Pharmacological chaperone design for reducing risk factor
of Parkinson’s disease from traditional Chinese medicine
Hung-Jin Huang a , Cheng-Chun Lee b* , Calvin Yu-Chian Chen b, c, d e *
a Department of Chinese Pharmaceutical Sciences and Chinese Medicine Resources, College of
Pharmacy, China Medical University, Taichung 40402, Taiwan.
b SchoolofMedicine,College of Medicine,ChinaMedicalUniversity,Taichung, 40402,Taiwan cDepartment of Biotechnology,AsiaUniversity,Taichung, 41354,Taiwan.
dChinaMedicalUniversityBeigangHospital,Yunlin,65152,Taiwan. eComputational and Systems Biology, Massachusetts Institute of Technology
Technology,Cambridge,MA02139,USA.
*Corresponding author (C. Y. C. Chen).Tel.: +886-4-2205-2121ext. 4306 E-mail address:[email protected](C.Y.C. Chen)
1 2 3 4 5 6 7 8 9 10 11 12 13
Abstract
Dysfunction of β-glucocerebrosidase (GCase) has no hydrolytic activity in patients of Gaucher's disease, and increasing the risk factor for Parkinson’s disease occurrence. Pharmacological chaperone design have been used to treat with misfolded protein in related disease, which utilized a small compound to cause protein folding correctly. This study employed the world largest traditional Chinese medicine (TCM) database for searching potential lead compound as pharmacological chaperone, and we also performed molecular dynamics (MD) simulations to observe the stability of binding conformation between ligands and active site of GCase structure. The docking results from database screening show that N-Methylmescaline and Shihunine have high binding ability to GCase than Tetrahydroxyazepanes. From MD simulation analysis, Tetrahydroxyazepanes displayed high opportunity of ligand migration instead of our TCM candidates, and H-bonds number were decreased in the end of MD snapshot. Our result indicated that binding conformation of N-Methylmescaline and Shihunine are remain stable during MD simulation, which demonstrating the two candidates are suitable for GCase binding and might be potential as pharmacological chaperone for GCase folding correctly.
Keywords:
traditional Chinese medicine (TCM); molecular dynamics; Parkinson’s disease; β-glucocerebrosidase; pharmacological chaperone14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
1. Introduction
Gaucher's disease (GD) is caused by mutations in the GBA gene encoding β-glucocerebrosidase (GCase), which is leading to inherited β-glucocerebrosidase deficiency. Due to the mutated GCase has no function of hydrolytic activity, the deficient activity of GCase cannot transport from the endoplasmic reticulum (ER) to lysosomes , this phenomenon impaired intracellular transport and contributes to defects of metabolism in fibroblasts of patients . Subsequently, both of glucosylceramide (GluCer) and glucosylsphingosine (GluSph) accumulate in macrophages of various organs , such as spleen, liver, lungs, bone marrow, and brain , clinical feature of GD disease reveals deposition of undigested subtracts in lysosome of macrophages. In several clinical cases of Gaucher's disease, symptoms of central nervous system (CNS) disorder were existed in patient's brain . GD is the most prevalent lysosomal storage diseases , some evidences were indicated that reducing GCase activity is associated with Parkinson’s disease (PD) , and the mutation of GCase has high risk factor for PD occurrence . Parkinson's disease (PD) is one of common CNS diseases of neurodegenerative disorders effecting 2% of older adults alter the age of 65 , which is the second common neuron degenerated disease after Alzheimer’s disease . Pathological hallmark of PD is loss of dopaminergic neuron in the pars compacta of the substantia nigra (SNc) , PD patients display reduction of 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51
dopamine levels in striatum and degeneration of the dopamine producing neurons, because of the catalytic rate limiting step of tyrosine hydroxylase in catalyzing the conversion of the amino acid tyrosine to 3,4-dihydroxyphenylalanine (DOPA). L-DOPA is a precursor for important neurotransmitters synthesis, including dopamine, noradrenaline, and adrenaline. PD disease can be considered as a tyrosine hydroxylase deficiency . Levodopa (L-DOPA) is wildly used for treatment of PD disease, improving the motor features of PD patients, but there are remain several side effects in clinical treatment such as fluctuating motor response, dyskinesia, and neuropsychiatric problems .
The aim of this study is focus on pharmacological chaperone (PC) design, the PC therapy is utilize a small molecular to cause misfolded protein structure to fold correctly. Because of mutant GCase structure cannot hydrolysis the beta-glucosidic linkage of the GluCer and GluSph, which has high risk factor of PD disease, designing small compounds to promote GCase structure folding correctly is necessary.
Computer-aided drug design (CADD) has been wildly used to design new drugs, TCM compounds are regarded as potential leading compounds have been reported in many studies . In our study, small molecular from the world largest traditional Chinese medicine (TCM) database were used to search potential compounds with 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70
high affinity in GCase active site, and we further utilized molecular dynamics (MD) simulation to observe the stability between protein and ligands for binding assay. Synthetic drug often with side-effect in clinical treatments, our results could provided nature product as drug candidate, which are more safer and reduce adverse reactions.
2. Materials and Methods
2.1. Database Screening
The crystal structure of β-glucocerebrosidase (GCase) was downloaded from PDB database (PDB code: 3RIK) , which were cleaned up mistakes of the crystal structure by Prepare Protein module of Accelrys Discovery Studio 2.5.5.9350 (DS 2.5) software , including inserting missing atoms, modeling missing loops, and removing crystal waters. The pH value of 7.4 is specifies to protonate all residues. Total number of 61000 TCM compounds were obtained from the TCM Database@Taiwan to dock into GCase binding site for binding analysis. All TCM compound were accessed by ADMET prediction for drug-like evaluating. We performed Ligand docking using LigandFit of DS 2.5, Monte-Carlo techniques was used to generate different ligand conformations and docks into the active site of GCase. CHARMm force field was applied to minimize all docked poses. We selected Smart minimizer as minimization algorithm for ligands minimization, this process 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89
contains Steepest Descent and Conjugate Gradient. The Steepest Descent performed 1,000 steps and followed by Conjugate Gradient.
2.2 MD simulation
The molecular dynamic simulation was performed using GROMACS 4.5.5 package with charmm27 force field. Time step of MD simulation was set as 0.002 ps. We used particle mesh Ewald (PME) method as coulomb type for treating electrostatics, and cut-off distance with 1.4 nm to define van der Waals (VDW) residues. The distance of real space cutoff was set to 1.2 nm in define box, LINCS algorithm was used restrain the lengths of all bonds. We utilized SwissParam to obtain topology file and parameters of small compounds for GROMACS simulation . TIP3P water model was employed in MD simulations, we also using the concentration of 0.145 M NaCl model in the solvent system, the solvent molecules were random replaced by Na and Cl ions. Energy minimization using Steepest Descent algorithm and performed 5,000 cycle steps. Equilibration was performed under position restraints for 100 ps. Following this step, production simulations was run for all system for 5000 ps, the temperature of all simulation system was set as 310K. MD conformations were collected every 20 ps for trajectory analysis.
90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107
Results and discussion
3.1. Docking analysis of database screening
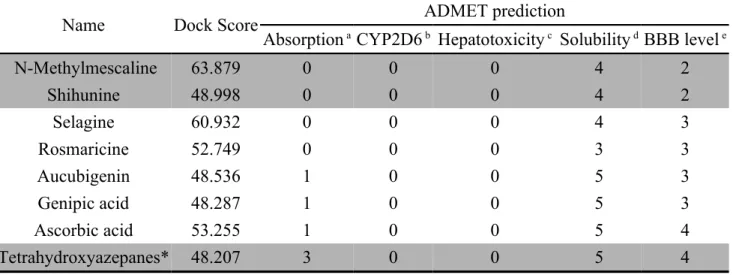
Docking result of TCM database screening were determined by Dock score and ADMET predictions, top candidates are filtered by comparing with Tetrahydroxyazepanes and listed in Table 1. From ADMET prediction, all TCM candidates reveal good or moderate absorption, but Tetrahydroxyazepanes has very low absorption, which indicated that our candidates have well movement into the bloodstream. For CYP2D6 and hepatotoxicity, the both values in candidates and control are less than 0.5, which indicated that they have no toxicity ability to CYP2D6 and no hepatotoxicity in liver. For solubility evaluation, optimal drug-likeness compounds are present in top three candidates with 4 value of solubility, and combining with BBB level analysis, N-Methylmescaline and Shihunine have the highest penetrate in blood brain barrier, Tetrahydroxyazepanes has too solubility and no penetration in BBB level. Based on solubility and BBB level, N-Methylmescaline and Shihunine may be potential lead compounds for CNS diseases, and the dock score are better than control; hence, we selected the top two candidates for further analysis in MD simulation. Chemical scaffold of the two candidates and control are displayed in Figure 1. The structure of Tetrahydroxyazepanes is taken from crystal structure, which has well binding affinity with GCase. N-Methylmescaline is come from Alhagi 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126
pseudalhagi , and Shihunine is available in Dendrobium loddigesii . To compare
docked pose of Tetrahydroxyazepanes with x-ray crystal structure by RMSD value of heavy atoms calculation (Figure 2), the two scaffolds is vary close with RMSD value of 0.7293 Å, indicating that the predicted binding poses from the LigandFit are matched with experimental poses, all of the compounds from TCM database are according to the binding pose of crystal Tetrahydroxyazepanes for binding poses generation. For docking poses, polar and van der Waals (VDW) interactions between residues of GCase and ligand are shown in Figure 3. We found that Glu284, Glu235, Asp127, Trp179, Trp381, Asn234, Glu340 displayed polar forces among all docking poses, indicated that N-Methylmescaline and Shihunine have similar binding residues with Tetrahydroxyazepanes in GCase active site. For residues of Asn396 and Asn392, which reveals polar interactions with Tetrahydroxyazepanes (Figure 3a), but displaying VDW force for N-Methylmescaline binding (Figure 3b). Asn396 forms VDW interaction for Shihunine interacting, but Asn392 are not shown in Figure 3c and has no interactions with the compound. Interestingly, pi interactions are displaced in the 2D diagram of N-Methylmescaline and Shihunine, which are interacted with residue His311 and Tyr313, respectively (Figure 3b, 3c), but no pi interactions are found in docking pose of Tetrahydroxyazepanes (Figure 3a). We also utilized Ligplot plus to access hydrophobic interactions between compounds and residues, the distinct 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145
interaction of residues are displaced by red circle (Figure 4). Comparing with hydrophobic residues for all docking poses, there are few amino acids are difference between the three docked compounds, suggesting that our candidates have similar hydrophobic interactions with the control set. We further analyze the disorder region of GCase protein, residue index of 44 - 56 and 196 - 201 are disorder region (Figure 5.), all of binding residues of GCase are not locate in disorder region, suggesting that residues of binding site are stable folded structure. The prediction of disorder structure shows that interaction between ligands and residues are not effect by protein structure of GCase .
3.2. Molecular dynamics simulation analysis
To determine stability of conformations among MD simulation, we employed root mean square deviation (RMSD), radius of protein gyration and total energy to analyze the deviation of all complexes with docked ligand. The RMSD values of protein structure was used to verified stability among MD simulation, Figure 6 shows all values within the range from 0.15 to 0.18 nm, which indicate that all protein structures from protein-ligand complexes display stable during 5000 ps simulation, and the 5000 ps simulation time is enough for decreasing the fluctuation of all complexes. For ligand RMSD (Figure 7), it is obvious that the conformation of 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164
Tetrahydroxyazepanes display high degree of difference during dynamic simulation, the value of ligand RMAD increased to 0.15 nm from 500 ps to 5000 ps. The ligand RMSD of TCM candidates are reveal slightly deviation, the increased values of N-Methylmescaline and Shihunine are 0.08 and 0.05, respectively. Radius of protein gyration (Rg) was used to observe the compactness of protein structure (Figure 8), the stability of the gyration plot is similar to protein RMSD, gyration values of the three complexes are remain at 2.30 nm for all simulation times, which showing that all of the protein complexes are more compacted among dynamic simulation. Total energy shown that N-Methylmescaline and Shihunine have lower energy values (below -1.252 × 10 6 KJ / mol) than Tetrahydroxyazepanes (above -1.252 × 10 6 KJ / mol) in GCase complexes (Figure 9.), suggesting that both of TCM compounds are more stable in protein structure. Because of the plots of ligand RMSD and total energy display significant change, observing the variation of Tetrahydroxyazepanes between initial pose and dynamic pose for comparing with TCM candidates is necessary.
3.3. Comparison between initial and dynamic conformations
In order to observe conformation variation between docking pose and the end of MD pose, snapshots of protein-ligand complexes were listed in Figure 10. Docking pose of Tetrahydroxyazepanes shows that H-bonds were formed with eight amino 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183
acids (Asp127, Trp179, Glu340, Asn234, Glu235, Gln284, Trp312 and Trp381), but the final of MD conformation shows lots of H-bonds are disappeared, the H-bond only displayed on Asp127, Ser345, and Glu340 in the active site, and this phenomenon are refer to the high degree value in ligand RMSD plot. Docking pose of N-Methylmescaline has pi-cation interaction with His311 and forms H-bond with Trp179, Asn234 and Glu235. Interestingly, the pi-cation are change to Trp179, and Glu235 are still has H-bond, which indicated that N-Methylmescaline reveals stable binding affinity in GCase active site after MD. For Shihunine snapshots analysis, pi-cation interaction are formed with Tyr313 and generated H-bond with Trp179 and Glu235, but in the end of MD conformation, H-bonds change from Trp179 to Asn234, Glu235 is still keep the H-bond with ligand. The binding interactions of Shihunine in final pose are less than in docking pose, however, the conformation of ligand is not change significantly after MD simulation. The slight increased value of ligand RMSD are also supported this finding, which indicated that Shihunine could form stable binding pose in GCase active site. We also measured the distance of H-bond during MD simulation, the distance are significantly increase to 0.7 nm between Tetrahydroxyazepanes and three residues : Gln284, Trp179 and Glu235 (Figure 11a), N-Methylmescaline and Shihunine are remain within 0.35 during all simulation time (Figure 11b, 11c). The variation of H-bond distance are correlated with comparison of 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202
snapshots between docking and MD poses.
3.4. Migration assay of ligands in GCase
Due to the Tetrahydroxyazepanes have significant difference between docking and MD poses, we further computed the mean square displacement (MSD) of ligand atoms to analyze the migration of docked ligands among all simulation times, MSD value of Tetrahydroxyazepanes are higher than our two candidates (Figure 12.). We utilized CAVER 3.0 software to predict the migrated ligand tunnels in GCase, the network of ligand pathway has been used to analyze the binding stability of docked compounds . The seven clusters of ligand channels are shown in Figure 13, it is worthy to note that there are three different directions of clusters for N-Methylmescaline and Shihunine migrate from starting point (Figure 13b, 13c), but Tetrahydroxyazepanes generated four different directions of clusters in GCase, the distinct tunnel is cluster 6 (Figure 13a), this result reveals that Tetrahydroxyazepanes has high opportunity than N-Methylmescaline and Shihunine to cause ligand move aware from the docked site.
3. Conclusion
In ADMET prediction, N-Methylmescaline and Shihunine have optimal drug-203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222
likeness features and medium blood brain barrier penetration ability, both of the two TCM candidates are more potential lead compound than Tetrahydroxyazepanes in CNS disease treatments. For docking analysis, both of N-Methylmescaline and Shihunine have higher Dock Score than control, which have pi-cation interactions in docking pose snapshots instead of Tetrahydroxyazepanes. All of binding residues reveal folding stable from protein disorder prediction, which indicated that the docked ligand have no effect by disorder region to avoid unstable binding. We further analyze the conformational changes of protein-ligand complexes after MD simulation, Tetrahydroxyazepanes displayed significant change on ligand RMSD plot analysis, but N-Methylmescaline and Shihunine reveal stable during MD simulation. Besides, the end of MD snapshots shown that Tetrahydroxyazepanes decreased H-bond number after MD simulation, the end of MD poses of our candidates are similar to docking poses, suggesting that N-Methylmescaline and Shihunine have high affinity with GCase among all MD simulation. For ligand migration analysis, Tetrahydroxyazepanes has the highest distance of movement in MSD evaluation. In addition, the ligand paths from channel prediction reveals more directions from initial binding position, this result are correlated with the comparison of snapshots. Our result indicated that N-Methylmescaline and Shihunine might be potential lead compound to design pharmacological chaperone for GCase folding correctly, and 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241
reducing the risk factor for PD occurrence.
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC102-2325-B039-001, NSC102-2221-E-468-027-), Asia University (ASIA101-CMU-2), and China Medical University Hospital (DMR-103-058, DMR-103-001, DMR-103-096). This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH102-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH102-TD-C-111-005). 242 243 244 245 246 247 248 249 250 251
References
[1] K. P. Zimmer, P. le Coutre, H. M. Aerts, et al. "Intracellular transport of acid beta-glucosidase and lysosome-associated membrane proteins is affected in Gaucher's disease (G202R mutation)," Journal of Pathology, vol. 188, no. 4, pp. 407-414, 1999. [2] L. M. Jonsson, G. J. Murray, S. H. Sorrell, et al. "Biosynthesis and maturation of glucocerebrosidase in Gaucher fibroblasts," European Journal of Biochemistry, vol. 164, no. 1, pp. 171-179, 1987.
[3] N. Dasgupta, Y. H. Xu, S. Oh, et al. "Gaucher Disease: Transcriptome Analyses Using Microarray or mRNA Sequencing in a Gba1 Mutant Mouse Model Treated with Velaglucerase alfa or Imiglucerase," PLoS One, vol. 8, no. 10, p. e74912, 2013. [4] E. Cassinerio, G. Graziadei, E. Poggiali. "Gaucher disease: A diagnostic challenge for internists," Evidence-based Complementary and Alternative Medicine, 2013.
[5] R. L. Lieberman. "A Guided Tour of the Structural Biology of Gaucher Disease: Acid-beta-Glucosidase and Saposin C," Enzyme Res, vol. 2011, p. 973231, 2011. [6] W. Schonemann, E. Gallienne, K. Ikeda-Obatake, et al. "Glucosylceramide Mimics: Highly Potent GCase Inhibitors and Selective Pharmacological Chaperones for Mutations Associated with Types 1 and 2 Gaucher Disease," ChemMedChem, 2013.
[7] H. Rosenbaum, J. Aharon-Peretz, B. Brenner. "Hypercoagulability, 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271
parkinsonism, and Gaucher disease," Seminars in Thrombosis and Hemostasis, vol. 39, no. 8, pp. 928-934, 2013.
[8] O. Neudorfer, N. Giladi, D. Elstein, et al. "Occurrence of Parkinson's syndrome in type I Gaucher disease," QJM, vol. 89, no. 9, pp. 691-694, 1996.
[9] Y. Li, T. Sekine, M. Funayama, et al. "Clinicogenetic study of GBA mutations in patients with familial Parkinson's disease," Neurobiology of Aging, 2013.
[10] M. Federoff, B. Jimenez-Rolando, M. A. Nalls, A. B. Singleton. "A large study reveals no association between APOE and Parkinson's disease," Neurobiology of
Disease, vol. 46, no. 2, pp. 389-392, 2012.
[11] K. Wirdefeldt, H. O. Adami, P. Cole, D. Trichopoulos, J. Mandel. "Epidemiology and etiology of Parkinson's disease: a review of the evidence,"
European Journal of Epidemiology, vol. 26 Suppl 1, pp. S1-58, 2011.
[12] R. L. Nussbaum, C. E. Ellis. "Alzheimer's disease and Parkinson's disease," New
England Journal of Medicine, vol. 348, no. 14, pp. 1356-1364, 2003.
[13] L. Prensa, M. Cossette, A. Parent. "Dopaminergic innervation of human basal ganglia," Journal of Chemical Neuroanatomy, vol. 20, no. 3-4, pp. 207-213, 2000. [14] J. R. Roede, K. Uppal, Y. Park, et al. "Serum Metabolomics of Slow vs. Rapid Motor Progression Parkinson's Disease: a Pilot Study," PLoS One, vol. 8, no. 10, p. e77629, 2013.
[15] J. Haavik, K. Toska. "Tyrosine hydroxylase and Parkinson's disease," Molecular 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291
Neurobiology, vol. 16, no. 3, pp. 285-309, 1998.
[16] C. W. Olanow, J. Jankovic. "Neuroprotective therapy in Parkinson's disease and motor complications: a search for a pathogenesis-targeted, disease-modifying strategy," Movement Disorders, vol. 20 Suppl 11, pp. S3-10, 2005.
[17] K. C. Chen, C. Y. C. Chen. "Stroke prevention by traditional Chinese medicine? A genetic algorithm, support vector machine and molecular dynamics approach," Soft
Matter, vol. 7, no. 8, pp. 4001-4008, 2011.
[18] T. Y. Tsai, K. W. Chang, C. Y. C. Chen. "iScreen: world's first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan," Journal of Computer-Aided Molecular Design, vol. 25, no. 6, pp. 525-531, 2011.
[19] S. S. Chang, H. J. Huang, C. Y. Chen. "Two birds with one stone? Possible dual-targeting H1N1 inhibitors from traditional Chinese medicine," PLoS Comput Biol,
vol. 7, no. 12, p. e1002315, 2011.
[20] S. C. Yang, S. S. Chang, H. Y. Chen, C. Y. Chen. "Identification of potent EGFR inhibitors from TCM Database@Taiwan," PLoS Comput Biol, vol. 7, no. 10, p. e1002189, 2011.
[21] K. C. Chen, K. W. Chang, H. Y. Chen, C. Y. Chen. "Traditional Chinese medicine, a solution for reducing dual stroke risk factors at once?," Mol Biosyst, vol. 7, no. 9, pp. 2711-2719, 2011. 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311
[22] K. C. Chen, M. F. Sun, S. C. Yang, et al. "Investigation into Potent Inflammation Inhibitors from Traditional Chinese Medicine," Chemical Biology & Drug Design,
vol. 78, no. 4, pp. 679-688, 2011.
[23] W. I. Tou, S. S. Chang, C. C. Lee, C. Y. Chen. "Drug design for neuropathic pain regulation from traditional Chinese medicine," Sci Rep, vol. 3, p. 844, 2013.
[24] C. Y. Chen. "TCM Database@Taiwan: the world's largest traditional Chinese medicine database for drug screening in silico," PLoS One, vol. 6, no. 1, p. e15939, 2011.
[25] S. D. Orwig, Y. L. Tan, N. P. Grimster, et al. "Binding of 3,4,5,6-tetrahydroxyazepanes to the acid-beta-glucosidase active site: implications for pharmacological chaperone design for Gaucher disease," Biochemistry, vol. 50, no. 49, pp. 10647-10657, 2011.
[26] Accelerys. Discovery Studio Client v2.5. In, Accelrys Inc. San Diego, CA, USA.: 2009.
[27] S. Pronk, S. Pall, R. Schulz, et al. "GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit," Bioinformatics, vol. 29, no. 7, pp. 845-854, 2013.
[28] T. E. Cheatham, J. L. Miller, T. Fox, T. A. Darden, P. A. Kollman. "Molecular-Dynamics Simulations on Solvated Biomolecular Systems - the Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, Rna, and Proteins," Journal of the 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331
American Chemical Society, vol. 117, no. 14, pp. 4193-4194, 1995.
[29] V. Zoete, M. A. Cuendet, A. Grosdidier, O. Michielin. "SwissParam: a fast force field generation tool for small organic molecules," J Comput Chem, vol. 32, no. 11, pp. 2359-2368, 2011.
[30] W. Sun, J. Sneng. Brief Handbook of Natural Active Compounds. Medicinal Science and Technology Press of China, Beijing, 1998.
[31] M. F. Li, Y. Hirata, G. J. Xu, M. Niwa, H. M. Wu. "[Studies on the chemical constituents of Dendrobium loddigesii rolfe]," Yao Xue Xue Bao, vol. 26, no. 4, pp. 307-310, 1991.
[32] R. A. Laskowski, M. B. Swindells. "LigPlot+: multiple ligand-protein interaction diagrams for drug discovery," J Chem Inf Model, vol. 51, no. 10, pp. 2778-2786, 2011.
[33] W. I. Tou, C. Y. Chen. "May disordered protein cause serious drug side effect?,"
Drug Discov Today, 2013.
[34] C. Y. Chen, W. I. Tou. "How to design a drug for the disordered proteins?,"
Drug Discov Today, vol. 18, no. 19-20, pp. 910-915, 2013.
[35] E. Chovancova, A. Pavelka, P. Benes, et al. "CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures," PLoS Comput Biol, vol. 8, no. 10, p. e1002708, 2012.
[36] K. C. Chen, S. S. Chang, H. J. Huang, et al. "Three-in-one agonists for PPAR-332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351
alpha, PPAR-gamma, and PPAR-delta from traditional Chinese medicine," J Biomol
Struct Dyn, vol. 30, no. 6, pp. 662-683, 2012.
352 353 354 355
Table 1. Results of TCM database screening from LigandFit Docking protocol, all
candidates are evaluated by ADMET prediction.
Name Dock Score ADMET prediction
Absorption aCYP2D6 b Hepatotoxicity c Solubility dBBB level e
N-Methylmescaline 63.879 0 0 0 4 2 Shihunine 48.998 0 0 0 4 2 Selagine 60.932 0 0 0 4 3 Rosmaricine 52.749 0 0 0 3 3 Aucubigenin 48.536 1 0 0 5 3 Genipic acid 48.287 1 0 0 5 3 Ascorbic acid 53.255 1 0 0 5 4 Tetrahydroxyazepanes* 48.207 3 0 0 5 4 *Control
a Absorption: Good absorption = 0; Moderate absorption = 1; Low absorption = 2; Very low absorption = 3
b CYP2D6: Non-inhibitor < 0.5; Inhibitor > 0.5 c Hepatotoxicity: Nontoxic < 0.5; Toxic > 0.5
d Solubility: Good drug-likeness = 3; Optimal drug-likeness = 4; Too solubility = 5 e BBB level (Blood Brain Barrier): Medium penetration = 2; Low penetration = 3; Undefined penetration = 4 356 357 358 359 360 361 362 363 364 365
Figure legends
Figure 1. Chemical scaffold of top2 TCM candidates (N-Methylmescaline and
Shihunine) and Tetrahydroxyazepanes.
Figure 2. RMSD values of Tetrahydroxyazepanes between Crystal structure and
docking pose.
Figure 3. Pi interaction analysis from docking poses of ligands in GBA binding site
by Discovery Studio 2.5: (a) Tetrahydroxyazepanes, (b) N-Methylmescaline and (c) Shihunine, polar and van der Waals of residues interaction are represented by purple circle and green circle, respectively.
Figure 4. H-bond and hydrophobic interactions analysis of docking poses by Ligplot
plus tool, the red circle in 2D plots indicate distinct residues from other docking poses. residues with solid surface represent hydrophobic interactions in 3D docking poses, H-bonds and ligands are colored in blue and green, respectively.
Figure 5. Disorder prediction of GCase by PONDR-FIT, the value above 0.5 in
disorder disposition are indicated disorder residues.
Figure 6. RMSD values of protein structures from the three complexes with docked
ligand: Tetrahydroxyazepanes, N-Methylmescaline and Shihunine among 5000 ps simulation.
Figure 7. RMSD values of three compounds: Tetrahydroxyazepanes,
N-Methylmescaline and Shihunine in complexes among 5000 ps simulation.
Figure 8. Protein gyration values from the three complexes with docked ligand:
Tetrahydroxyazepanes, N-Methylmescaline and Shihunine among 5000 ps simulation
Figure 9. Total energy of three complexes with docked ligand:
Tetrahydroxyazepanes, N-Methylmescaline and Shihunine among 5000 ps simulation
Figure 10. Docking poses compare to the end of MD conformation, Pi stack
interactions and hydrogen bonds are colored in origin and green, respectively. 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403
Figure 11. H-bond distance between residues and ligands among all simulation
times : (a) Tetrahydroxyazepanes, (b) N-Methylmescaline and (c) Shihunine
Figure 12. Mean square displacement (MSD) of different ligands during all
simulation times, more increased value of MSD indicates higher migration of docked ligand aware from initial site.
Figure 13. Ligand pathway of (a) Tetrahydroxyazepanes, (b) N-Methylmescaline and
(c) Shihunine in GCase binding site by CAVER 3.0, starting point is the initial place for ligand binding, all generated channels are grouped to seven clusters.
404 405 406 407 408 409 410 411 412 413
Figure 1. 414 415 416 417 418
Figure 2. 419 420 421 422 423
Figure 3. 424 425 426 427 428 429
Figure 4.
430 431
Figure 5. 432 433 434 435 436
Figure 6. 437 438 439 440 441
Figure 7. 442 443 444 445 446
Figure 8. 447 448 449 450 451
Figure 9. 452 453 454 455 456 457 458