: TLC (EtOAc/hexane, 3:7) Rf= 0.22; [a]D 23 = +26.9 (c = 0.1, CH2Cl2); IR (KBr, cm-1) 2958, 1733, 1694, 1594, 1439, 1318, 1267, 1053;1H NMR (CDCl3, 400 MHz)d 3.74 (s, 3H), 4.53 (dd, J = 8.8, 10.6 Hz, 1H), 4.63 (dd, J = 8.2, 8.6 Hz, 1H), 4.92 (dd, J = 8.1, 10.7 Hz, 1H), 7.21-7.39 (m, 3H), 7.72 (dd, J = 1.7, 7.7 Hz, 1H);13C NMR (CDCl3, 100 MHz)d 52.6, 68.6, 69.5, 126.4, 126.5, 130.6, 131.5, 131.9, 133.4, 165.1, 171.2; HR-FAB-MS calcd for C11H11ClNO3
(M++ H): 240.0349, found: m/z 240.0427.
Methyl (S)-2-(2-bromobenzamido)-3-hydroxypropanoate (11)
Amidation of 2-bromobenzoic acid withL-serine meth-yl ester hydrochloride, by a procedure similar to that for 7, gave 11 in 85% yield. TLC (EtOAc/hexane, 1:1) Rf= 0.50; [a]D23= +26.9 (c = 1.0, CH2Cl2); IR (KBr, cm-1 ) 3401, 3345, 2961, 1750, 1619, 1542, 1376, 1057;1H NMR (CDCl3, 400 MHz)d 3.09 (br s, OH), 3.73 (s, 3H), 3.92-4.07 (m, 2H), 4.76 (m, 1H), 7.06 (d, J = 5.6 Hz, NH), 7.18-7.30 (m, 2H), 7.47 (dd, J = 1.8, 7.5 Hz, 1H), 7.53 (dd, J = 1.2, 7.9 Hz, 1H);13C NMR (CDCl3, 100 MHz)d 52.6, 55.0, 62.7, 119.3, 127.4, 129.4, 131.4, 133.3, 136.7, 167.7, 170.5; HR-FAB-MS calcd for C11H1279BrNO4(M++ H): 301.9950, found: m/z 302.0028.
(
S)-2-(2-Bromophenyl)-4-methoxycarbonyl-4,5-dihydro-1,3-oxazole (12)
A solution of hydroxylamide 11 (232 mg, 0.77 mmol) and DAST (0.12 mL, 0.93 mmol) in CH2Cl2(30 mL) was stirred at -78°C for 1 h and then to which was added K2CO3 (0.21 g, 1.6 mmol). The mixture was stirred for 3 h at -78°C, warmed to room temperature, and quenched by slow addition of an aqueous NaHCO3solution (5%, 50 mL). The mixture was extracted with CH2Cl2(30 mL´ 2). The combined or-ganic phase was dried (MgSO4), filtered, concentrated, and chromatographed on a silica gel column with elution of EtOAc/hexane (3:7) to give oxazoline 12 (216 mg, 99%). TLC (EtOAc/hexane, 1:1) Rf= 0.71; [a]D23= +90.6 (c = 0.1, CH2Cl2); IR (KBr, cm-1) 2957, 1745, 1648, 1592, 1437, 1361, 1211, 1027;1H NMR (CDCl3, 400 MHz)d 3.80 (s, 3H), 4.60 (m, 1H), 4.71 (m, 1H), 4.98 (dd, J = 8.0, 10.6 Hz, 1H), 7.24-7.34 (m, 2H), 7.61 (dd, J = 1.0, 8.0 Hz), 7.72 (dd, J = 1.8, 7.6 Hz, 1H);13C NMR (CDCl3, 100 MHz)d 52.8, 68.7, 69.8, 121.9, 127.1, 128.9, 131.7, 132.1, 133.9, 165.9, 171.3; HR-FAB-MS calcd for C11H11BrNO3(M++ H): 283.9844, found: m/z 283.9922.
(
S)-2-(2-Bromophenyl)-4-hydroxymethyl-4,5-dihydro-1,3-oxazole (13)
A solution of ester 12 (4.41 g, 15.6 mmol) in THF (150 mL) was treated with LiAlH4(18.7 mL of 1 M solution in Et2O) at room temperature for 1 h. After sequential addition of water (1 mL), aqueous NaOH (15%, 1 mL) and water (3 mL), the mixture was filtered through a pad of Celite, and washed with CH2Cl2(50 mL) and MeOH/CH2Cl2(5:95, 50 mL). The organic phase was concentrated and then chro-matographed on a silica gel column with elution of EtOAc to
give alcohol 13 (3.78 g, 95%). TLC (EtOAc/hexane, 8:2) Rf= 0.26; [a]D22= +58.3 (c = 1.0, CH2Cl2); IR (KBr, cm-1 ) 3351, 2939, 1653, 1591, 1434, 1362, 1250, 1027;1H NMR (CDCl3, 400 MHz)d 3.55 (m, 1H), 3.57 (br s, OH), 3.72 (m, 1H), 4.20 (m, 1H), 4.35 (m, 1H), 7.16-7.24 (m, 2H), 7.52-7.56 (m, 2H); 13 C NMR (CDCl3, 100 MHz)d 63.8, 68.3, 69.6, 121.6, 127.0, 128.2, 129.4, 131.1, 131.7, 133.6, 164.8; HR-FAB-MS calcd for C10H11BrNO2(M++ H): 255.9895, found: m/z 255.9974.
(
R)-4-Methoxymethyl-2-(2-bromophenyl)-4,5-dihydro-1,3-oxazole (14)
A solution of alcohol 13 (3.00 g, 11.8 mmol) in THF (30 mL) was stirred with NaH (14.2 mmol, 60% dispersion in mineral oil washed with anhydrous hexane before use) at room temperature for 10 min. A solution of MeI (1.68 g, 11.8 mmol) in THF (30 mL) was added dropwise. The mixture was stirred for 3 h, filtered through a pad of Celite, and washed with EtOAc (50 mL). The organic phase was concentrated and then chromatographed on a silica gel column with elution of EtOAc/hexane (3:7) to give ether 14 (3.04 g, 96%). TLC (EtOAc/hexane, 1:1) Rf = 0.33; [a]D 22 = +64.4 (c = 1.0, CH2Cl2); IR (KBr, cm-1) 2931, 1651, 1593, 1472, 1358, 1245, 1031;1H NMR (CDCl3, 400 MHz)d 3.37 (s, 3H), 3.45 (m, 1H), 3.66 (m, 1H), 4.29 (m, 1H), 4.42-4.53 (m, 2H), 7.22 (m, 1H), 7.29 (m, 1H), 7.59 (dd, J = 7.9, 1.3 Hz, 1H), 7.66 (dd, J = 7.6, 1.8 Hz, 1H);13C NMR (CDCl3, 100 MHz)d 58.9, 66.3, 70.3, 74.1, 121.3, 126.6, 129.4, 131.0, 131.3, 133.3, 163.8; HR-FAB-MS calcd for C11H13BrNO2(M++ H): 270.0051, found: m/z 270.0129.
(
R)-4-{[3-(4-benzyloxyphenyl)propoxy]methyl}-2-(2-bromophenyl)-4,5-dihydro-1,3-oxazole (15)
Alcohol 13 treated with NaH, Ag2CO3(1 equiv) and 1-benzyloxy-4-(3-bromopropyl)benzene in THF/DMF (5:1) solution for 5 h at room temperature, by a procedure similar to that for 14, gave ether 15 in 58% yield. TLC (EtOAc/hex-ane, 3:7) Rf= 0.45; [a]D 24= +20.0 (c = 1.0, CH2Cl2); IR (KBr, cm-1) 2928, 1652, 1512, 1456, 1356, 1240, 1025;1H NMR (CDCl3, 400 MHz)d 1.87 (m, 2H), 2.63 (t, J = 7.3 Hz, 2H), 3.47-3.52 (m, 3H), 3.74 (dd, J = 7.7, 2.5 Hz, 1H), 4.35-4.40 (m, 1H), 4.46-4.55 (m, 2H), 5.02 (s, 2H), 6.89 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.6 Hz, 2H), 7.24-7.45 (m, 7H), 7.62 (dd, J = 7.7, 1.0 Hz, 1H), 7.69 (dd, J = 7.7, 1.7 Hz, 1H);13C NMR (CDCl3, 100 MHz)d 31.24, 31.28, 66.7, 69.9, 70.5, 70.7, 72.5, 114.6, 121.6, 126.9, 127.3, 127.7, 128.4, 129.2, 129.7, 131.2, 131.5, 133.6, 134.1, 137.1, 156.9, 164.2; HR-FAB-MS calcd for C26H27BrNO3 (M+ + H): 480.1096, found: m/z 480.1165.
(
R)-4-Methoxymethyl-2-(2-diphenylphosphino)phenyl-4,5-dihydro-1,3-oxazole (3)
Under an atmosphere of argon, KPPh2(1.05 mmol, 2.1 mL of 0.5 M solution in THF) was added to a solution of the bromophenyl compound 14 (56 mg, 0.21 mmol) in THF (3 mL). The mixture was stirred at room temperature for 24 h, and then to which was added water (20 mL) and CH2Cl2(20 mL). The aqueous phase was separated and then extracted with CH2Cl2(30 mL). The combined organic phase was dried (MgSO4), filtered, concentrated, and chromatographed on a silica gel column with elution of EtOAc/pentane (1:9) to give 3 (68 mg, 86%). TLC (EtOAc/hexane, 3:7) Rf= 0.34; [a]D22= +33.3 (c = 1.0, CH2Cl2); IR (KBr, cm-1) 2928, 1643, 1536, 1486, 1299, 1120;1H NMR (CDCl3, 400 MHz)d 2.44 (dd, J = 13.9, 7.4 Hz, 1H), 2.60 (dd, J = 13.9, 6.7 Hz, 1H), 3.27 (s, 3H), 3.50 (dd, J = 9.4, 4.1 Hz, 1H), 3.62 (dd, J = 9.4, 3.3 Hz, 1H), 4.42 (m, 1H), 6.49 (d, J = 8.0 Hz, 1H), 7.24-7.65 (m, 13H);13C NMR (CDCl3, 100 MHz)d 48.0, 58.8, 73.6, 73.7, 126.8, 128.33, 128.38, 128.4, 128.51, 128.58, 128.6, 128.7, 131.3, 132.61, 132.66, 132.80, 132.85, 134.3, 137.6, 137.7, 138.3, 138.4, 166.5;31P NMR (CDCl3, 162 MHz)d -22.87; HR-FAB-MS calcd for C23H23NO2P (M+ + H): 376.1388, found: m/z 376.1618.
(
R)-4-[3-(4-Benzyloxyphenyl)propoxy]methyl-2-(2-diphen-ylphosphino)phenyl-4,5-dihydro-1,3-oxazole (4)
Treatment of the bromophenyl compound 15 with KPPh2, by a procedure similar to that for 3, gave compound 4 in 75% yield. TLC (EtOAc/hexane, 3:7) Rf= 0.54; [a]D 23 = -12.5 (c = 1.0, CH2Cl2); IR (KBr, cm-1) 3064, 2926, 1641, 1512, 1485, 1241, 1027;1H NMR (CDCl3, 400 MHz)d 1.83 (m, 2H), 2.47 (dd, J = 7.6, 13.8 Hz, 1H), 2.53-2.69 (m, 3H), 3.32-3.45 (m, 2H), 3.54 (m, 1H), 3.72 (m, 1H), 4.42 (m, 1H), 5.01 (s, 2H), 6.86 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 7.27-7.63 (m, 18H); 13C NMR (CDCl3, 100 MHz)d 31.2, 31.3, 48.2, 70.0, 70.3, 71.6, 71.7, 114.7, 126.8, 127.4, 127.8, 128.4, 128.49, 128.55, 128.62, 128.65, 128.7, 129.2, 131.3, 132.6, 132.7, 132.8, 132.9, 137.1, 138.4, 157.0, 166.6;31P NMR (CDCl3, 162 MHz)d -22.96; HR-FAB-MS calcd for C38H37NO3P (M+ + H): 586.2433, found: m/z 586.2503.
(
R)-4-[3-(4-Benzyloxyphenyl)propoxy]methyl-2-(2-diphen-ylphosphorino)phenyl-4,5-dihydro-1,3-oxazole (16) Phosphine 4 in MeOH/AcOH (10:1) solution was stirred in the air for 18 h to give phosphine oxide 16. TLC (EtOAc/ hexane, 8:2) Rf= 0.34; [a]D23= -13.7 (c = 1.0, CH2Cl2); IR (KBr, cm-1) 3063, 1658, 1514, 1439, 1322, 1236, 1177, 1117;
1 H NMR (CDCl3, 400 MHz)d 1.79 (m, 2H), 2.53 (t, J = 7.6 Hz, 2H), 2.68 (m, 1H), 2.95 (m, 1H), 3.12 (m, 1H), 3.26 (m, 1H), 3.40 (t, J = 8.6 Hz, 1H), 3.68 (m, 1H), 4.52 (m, 1H), 5.01 (s, 2H), 6.85 (d, J = 8.5 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 7.15 (m, 1H), 7.24-7.50 (m, 10H), 7.62-7.89 (m, 7H), 8.02 (m, 1H);13C NMR (CDCl3, 100 MHz)d 31.3, 31.4, 46.9, 70.0, 70.2, 71.0, 71.1, 114.7, 127.1, 127.4, 127.8, 128.24, 128.36, 128.43, 128.53, 128.67, 128.7, 128.8, 128.9, 129.2, 130.3, 130.4, 130.7, 130.8, 131.1, 131.2, 131.4, 131.9, 133.9, 134.1, 137.1, 156.9, 166.8;31P NMR (CDCl3, 162 MHz)d 32.76 ppm; HR-FAB-MS calcd for C38H37BrNO4P (M++ H): 602.2382, found: m/z 602.2610; Anal. Calcd for C38H36NO4BrP: C, 75.86; H, 6.03; N, 2.33. Found: C, 75.97; H, 6.12; N, 1.98.
(
R)-4-[3-(4-Benzyloxyphenyl)propoxy]methyl-2-(2-chloro-6-diphenylphosphino)phenyl-4,5-dihydro-1,3-oxazole (17) To a solution of 9 (502 mg, 2.1 mmol) in Et2O (20 mL) was added dropwise t-BuLi (1.6 mL of 1.5 M solution in THF) at -78°C. The mixture was stirred for 3 h; a solution of ClPPh2 (946 mg, 4.3 mmol) in Et2O (10 mL) was added dropwise at -78°C. The reaction mixture was stirred at room temperature for 18 h, quenched by addition of water (30 mL), and extracted with CH2Cl2(40 mL´ 2). The organic phase was dried (MgSO4), filtered, concentrated, and chromato-graphed on a silica gel column with elution of EtOAc/hexane (2:8) to give compound 17 (416 mg, 32%). TLC (EtOAc/hex-ane, 3:7) Rf= 0.54; [a]D 22 = +40.1 (c = 1.0, CH2Cl2); IR (KBr, cm-1) 2965, 1652, 1513, 1460, 1222, 1037;1H NMR (CDCl3, 400 MHz)d 1.85 (m, 2H), 2.62 (t, J = 7.3 Hz, 2H), 3.36 (t, J = 8.4 Hz, 1H), 3.45 (m, 2H), 3.65 (dd, J = 4.6, 9.4 Hz, 1H), 4.17 (m, 2H), 4.38 (m, 1H), 5.03 (s, 2H), 6.89-6.92 (m, 3H), 7.08-7.11 (m, 2H), 7.22-7.24 (m, 2H), 7.31-7.43 (m, 15H);13 C NMR (CDCl3, 100 MHz)d 31.2, 31.3, 66.6, 69.9, 70.4, 70.9, 72.5, 114.6, 127.4, 127.8, 128.42, 128.46, 128.48, 128.49, 128.5, 128.9, 129.2, 129.3, 129.6, 130.5, 131.7, 133.4, 133.71, 133.73, 133.81, 133.88, 133.92, 133.94, 134.1, 136.09, 136.11, 136.21, 136.22, 137.1, 140.5, 140.7, 156.9, 162.4;31P NMR (CDCl3, 162 MHz)d -8.33; HR-FAB-MS calcd for C38H36ClNO3P (M++ H): 620.2043, found: m/z 620.2120.
Representative Procedure for the Palladium Catalyzed Asymmetric Allylic Substitution Reaction (Table 1)
A mixture of ligand 4 (24 mg, 0.04 mmol) and [Pd(allyl)Cl]2(7.2 mg, 0.02 mmol) in CH2Cl2(10 mL) was stirred for 10 min. A solution of 1,3-diphenyl-2-propenyl ac-etate (50 mg, 0.2 mmol) in CH2Cl2(10 mL) was added, fol-lowed by dimethyl malonate (78 mg, 0.6 mmol), BSA (122
mg, 0.6 mmol) and KOAc (1 mg, 0.01 mmol). The mixture was stirred at room temperature for 40 h, and taken up with Et2O (80 mL). The ethereal solution was washed with satu-rated NH4Cl (20 mL´ 2), dried (MgSO4), filtered, concen-trated, and chromatographed on a silica gel column with elu-tion of EtOAc/hexane (1:9) to give dimethyl 2-(1,3-diphen-yl-2-propenyl)malonate (19). A sample of 19 was analyzed by HPLC on a Chiralcel column (25 cm´ 0.46 cm) with elu-tion of 2-propanol/hexane (1:120, flow rate 0.9 mL) using RI and UV (254 nm) detectors. tR= 13.7 min (R-19) and 14.6 min (S-19).
(
R)-4-[3-(4-Hydroxyphenyl)propoxy]methyl-2-(2-diphenyl-phosphino)phenyl-4,5-dihydro-1,3-oxazole (20)
To a solution of 4 (500 mg, 0.85 mmol) and AlCl3(0.34 g, 2.56 mmol) in CH2Cl2(30 mL) was added N,N-dimethyl-aniline (1 g, 8.5 mmol). The mixture was stirred at room tem-perature for 1 h, filtered through a pad of Celite, and washed with CH2Cl2(20 mL) and EtOAc/hexane (1:1, 30 mL). The combined organic phase was concentrated and chromato-graphed on a silica gel column with elution of EtOAc/hexane (1:9) to give phenol 20 (328 mg, 78%). TLC (EtOAc/hexane, 3:7) Rf= 0.33; [a]D 23 = -23.0, (c = 1.0, CH2Cl2); IR (KBr, cm-1) 3343, 3022, 1653, 1516, 1439, 1352, 1218;1H NMR (CDCl3, 400 MHz)d 1.78-1.85 (m, 2H), 2.48 (dd, J = 7.4, 14.0 Hz, 1H), 2.55 (m, 2H), 2.84 (m, 1H), 3.27-3.42 (m, 2H), 3.54 (m, 1H), 3.67 (dd, J = 3.3, 9.5 Hz, 1H), 4.40-4.47 (m, 1H), 6.37 (br s, OH), 6.58 (d, J = 8.2 Hz, 1H), 6.73 (d, J = 8.2 Hz, 2H), 6.90-6.98 (m, 2H), 7.23-7.63 (m, 13H);13C NMR (CDCl3, 100 MHz)d 30.8, 31.2, 48.0, 70.4, 71.6, 71.7, 115.2, 126.9, 128.4, 128.50, 128.57, 128.62, 128.69, 128.8, 128.9, 129.3, 131.5, 132.6, 132.8, 133.2, 134.2, 154.2, 166.9;31 P NMR (CDCl3, 162 MHz)d -22.45; HR-FAB-MS calcd for C31H31NO3P (M+ + H): 496.1963, found: m/z 496.2187.
A Polymer-Supported Ligand 21
Merrifield resins (2% cross-linked, 2-2.5 meq Cl/g, 200-400 mesh) were dried under reduced pressure before use. Under an atmosphere of N2, a mixture of alcohol 20 (500 mg, 1.0 mmol), Merrifield resins (367 mg, 0.73-0.91 mmol) and Cs2CO3(359 mg, 1.1 mmol) in DMF (10 mL) was agitated at 60°C for 24 h. The mixture was cooled and the resins were collected by filtration and washed successively with 1:1 H2O/THF (3´ 25 mL), MeOH (3 ´ 25 mL), and THF (3 ´ 25 mL). The product was dried to yield 710 mg of 21. Elemental analysis showed the average nitrogen content of 0.46 ± 0.09% (three measurements), equivalent to an average load-ing of 0.33 mmol/g.
ACKNOWLEDGEMENT
We thank the National Science Council for financial support.
Received February 21, 2005.
REFERENCES
1. For reviews, see: (a) Helmchen, G.; Pfaltz, A. Acc. Chem.
Res. 2000, 33, 336-345. (b) Pfaltz, A.; Drury, W. J., III. Proc. Natl. Acad. Sci. USA 2004, 101, 5723-5726. For related
rep-resentative ligands, see: (c) Strangeland, E. L.; Sammakia, T.
Tetrahedron 1997, 53, 16503-16510. (d) Gilbertson, S. R.;
Chang, C.-W. T. J. Org. Chem. 1998, 63, 8424-8431. (e) Nishibayashi, Y.; Takei, I.; Uemura, S.; Hidai, M.
Organo-metallics 1998, 17, 3420-3422. (f) Zhang, W.; Yoneda, Y.;
Kida, T.; Naktshji, Y.; Ikeda, I. Tetrahedron: Asymmetry 1998, 9, 3371-3380. (g) Yonehara, K.; Hashimume, T.; Mori, K.; Ohe, K.; Uemura, S. J. Org. Chem. 1999, 64, 9374-9380. (h) Jiang, Y.; Jiang, Q.; Zhu, G.; Zhang, X. Tetrahedron Lett. 1997, 38, 215-218. (i) Ahn, K. H.; Cho, C.-W.; Park, J.; Lee, S. Tetrahedron: Asymmetry 1997, 8, 1179-1185. (j)
Ogasawara, M.; Yoshida, K.; Kamei, H.; Kato, K.; Uozumi, Y.; Hayashi, T. Tetrahedron: Asymmetry 1998, 9, 1779-1787. (k) Wiese, B.; Helmchen, G. Tetrahedron Lett. 1998,
39, 5727-5730. (l) Hou, D.-R.; Reibenspies, J. H.; Burgess,
K. J. Org. Chem. 2001, 66, 206-215. (m) Blankenstein, J.; Pfaltz, A. Angew. Chem., Int. Ed. 2001, 40, 4445-4447. (n) Bernardinelli, G. H.; Kundig, E. P.; Meier, P.; Pfaltz, A.; Radkowski, K.; Zimmermann, N.; Neuburger-Zehnder, M.
Helv. Chim. Acta 2001, 84, 3233-3246. (o) Cozzi, P. G.;
Zim-mermann, N.; Hilgraf, R.; Schaffner, S.; Pfaltz, A. Adv.
Synth. Cat. 2001, 343, 450-454.
2. von Matt, P.; Pfaltz, A. Angew. Chem., Int. Engl. 1993, 32, 566-568.
3. Sprinz, J.; Helmchen, G. Tetrahedron Lett. 1993, 34,
1769-1772.
4. Dawson, G. J.; Frost, C. G.; Williams, J. M. J. Tetrahedron
Lett. 1993, 34, 3149-3150.
5. (a) Helmchen, G. J. Organomet. Chem. 1999, 576, 203-214. Steinhagen, H.; Reggelin, M.; Helmchen, G. Angew. Chem.,
Int. Ed. Engl. 1997, 36, 2108-2110. (b) Kollmar, M.; Goldfuss,
B.; Reggelin, M.; Rominger, F.; Helmchen, G. Chem. Eur. J. 2001, 7, 4913-4927. (c) Kollmar, M.; Steinhagen, H.; Janssen, J. P.; Goldfuss, B.; Malinovskaya, S. A.; Vazquez, J.; Rominger, F.; Helmchen, G. Chem. Eur. J. 2002, 8, 3103-3114. (d) Vazquez, J.; Goldfuss, B.; Helmchen, G. J.
Organomet. Chem. 2002, 641, 67-70. (e) Apfelbacher, A.;
Braunstein, P.; Brissieux, L.; Welter, R. J. Chem. Soc.,
Dal-ton Trans. 2003, 1669-1674.
6. Peer, M.; de Jong, J. C.; Kiefer, M.; Langer, T.; Rieck, H.; Schell, H.; Sennhenn, P.; Sprinz, J.; Steinhagen, H.; Wiese, B.; Helmchen, G. Tetrahedron 1996, 52, 7547-7583. 7. Phillips, A. J.; Uto, Y.; Wipf, P.; Reno, M. J.; Williams, D. R.
Org. Lett. 2000, 2, 1165-1168.
8. (a) Brown, J. M.; Baker, K. V.; Hughs, N.; Skarnulis, A. J.; Sexton, A. J. Org. Chem. 1991, 56, 698-703. (b) Koch, G.; Lloyd-Jones, G. C.; Loiseleur, O.; Pfaltz, A.; Pretot, R.; Schaffner, S.; Schnider, P.; von Matt, P. Recl. Trav. Chim.
Pays-Bas 1995, 114, 206-210. (c) Overmann, L. E.; Owen, C.
E.; Zipp, G. G. Angew. Chem. Int. Engl. 2002, 41, 3884-3887.
9. (a) Allen, J. V.; Dawson, G. J.; Frost, C. G.; Williams, J. M. J.
Tetrahedron 1994, 50, 799-808. (b) Nagel, U.; Nedden, H. G. Chem. Ber. Recueil 1997, 130, 385-397. (c) Haenel, M. W.;
Oevers, S.; Bruckmann, J.; Kuhnigk, J.; Krueger, C. Synlett 1998, 301-303.
10. Read, G.; Urgelles, M. J. Chem. Soc., Dalton Trans. 1986, 1383-1388.
11. (a) Snieckus, V. Chem. Rev. 1990, 90, 879-933. (b) Snieckus, V. NATO ASI Series E: Applied Sciences 1996, 320, 191-221. (c) Hartung, C. G.; Snieckus, V. (Ed., Astruc, D.)
Mod-ern Arene Chemistry 2002, 330-367.
12. For review of palladium-catalyzed enantioselective allylic substitution reactions: (a) Sesay, S. J.; Williams, J. M. J. Adv.
Asym. Syn. 1998, 3, 235-271. (b) Acemoglu, L.; Williams, J.
M. J. (Ed., Negishi, E.) Handbook of Organopalladium
Chemistry for Organic Synthesis 2002, 2, 1945-1979. For
re-lated examples: (c) Trost, B. M.; Murphy, D. J.
Organo-metallics 1985, 4, 1143. (d) Loiseleur, O.; Elliott, M. C.; von
Matt, P.; Pfaltz, A. Helv. Chim. Acta 2000, 83, 2287-2294. (e) Gibson, C.; Rebek, J., Jr. Org. Lett. 2002, 4, 1887-1890. 13. (a) Evans, D. A.; Campos, K. R.; Tedrow, J. S.; Michael, F.
E.; Gagné, M. R. J. Am. Chem. Soc. 2000, 122, 7905-7920. (b) Fukuda, T.; Takehara, A.; Iwao, M. Tetrahedron:
Asym-metry 2001, 12, 2793-2799.
14. For the polymer-supported oxazoline ligands: (a) Bolm, C.; Hermanns, N.; Claßen, A.; Muñiz, K. Bioorg. Med. Chem.
Lett. 2002, 12, 1795-1798. (b) Burguete, M. I.; Díez-Barra,
E.; Fraile, J. M.; García, J. I.; García-Verdugo, E.; Garcia-Verdugo, E.; Gonzalez, R.; Herrerias, C. I.; Luis, S. V.; May-oral, J. A. Bioorg. Med. Chem. Lett. 2002, 12, 1821-1824. (c) Jonsson, C.; Hallman, K.; Andersson, H.; Stemme, G.; Malkoch, M.; Malmstrom, E.; Hult, A.; Moberg, C. Bioorg.
Med. Chem. Lett. 2002, 12, 1857-1861.
15. (a) Akiyama, T.; Hirofuji, H.; Ozaki, S. Tetrahedron Lett. 1991, 32, 1321-1324. (b) Akiyama, T.; Hirofuji, H.; Hirose, A.; Ozaki, S. Synth. Commun. 1994, 24, 2179-2185. 16. (a) Dorwald, F. Z. Organic Synthesis on Solid Phase:
Sup-ports, Linkers, Reactions; Wiley-VCH: Weinheim, 2002. (b)
Talukdar, S.; Chen, R.-J.; Chen, C.-T.; Lo, L.-C.; Fang, J.-M.