Autophagy 6:8, 1057-1065; November 16, 2010; © 2010 Landes Bioscience
BAsic ReseARch PAPeR BAsic ReseARch PAPeR
*Correspondence to: Ching-Chow Chen; Email: [email protected] Submitted: 04/04/10; Revised: 07/30/10; Accepted: 08/19/10
Previously published online: www.landesbioscience.com/journals/autophagy/article/13365 DOI: 10.4161/auto.6.8.13365
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common can-cer and the third leading cause of cancan-cer mortality worldwide.1
To date, surgical resection and liver transplantation are regarded as the main curative treatments for HCC which is highly refrac-tory to cytotoxic chemotherapy.2 During the course of
hepato-carcinogenesis, heterogeneity of the genetic abnormalities and activation of Ras/Raf signaling pathway may contribute to chemo-resistance through promotion of HCC cells to prolifera-tion and survival. For example, increased levels of anti-apoptotic proteins such as FLIP and survivin or decreased levels of pro-apoptotic proteins such as FADD have been described in HCC.3,4
Although the multi-kinase inhibitor sorafenib has been approved for advanced HCC in 2007,5 and molecular targeted agents such
as imatinib, trastuzumab and gefitinib have been reported to be possible new treatments,6 there is still an urgent need to develop
an effective systemic therapy for patients with advanced HCC. Histone deacetylases (HDACs) overexpression has been found in tumors and thus inhibits the expression of tumor suppressor genes. HDAC inhibitors could reactivate these genes and serve as potential anti-cancer drugs.7 Substantial evidence indicate that
HDAC inhibitors can induce cell cycle arrest, differentiation and apoptosis in HCC, and can increase the sensitivity of hepatoma
hepatocellular carcinoma (hcc) is the fifth most common cancer and the third leading cause of cancer death worldwide. Drug treatments for hcc have been largely unsuccessful. histone deacetylase inhibitors can reactivate tumor suppressor genes in cancer cells and serve as potential anti-cancer drugs. Two potent hDAc inhibitors OsU-hDAc42 and sAhA induced autophagy in hcc cells as revealed by transmission electron microscopy, immunofluorescence and Lc3-ii accumulation. We found that sAhA and OsU-hDAc42 induced autophagy through downregulation of Akt/mTOR signaling and induction of eR stress response. inhibition of autophagy by 3-MA or Atg5 knockout reduced sAhA-induced cytotoxicity, indicating that sAhA-induced autophagy led to cell death. Our results show that the combination of autophagy inducers with sAhA might be attractive for the treatment of hcc and pharmacological targeting of autophagy provides promise for the management of cancer therapy.
Autophagy potentiates the anti-cancer effects
of the histone deacetylase inhibitors
in hepatocellular carcinoma
Yuan-Ling Liu,1,† Pei-Ming Yang,1,† chia-Tung shun,2 Ming-shiang Wu,3 Jing-Ru Weng4 and ching-chow chen1,* 1Department of Pharmacology; college of Medicine; 2Department of Forensic Medicine and Pathology; 3Division of Gastroenterology; Department of internal Medicine;
National Taiwan University hospital; Taipei; 4Department of Biological science and Technology; china Medical University; Taichung, Taiwan †These authors contributed equally to this work.
Key words: histone deacetylase inhibitor, hepatocellular carcinoma, autophagy, apoptosis, ER stress
cells to the Fas-induced cell death.8-11 Therefore, pleiotropic
cel-lular effects let HDAC inhibitors be exciting agents for HCC treatment.
Autophagy is a physiological process involved in the routine turnover of proteins or intracellular organelles.12 The process of
autophagy starts by sequestering cytosolic proteins or organelles into autophagosomes, which then fuse with lysosomes to form autolysosomes for the degradation of sequestered contents by lysosomal hydrolases.13 A group of autophagy-related proteins
(Atg) involved in autophagy has been isolated and character-ized.14 Autophagosome nucleation is mediated by a class III
phos-phatidylinositol 3-kinase complex (Beclin 1, Vps15, Vps34).15,16
The production of phosphatidylinositol 3-phosphate is essential for the recruitment of Atg12-Atg5 complex and microtubule-associated protein 1 light chain 3 (LC3), which are required for the elongation of autophagosomes. Fusion of autophago-somes and lysoautophago-somes is mediated by microtubules.15,17 LC3 has
two forms: type I is cytosolic and type II is membrane-bound. During autophagy, LC3-II is increased from the conversion of LC3-I. LC3-II is localized in both inside and outside membrane of autophagosomes, making itself an autophagosomal marker.18
Autophagy may protect against cancer by promoting auto-phagic cell death (type II programmed cell death) or contribute to cancer by promoting the survival of nutrient-starved cells.13
MEF, HeLa and malignant rhabdoid tumor (MRT) cells.19,20 We would like to investigate its effect on
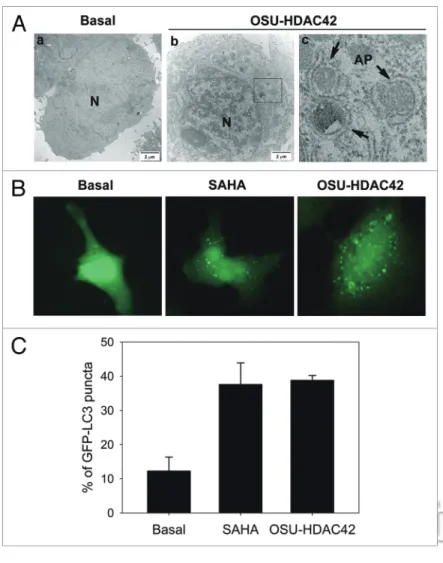
HCC and explore possibly new treatment. By trans-mission electron microscopy, Huh7 cells exhibited obvious autophagic vacuoles in the cytoplasm after treatment with 1 μM OSU-HDAC42 for 24 h (Fig. 1A). OSU-HDAC42 is a novel phenylbutyrate-derived HDAC inhibitor which shows potent and pleiotropic anti-cancer activities in human prostate and hepatic cancers.21 After treatment with OSU-HDAC42 or
another HDAC inhibitor SAHA, an increase of GFP-LC3 puncta representing autophagic vacuoles was formed in the cytoplasm (Fig. 1B and C). SAHA- and OSU-HDAC42-induced autophagosome for-mation was confirmed by the conversion of cytosolic LC3-I to autophagosomal membrane-bound LC3-II. Because LC3-I expression can be transcriptionally regulated and LC3-II tends to be more sensitive than LC3-I in immunoblotting, comparison of the ratio of LC3-II/β-actin would be more reliable.22-24 SAHA- or
OSU-HDAC42-induced LC3-II accumulation was found in Huh7, HepG2 and Hep3B cells (Fig. 2A). Other HDAC inhibitors including TSA and MS-275 increased the LC3-II accumulation as well (Fig. 2B). LC3-II can accumulate as a result of increased upstream autophagosome formation or impaired downstream autophagosome-lysosome fusion. To distinguish these two possibilities, the LC3 flux was assayed in the pres-ence of 3-MA (a class III PI3K inhibitor that blocks autophagosome formation) or bafilomycin A1 (a vacuolar-type H+-ATPase inhibitor that blocks
auto-phagosome-lysosome fusion). SAHA-induced LC3-II accumulation was attenuated by 3-MA (Fig. 2C). However, SAHA could still lead to the accumulation of LC3-II in the presence of bafilomycin A1 (Fig. 2C), suggesting that the increase of LC3-II was not due to the blockade of autophagic degradation. The increase of LC3-I by bafilomycin A1 might be due to the recy-cling of LC3-II.15 The autophagic flux was further
con-firmed by the decrease of p62 that can be degraded by autophagy (Fig. 2D).25
SAHA induced autophagy through inhibition of Akt/ mTOR pathway. Interruption of mTOR signaling is known to stimulate autophagy.12 To investigate the molecular mechanisms
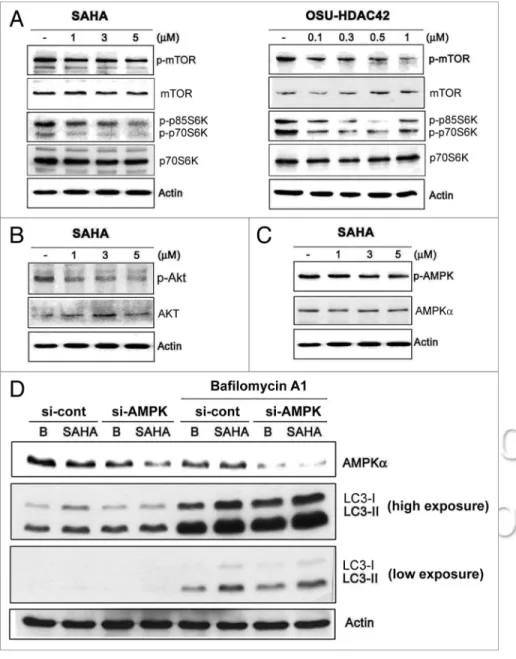
of SAHA-induced autophagy, mTOR kinase activity measured by its phosphorylation was first examined. SAHA and OSU-HDAC42 inhibited the phosphorylation of mTOR kinase as well as its downstream substrate p70/p85S6 kinase (p70/p85S6K) (Fig. 3A). The class I PI3K/Akt is reported to phosphorylate mTOR.26 Activation of AMPK could induce autophagy through
phosphorylation and activation of the TSC1/TSC2 complex, a negative regulator of mTOR.27,28 SAHA inhibited Akt
activ-ity (phosphorylation) in a dose-dependent manner (Fig. 3B). However, SAHA did not increase AMPK phosphorylation and knockdown of AMPK did not affect SAHA-induced LC3-II accumulation in the presence or absence of bafilomycin A1, either Appropriate modification of autophagy, i.e., inhibition of
cyto-protective autophagy or promotion of autophagic cell death, might enhance cytotoxicity of anti-cancer therapy. In this study, we showed that SAHA induced autophagy in HCC cells through inhibition of Akt/mTOR pathway and induction of ER stress response. Inhibition of autophagy reduced SAHA-induced cyto-toxicity, indicating that SAHA elicited autophagic cell death. Our results show that SAHA is an attractive candidate for the treatment of HCC and that pharmacological targeting of auto-phagy provides promise for the management of cancer therapy.
Results
HDAC inhibitors induced autophagy. It has been reported that HDAC inhibitors can induce not only mitochondria-mediated apoptosis, but also caspase-independent autophagic cell death in
Figure 1. hDAc inhibitors induced autophagosome formation. (A) The
ultrastruc-tures of huh7 cells treated with or without 0.5 μM OsU-hDAc42 for 24 h were analyzed by electron microscopy. (a) no treatment. (b) OsU-hDAc42-treated cells. (c) amplification from the square region of (b). N, nucleus. Arrowheads, autophago-somes (AP). Bars, 2 μm. (B) huh7 cells expressing eGFP-Lc3 were treated with 5 μM sAhA or 0.5 μM OsU-hDAc42 for 24 h and then fixed by 3.7% paraformaldehyde and mounted for fluorescence microscopy analysis. (c) Quantitative data calculated the percentage of huh7 cell expressing eGFP-Lc3 puncta. At least 100 cells from each treatment group were examined under fluorescence microscopy.
p21 was not involved in HDAC inhibitor-induced auto-phagy. The cyclin-dependent kinase (CDK) inhibitor p21 has been identified as a tumor suppressor involved in cell cycle arrest. HDAC inhibitors have been shown to induce p21 expres-sion.30 A recent study indicates that p21 acts as a mediator of the
apoptotic pathway and a negative regulator of autophagy.31 We
previously showed that statins increased p21 expression through inhibition of HDAC activity.32 Furthermore, statins were found
to induce p21-dependent autophagy.33 Therefore, the role of
p21 in SAHA-induced autophagy was also evaluated. SAHA and OSU-HDAC42 both induced the expression of p21 and LC3-II accumulation in Huh7 and HepG2 cells (Fig. 5A and B). However, inhibition of p21 by siRNA did not block the SAHA- and OSU-HDAC42-induced LC3-II accumulation in the presence of absence of bafilomycin A1 (Fig. 5C), demon-strating that HDAC inhibitor-induced autophagy is not depen-dent on p21.
(Fig. 3C and D). These results suggest that SAHA-induced autophagy is through inhibition of Akt/mTOR signaling but is not dependent on AMPK.
ER stress was involved in autophagy induction. Endoplasmic reticulum (ER) stress has been reported to connect autophagy through PERK/eIF2α and IRE1/TRAF/JNK signaling path-ways.29 We examined whether ER stress signaling was involved
in SAHA-induced autophagy in Huh7 cells. Phosphorylation of PERK and eIF2α was first examined. PERK phosphorylation was induced by treatment with SAHA for 1 to 4 h, and phos-phorylation of eIF2α was prolonged to 24 h (Fig. 4A). Following ER stress, the AKT activity was reduced and LC3-II protein level was accumulated (Fig. 4A). Consistently, OSU-HDAC42 induced phosphorylation of eIF2α in a dose-dependent man-ner (Fig. 4B). The ER stress inducer tunicamycin also induced LC3-II accumulation in Huh7 cells (Fig. 4C), suggesting that ER stress was involved in SAHA-induced autophagy.
Figure 2. hDAc inhibitors induced Lc3-ii accumulation. in (A) huh7, hep3B and hepG2 cells were treated with 1, 3, 5 μM sAhA or 0.3, 0.5, 1 μM
OsU-hDAc42 for 24 h. in (B) huh7 cells were treated with 5 μM sAhA, 1 μM OsU-OsU-hDAc42, 1 μM TsA or 5 μM Ms275 for 24 h. in (c) huh7 cells were treated with 5 μM sAhA alone or in combination with 5 mM 3-MA or 50 nM bafilomycin A1 for 24 h. in (D) huh7 cells were treated with 1, 3, 5 μM sAhA for 24 h and then harvested for protein analysis. cell lysates were resolved in sDs-PAGe and probed with specific antibodies against Lc3, p62 and β-actin. Quantification of each band by densitometry was performed and the Lc3-ii/Actin ratio was indicated.
or bafilomycin A1, then exposed to SAHA for 3 days and subjected to MTT assay. SAHA reduced the cell viability by 58.44%, which was rescued by 3-MA but not bafilo-mycin A1 (Fig. 6B). To confirm the effect of 3-MA, the Atg5 knockout MEFs, in which the early stage of autophagy was suppressed, were used. The cytotoxicity induced by SAHA was more sensitive in wild-type (WT) compared to Atg5-/- MEF cells (Fig.
6C). These results suggest that SAHA-induced autophagy leads to cell death.
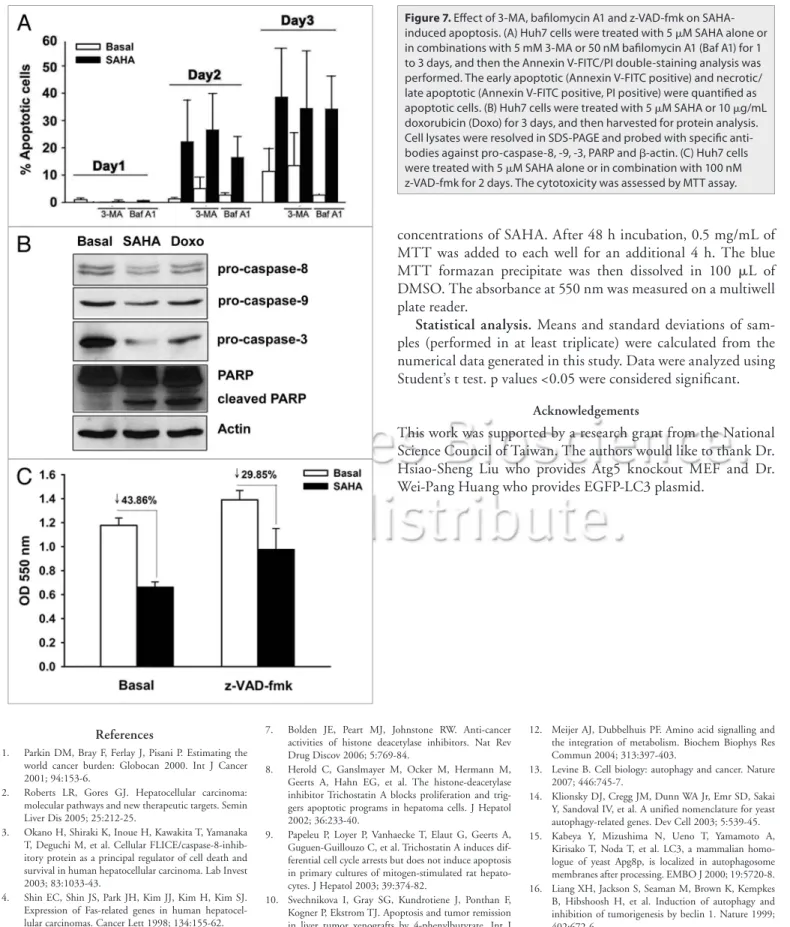
SAHA induced autophagy and apop-tosis in a parallel manner. Autophagy and apoptosis may be triggered in an inde-pendent or mutually exclusive manner, and these two phenomena jointly decide the cell fate.35 Thus, Annexin V-FITC/PI
double staining was used to further exam-ine whether SAHA also induced apoptosis. Apoptosis was seen after treatment with SAHA for 2 and 3 days (Fig. 7A). The cleav-age of caspase-8, -9, -3 and PARP was also detected in Huh7 cells treated with SAHA and doxorubicin that acts as a positive con-trol (Fig. 7B). The pan-caspase inhibitor z-VAD-fmk reversed SAHA-induced cyto-toxicity (Fig. 7C). SAHA-induced apoptosis was not affected by 3-MA or bafilomycin A1 (Fig. 7A), indicating that SAHA simultane-ously induced autophagy and apoptosis, and these two events occurred independently.
Discussion
Hepatocellular carcinoma (HCC) is rela-tively chemo-resistant and highly refrac-tory to cytotoxic chemotherapy. A number of chemotherapies have been evaluated in clinical trials, but none of them show signifi-cant efficacy.2 Defect in autophagy is
associ-ated with a malignant phenotype and poor prognosis of HCC.36 Thus, reactivation
of autophagy might be an effective strat-egy for the treatment of HCC. Our results demonstrated that HDAC inhibitor SAHA induced autophagy in HCC, similar to that reported in MEF, HeLa and MRT cells.19,20 Combination with 3-MA or
knock-down of Atg5 reduced the cytotoxicity of SAHA, suggesting that SAHA induced autophagic cell death.
Although autophagy is monitored by the signal ratio of LC3-I to LC3-II detected by immunoblot, false-positive or false-nega-tive results have been reported due to the lability of LC3-I.22-24
Therefore, it is suggested to measure the overall levels of LC3-II normalized to a loading control such as β-actin or α-tubulin.23,37
We found that SAHA increased the expression of both LC3-I and Inhibition of autophagy reversed SAHA-induced
cytotoxic-ity. To evaluate the cytotoxicity of SAHA, cells were treated with 5 μM SAHA for 1, 3, 5 days and MTT assay was performed. As shown in Figure 6A, SAHA reduced the growth rate of cells in a time-dependent manner. The declines of cell population at day 5 might represent the increases of dead cells. Autophagy is thought to be a cytoprotective process in starving cells. However, excess autophagy can induce type II programmed cell death (auto-phagic cell death).34 To study the role of autophagy in HDAC
inhibitor-induced cytotoxicity, cells were pretreated with 3-MA
Figure 3. sAhA activated Pi3K/Akt/mTOR signaling. huh7 cells were treated with 1, 3, 5 μM
sAhA or 0.1, 0.3, 0.5, 1 μM OsU-hDAc42 for 24 h and then harvested for protein analysis. cell lysates were resolved in sDs-PAGe and probed with specific antibodies against (A) p-mTOR (ser2448), mTOR, p-p70s6K (Thr389), p70s6K and (B) p-Akt, AKT and (c) p-AMPK (Thr172), AMPKα and β-actin. (D) The expression of AMPK in huh7 cells was knocked down by 24 h-siRNA transfection as described in Materials and Methods. The cells were treated with 5 μM sAhA for 24 h in the absence or presence of 50 nM bafilomycin A1, and then harvested for protein analy-sis. cell lysates were resolved in sDs-PAGe and probed with specific antibodies against AMPKα, Lc3 and β-actin.
stress: ER transmembrane eIF2α protein kinase (PERK), ser-ine/threonine kinases inositol-requiring enzyme-1 (IRE1) and transcription factor 6 (ATF6).47 An immediate response to
pro-tein overload in ER is the activation of PERK to phosphorylate eukaryotic translation initiation factor alpha (eIF2α) which then induces expression of a transcription factor CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP/ GADD153).48 Disturbance of ER homeostasis has been reported
to induce autophagy and cell death.49 Our results indicated that
SAHA induced ER stress via activation of PERK and phosphory-lation of eIF2α. Inhibition of Akt/mTOR activity by SAHA was also found. It is possible that SAHA induced ER stress linked to downregulation of Akt/mTOR signaling and led to autophagy. This was supported by the report that ER stress inducers including tunicamycin, DTT and MG132 negatively regulates Akt/TSC/ mTOR pathway to enhance autophagy.50 Therefore, ER might be
LC3-II. It has been reported that the expres-sion of LC3 mRNA increases during amino acid starvation, indicating that the induction of autophagy is accompanied by the increases of LC3 synthesis.38 To confirm that SAHA
indeed induced autophagy, GFP-LC3 puncta and ultrastructural analysis by transmission electron microscopy were performed to moni-tor the autophagosome formation. Additionally, autophagic flux was estimated by treatment with 3-MA or bafilomycin A1 and by measure-ment of p62 degradation.
It has been reported that autophagy not only plays a cytoprotective role but also induces cell death.39 A recent study indicates that
p62 might act as a key factor that modulates autophagy to cell death or survival.40 p62
inter-acts with TRAF6, which is a lysine 63 (K63) E3 ubiquitin ligase, then promotes TRAF6 oligomerization and results in the activation of NFκB.41 Thus, accumulation of p62 promotes
cell survival and tumorigenesis.40 However, p62
binds LC3 through the LC3-interacting region (LIR), and was degraded after fusing with the lysosome.42 The elimination of p62 suppresses
tumorigenesis.40 Our results found that SAHA
decreased the expression of p62 in a dose-dependent manner, consistent with the fact that SAHA induced autophagic cell death.
Autophagy is believed to have important but conflicting effects on tumor development. Experimental evidence supports a role of late stage in cancer promotion and early stage in can-cer suppression.43 It has been found that the late
stage promotes tumorigenesis by helping tumor cells located in the central area of the mass to survive under hypoxia and nutrient depriva-tion.13,43 However, autophagy shows its effects
on inhibition of cell growth at the early stage. Overexpression of beclin 1 in MCF7 breast cells
promoted autophagy and led to inhibition of cell proliferation. Decreased expression of beclin 1 has been reported in human breast, ovarian and brain tumors.16,44 Moreover, rapamycin,
which induces autophagy by inhibiting the mTOR kinase, has been demonstrated to be a potent therapeutic strategy for many tumor types in clinical studies.45 We also found SAHA induced
autophagy through inhibition of mTOR activity. Consistently, suppression of the early stage by 3-MA or Atg5 knockout reduced SAHA-induced cytotoxicity. Although SAHA-induced cyto-toxicity was attenuated in Atg5-/- MEF cells, it is probable that
wild-type and Atg5-/- cells have differential sensitivity to HDAC
inhibitors, which awaits further investigation.
The endoplasmic reticulum (ER) plays an important role as a sensor for cellular stress to detect the changes in cell homeo-stasis and respond to different signal pathways.46 There are three
ER transmembrane proteins acting as proximal sensors for ER
Figure 4. eR stress was involved in autophagy. (A) huh7 cells were treated with 5 μM sAhA
for 0–48 h and then harvested for protein analysis. cell lysates were resolved in sDs-PAGe and probed with specific antibodies against p-PeRK, PeRK, p-eiF2α, eiF2α, p-AKT, AKT, Lc3 and β-actin. Quantification of each band by densitometry was performed and the ratio of phosphoprotein and total protein was indicated. in (B) huh7 cells were treated with 0.1, 0.3, 0.5, 1 μM OsU-hDAc42 for 24 h. in (c) huh7 cells were treated with 1 μM OsU-hDAc42 (OsU42) or 5 μM tunicamycin (TUN) for 24 h. cell lysates were resolved in sDs-PAGe and probed with specific antibodies against p-eiF2α, eiF2α, Lc3 and β-actin.
Material and methods Materials. SAHA (Vorinostat) was gener-ously provided by Merck. TSA (T8552), MS-275 (M5568), 3-MA (M9281) and bafilomycin A1 (B1793) were purchased from Sigma. The antibodies specific for p21 (sc-397), PERK (sc-13073), p70S6K (sc-230), PARP (sc-25780) and β-actin (sc-161) were purchased from Santa Cruz Biotechnology. The antibodies specific for LC3B (2775), phospho-Thr980-PERK (3179), phospho-Ser2481-mTOR (2974), mTOR (2972), phospho-Thr389-p70S6K (9206), AMPKα1/2 (2532), phospho-Thr172-AMPKα1/2 (2531), pase-8 (9746), caspase-9 (9501) and cas-pase-3 (9662) were purchased from Cell Signaling Technology. Phospho-Thr308-Akt (2214-1) and phospho-Ser51-eIF2α (1090-1) antibodies were purchased from EPITOMICS. The EGFP-LC3 plasmid was kindly provided by Dr. Wei-Pang Huang (Department of Life Science, National Taiwan University). The AMPK (HSS108454) and p21 (VHS40202) siRNA were purchased from Invitrogen. The control scramble siRNA was pur-chased from MDBio, Inc. The Annexin V-FITC Apoptosis Detection Kit (AVK050) was purchased from Strong Biotech Corporation.
Cell culture. The human HCC cell lines Hep3B, HepG2 and Huh7 were kindly provided by Dr. Ann-Lii Cheng (Department of Internal Medicine, National Taiwan University Hospital). Atg5-/- MEF cells were kindly provided
by Dr. Hsiao-Sheng Liu (Department of Microbiology and Immunology, College of Medicine, National Cheng-Kung University). These cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, 12800-017), supplemented with 10% heat-inactivated fetal bovine serum (FBS; GIBCO, 26140-079), 50 units/mL penicillin and 50 μg/mL streptomycin and incubated at 37°C in a humidified incubator containing 5% CO2.
Western blot analysis. The cells were lysed with lysis buffer (50 mM Tris-HCl, 1 mM EGTA, 1 mM NaF, 150 mM NaCl, 1 mM Na3VO4, 1% TritonX-100, 1 mM phenylmethylsulfonyl fluoride, 1X protease cocktail) on ice for 30 min. Cell lysates were then centrifuged at 13,000x g for 15 min at 4°C. Supernatant was collected and the protein concentration was determined by the Bio-Rad Protein Assay (Bio-Rad Laboratories, 500-0006). Equal amounts of protein (50 μg) were resolved in 7.5–13% SDS-polyacrylamide gel and then transferred to nitrocellulose an important target for SAHA to induce autophagy. The 78 kDa
glucose-regulating protein (GRP78/BiP), an ER chaperone, is a key regulator of ER stress transducers, which binds and inhibits PERK, IRE1 and ATF6 in nonstressed cells. Upon ER stress, PERK, IRE1 and ATF6 are released from GRP78 and become activated.51 A recent study indicates that SAHA induces
acetyla-tion of GRP78, leading to dissociaacetyla-tion and activaacetyla-tion of PERK,52
providing a direct regulation of ER stress by HDAC inhibition. Our study showed that HDAC inhibitors SAHA and OSU-HDAC42 induced autophagy through downregulation of Akt/ mTOR pathway and elicitation of ER stress. SAHA simultane-ously induced apoptosis in HCC cells, which was parallel with autophagy. Inhibition of autophagy suppressed SAHA-induced cytotoxicity. Therefore, combination with autophagic inducers may enhance the efficacy of SAHA on cancer therapy.
Figure 5. Autophagy induction was p21-independent. in (A) huh7 cells were treated with 1, 3, 5
μM sAhA or 0.1, 0.3, 0.5, 1 μM OsU-hDAc42 for 24 h. in (B) huh7 and hepG2 cells were treated with 5 μM sAhA or 1 μM OsU-hDAc42 for 24 h. in (c) The induction of p21 in huh7 cells was knockdown by 24 h-siRNA transfection as described in Materials and Methods. The cells were treated with 5 μM of sAhA for 24 h in the absence or presence of 50 nM bafilomycin A1, and then harvested for protein analysis. cell lysates were resolved in sDs-PAGe and probed with specific antibodies against p21, Lc3 and β-actin.
1% tannic acid and 30 mM sodium cacodylate for 1 h. The fixed cells were suspended in a buffered solution containing 1% osmic acid for 1 h, followed by dehydration in a graded ethanol series, washing with acetone and embedding into EPON epoxy resin. Ultrathin sections (60–80 nm) were prepared on an ultramicro-tome and double-stained with uranyl acetate and lead citrate. All sections were examined and photographed with a Philips EM300 transmission electron microscope.
Flow cytometry. Huh7 cells were grown to 50% confluence, incubated with 5 μM SAHA for indicated time intervals. The cells were trypsinized, washed in PBS and centrifuged at 200x g for 5 min. Then, the pellet was resuspended in 100 μL binding buffer and added 2 μL Annexin V-FITC and 2 μL propidium iodide (PI). After 15 min incubation at room temperature, FITC and PI fluorescences were detected by FACScalibur flow cytom-eter (Becton Dickinson, San Diego, CA) and subsequently ana-lyzed by CellQuest software.
MTT assay. The cell viability after SAHA treatment was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT; Sigma, M2128) assay. Cells were plated in at least triplicate in 96-well plates and treated with increasing membrane (Amersham, RPN303E). The membrane was
incu-bated with the appropriate primary antibody at 4°C overnight. Then, the membrane was washed and incubated with a horse-radish peroxidase (HRP)-conjugated secondary antibody for 30 min at room temperature. The immunoblots were visualized by WESTERN LIGHTNING Plus-ECL (PerkinElmer, NEL105).
Immunofluorescence. Huh7 cells, grown on coverslips, were transfected with EGFP-LC3 plasmid, followed by HDAC inhibi-tors treatments. The cells were then rapidly washed with PBS and fixed at room temperature for 15 min with 3.7% paraformalde-hyde. After being washed with PBS twice, the cells were blocked with 1% bovine serum albumin in TTBS and then mounted. The subcellular distribution of EGFP-LC3 was observed under a fluorescence microscope (Zeiss Axiophot2).
Transient transfection. The plasmid (EGFP-LC3) or siRNA (AMPK and p21) was transiently transfected into cells with Lipofetamine 2000 Reagent (11668-019) from Invitrogen. After 24 h, the cells were treated with SAHA for 24 h and subjected to fluorescent analysis or western blotting assay.
Transmission electron microscopy. Cells were then fixed in a solution containing 8% paraformaldehyde, 5% glutaraldehyde,
Figure 6. effects of 3-MA and bafilomycin A1 on sAhA-induced autophagy and cytotoxicity. in (A), huh7 cells were treated with 5 μM sAhA for 1, 3, 5
days. in (B), huh7 cells were treated with 5 μM sAhA alone or in combination with 5 mM 3-MA or 50 nM bafilomycin A1 for 3 days. in (c), wild-type (WT) or Atg5-/- MeFs were treated 1, 3, 5 μM sAhA for 3 days. The cytotoxicity was assessed by MTT assay. *p < 0.05, **p < 0.01, ***p < 0.005.
concentrations of SAHA. After 48 h incubation, 0.5 mg/mL of MTT was added to each well for an additional 4 h. The blue MTT formazan precipitate was then dissolved in 100 μL of DMSO. The absorbance at 550 nm was measured on a multiwell plate reader.
Statistical analysis. Means and standard deviations of sam-ples (performed in at least triplicate) were calculated from the numerical data generated in this study. Data were analyzed using Student’s t test. p values <0.05 were considered significant.
Acknowledgements
This work was supported by a research grant from the National Science Council of Taiwan. The authors would like to thank Dr. Hsiao-Sheng Liu who provides Atg5 knockout MEF and Dr. Wei-Pang Huang who provides EGFP-LC3 plasmid.
Figure 7. effect of 3-MA, bafilomycin A1 and z-VAD-fmk on
sAhA-induced apoptosis. (A) huh7 cells were treated with 5 μM sAhA alone or in combinations with 5 mM 3-MA or 50 nM bafilomycin A1 (Baf A1) for 1 to 3 days, and then the Annexin V-FiTc/Pi double-staining analysis was performed. The early apoptotic (Annexin V-FiTc positive) and necrotic/ late apoptotic (Annexin V-FiTc positive, Pi positive) were quantified as apoptotic cells. (B) huh7 cells were treated with 5 μM sAhA or 10 μg/mL doxorubicin (Doxo) for 3 days, and then harvested for protein analysis. cell lysates were resolved in sDs-PAGe and probed with specific anti-bodies against pro-caspase-8, -9, -3, PARP and β-actin. (c) huh7 cells were treated with 5 μM sAhA alone or in combination with 100 nM z-VAD-fmk for 2 days. The cytotoxicity was assessed by MTT assay.
References
1. Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer 2001; 94:153-6.
2. Roberts LR, Gores GJ. Hepatocellular carcinoma: molecular pathways and new therapeutic targets. Semin Liver Dis 2005; 25:212-25.
3. Okano H, Shiraki K, Inoue H, Kawakita T, Yamanaka T, Deguchi M, et al. Cellular FLICE/caspase-8-inhib-itory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest 2003; 83:1033-43.
4. Shin EC, Shin JS, Park JH, Kim JJ, Kim H, Kim SJ. Expression of Fas-related genes in human hepatocel-lular carcinomas. Cancer Lett 1998; 134:155-62. 5. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E,
Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378-90. 6. Kudo M. Hepatocellular carcinoma 2009 and beyond:
from the surveillance to molecular targeted therapy. Oncology 2008; 75:1-12.
7. Bolden JE, Peart MJ, Johnstone RW. Anti-cancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5:769-84.
8. Herold C, Ganslmayer M, Ocker M, Hermann M, Geerts A, Hahn EG, et al. The histone-deacetylase inhibitor Trichostatin A blocks proliferation and trig-gers apoptotic programs in hepatoma cells. J Hepatol 2002; 36:233-40.
9. Papeleu P, Loyer P, Vanhaecke T, Elaut G, Geerts A, Guguen-Guillouzo C, et al. Trichostatin A induces dif-ferential cell cycle arrests but does not induce apoptosis in primary cultures of mitogen-stimulated rat hepato-cytes. J Hepatol 2003; 39:374-82.
10. Svechnikova I, Gray SG, Kundrotiene J, Ponthan F, Kogner P, Ekstrom TJ. Apoptosis and tumor remission in liver tumor xenografts by 4-phenylbutyrate. Int J Oncol 2003; 22:579-88.
11. Ogawa K, Yasumura S, Atarashi Y, Minemura M, Miyazaki T, Iwamoto M, et al. Sodium butyrate enhances Fas-mediated apoptosis of human hepatoma cells. J Hepatol 2004; 40:278-84.
12. Meijer AJ, Dubbelhuis PF. Amino acid signalling and the integration of metabolism. Biochem Biophys Res Commun 2004; 313:397-403.
13. Levine B. Cell biology: autophagy and cancer. Nature 2007; 446:745-7.
14. Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003; 5:539-45. 15. Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, et al. LC3, a mammalian homo-logue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8. 16. Liang XH, Jackson S, Seaman M, Brown K, Kempkes
B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6.
17. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007; 6:304-12.
18. Tanida I, Ueno T, Kominami E. LC3 conjugation sys-tem in mammalian autophagy. Int J Biochem Cell Biol 2004; 36:2503-18.
42. Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/ SQSTM1 into autophagosomes. J Cell Sci 2008; 121:2685-95.
43. Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res 2007; 67:9677-84. 44. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel
A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809-20.
45. Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res 2006; 66:10040-7.
46. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008; 454:455-62.
47. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519-29.
48. Wu J, Kaufman RJ. From acute ER stress to physiologi-cal roles of the Unfolded Protein Response. Cell Death Differ 2006; 13:374-84.
49. Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281:30299-304.
50. Qin L, Wang Z, Tao L, Wang Y. ER stress nega-tively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010; 6:1-9.
51. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2:326-32.
52. Kahali S, Sarcar B, Fang B, Williams ES, Koomen JM, Tofilon PJ, et al. Activation of the unfolded protein response contributes toward the antitumor activity of vorinostat. Neoplasia 2010; 12:80-6.
31. Fujiwara K, Daido S, Yamamoto A, Kobayashi R, Yokoyama T, Aoki H, et al. Pivotal role of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 in apoptosis
and autophagy. J Biol Chem 2008; 283:388-97. 32. Lin YC, Lin JH, Chou CW, Chang YF, Yeh SH, Chen
CC. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res 2008; 68:2375-83.
33. Yang PM, Liu YL, Lin YC, Shun CT, Wu MS, Chen CC. Inhibition of autophagy enhances anti-cancer effects of atorvastatin in digestive malignancies. Cancer Res 2010; 70:7699-709.
34. Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann R. Programmed cell death (PCD). Apoptosis, autophagic PCD or others? Ann NY Acad Sci 2000; 926:1-12.
35. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741-52. 36. Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, et al.
Association of autophagy defect with a malignant phe-notype and poor prognosis of hepatocellular carcinoma. Cancer Res 2008; 68:9167-75.
37. Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009; 452:1-12. 38. Nara A, Mizushima N, Yamamoto A, Kabeya Y, Ohsumi
Y, Yoshimori T. SKD1 AAA ATPase-dependent endo-somal transport is involved in autolysosome formation. Cell Struct Funct 2002; 27:29-37.
39. Bursch W, Ellinger A, Kienzl H, Torok L, Pandey S, Sikorska M, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis 1996; 17:1595-607. 40. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G,
Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062-75. 41. Moscat J, Diaz-Meco MT, Wooten MW. Signal
inte-gration and diversification through the p62 scaffold protein. Trends Biochem Sci 2007; 32:95-100. 19. Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and
autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA 2004; 101:18030-5. 20. Watanabe M, Adachi S, Matsubara H, Imai T, Yui Y,
Mizushima Y, et al. Induction of autophagy in malig-nant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int J Cancer 2009; 124:55-67.
21. Lu YS, Kashida Y, Kulp SK, Wang YC, Wang D, Hung JH, et al. Efficacy of a novel histone deacetylase inhibi-tor in murine models of hepatocellular carcinoma. Hepatology 2007; 46:1119-30.
22. Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol 2010; 221:117-24.
23. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75. 24. Mizushima N, Yoshimori T. How to interpret LC3
immunoblotting. Autophagy 2007; 3:542-5. 25. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander
M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14.
26. Manning BD, Cantley LC. United at last: the tuberous sclerosis complex gene products connect the phos-phoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans 2003; 31:573-8.
27. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577-90.
28. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100:914-22.
29. Hoyer-Hansen M, Jaattela M. Connecting endoplas-mic reticulum stress to autophagy by unfolded pro-tein response and calcium. Cell Death Differ 2007; 14:1576-82.
30. Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem