ELSEVIER
Journal of Orthopaedic Research 23 (2005) 462468Journal
of
Orthopaedic
Research
www.elsevier.com/locate/orthresMolecular mechanism
of
nitric oxide-induced osteoblast apoptosis
*
Ruei-Ming Chen
a$b2*,
Ta-Liang Chen
a,Wen-Ta Chiu
',
Chia-Chen Chang
a Department of Anesthesiology, Wan-Fang Hospital, College of Medicine, Taipei Medical University.
No. 11 1 , Hsing-Lung Rd.. Sec. 3, Taipei 116. Taiwan
Graduate Institute of Medical Sciences, College of Medicine, Taipei Medical University, No. 250, Wu-H.sing St.. Taipei 110, Taiwan
Department of Surgery, Division of Neurosurgery, Wan-Fang Hospital, College of Medicine, Taipei Medical University, Taipei 11 6, Taiwan
Accepted 5 August 2004
Abstract
Nitric oxide (NO) can regulate osteoblast activities. Our previous study showed that NO induced osteoblast apoptosis [Chen
RM, Liu HC, Lin YL, Jean WC, Chen
JS, Wang JH. Nitric oxide induces osteoblast apoptosis through the de novo synthesis
of Bax protein. J Orthop Res 2002;20:295-3021. This study was further aimed to evaluate the mechanism of NO-induced osteoblast
apoptosis from the viewpoints of mitochondrial functions, intracellular oxidative stress, and the anti-apoptotic Bcl-2 protein using
neonatal rat calvarial osteoblasts as the experimental model. Exposure of osteoblasts to sodium nitroprusside (SNP), an NO donor,
significantly increased amounts of lactate dehydrogenase in the culture medium, and decreased cell viability in concentration- and
time-dependent manners. Administration of SNP in osteoblasts time-dependently led to DNA fragmentation. The mitochondrial
membrane potential was significantly reduced following SNP administration. SNP decreased complex
I NADH dehydrogenase
activity in a time-dependent manner. Levels of cellular adenosine triphosphate (ATP) were suppressed by SNP. In parallel with
the mitochondrial dysfunction, SNP time-dependently increased levels
of intracellular reactive oxygen species. Immunoblotting
analysis revealed that SNP reduced Bcl-2 protein levels. Exposure to lipopolysaccharide (LPS) and IFN-y significant increased
endogenous nitrite production. In parallel with the increase in endogenous NO, administration of LPS and IFN-y suppressed cell
viability, mitochondrial membrane potential, and ATP synthesis. Results of this study show that NO released from SNP can induce
osteoblast insults and apoptosis, and the mechanism may involve the modulation of mitochondrial functions, intracellular reactive
oxygen species, and Bcl-2 protein.
0
2004 Orthopaedic Research Society. Published by Elsevier Ltd.
All rights reserved.
Keywords: Osteoblasts; Nitric oxide; Apoptosis; Mitochondria1 functions; Reactive oxygen species; Bcl-2 protein
Introduction
*
Part of this study was presented at the 47th Annual Meeting of the Orthopedic Research Society, February 2001, San Francisco, CA, USA.Nitric oxide (NO), synthesized from L-argenine by
NO
synthases, contributes to the regulation of tissue/
* Corresponding author. Address: Graduate Institute of MedicalSciences, College of Medicine, Taipei Medical University, No. 250, Wu-Hsing St., Taipei 110, Taiwan, ROC. Tel.: +886 2 29307930x21 59; fax: +886 2 86621150.
cell activities, including vasodilation, neurotransmis-
sion, immunoresponses,
and death
control
~4,271. NO
E-mail addresses: [email protected], [email protected]
osteoblasts, N O is constitutively produced
I1
l1.
can also modulate bone remodeling [ l l ] . In untreated
Following pretreatment with inflammatory cytokines
(R.-M. Chen).
0736-0266/$ - see front matter 0 2004 Orthopaedic Research Society. Published by Elsevier Ltd. All rights reserved. doi:lO. 1016/j.orthres.2004.08.011
R.-M. Chen et al. I Journal of’ Orthopaedic Research 23 (2005) 462-468 463
or mechanical stress, high levels
of NO
in osteoblasts
are synthesized [8,26].
NO
has biphasic effects on
osteoblast activities [11,27]. Constitutive NO can be
an effective mediator to regulate osteoblast prolifera-
tion and differentiation [29]. However, overproduction
of NO leads to osteoblast injuries [12,24,26].
Apoptosis, an energy-dependent type of pro-
grammed cell death, has a critical role in evolutionarily
conserving physiologic cell death [14,15]. Hock et al. re-
ported that apoptosis determines osteoblast popula-
tions in the postanal and adult skeleton [18]. A
variety of intrinsic and extrinsic factors are involved
in the regulation of cell apoptosis [10,15,28].
NO
can
be an effector for death regulation [4,9]. In inflamma-
tion-induced osteoporosis, elevated levels of
NO
have
been shown to induce osteoblast apoptosis, and to
decrease bone mineral density [l]. Several lines of
evidence were provided by our previous study to
demonstrate that the NO-induced osteoblast insult
occurs via an apoptotic mechanism [6].
Mitochondria are energy-producing organelles.
Maintenance of the mitochondrial membrane potential
and metabolizing enzyme activities is critical to
adenosine triphosphate (ATP) synthesis [32,38]. Depo-
larization of the mitochondrial membrane potential in-
creases the release of apoptotic factors from the
mitochondria to the cytoplasm and leads to cell apopto-
sis [3,19,34]. Intracellular reactive oxygen species
(ROS),
one of several apoptotic factors, can augment oxidative
stress and damage cells [9,17,22]. Bcl-2 is an anti-apop-
totic protein [20,35]. A decrease in the ratio of Bcl-2
over Bax, an apoptotic protein, increases the risk that
cells will undergo apoptosis [16]. Our previous study
showed that NO increases Bax protein production and
induces osteoblast apoptosis [6]. In this study, we fur-
ther hypothesized that NO-induced osteoblast apoptosis
may occur through modulation of the mitochondrial
functions, intracellular oxidative stress, and Bcl-2
protein levels.
Materials and methods
Cell isolation and drug treatment
Rat osteoblasts were prepared from 3-day-old Wistar rat calvaria according to the method of Partridge et al. [31]. Osteoblasts were seeded in Dulbecco’s modified Eagle’s medium (Gibco-BRL, Grand Island,
NY,
USA) supplemented with 10% heat-inactivated fetal bo- vine serum, L-glutamine, penicillin (1 00 IUIml), and streptomycin (lOOpg/ml) in 75-cm2 flasks at 37°C in a humidified atmosphere of 5% C02. Osteoblasts were grown to confluence prior to drug treat- ment. Only first passage of rat osteoblasts was used in the present study. Each osteoblast isolation represents a determination. Each experiment was repeated at least three times.Sodium nitroprusside (SNP) purchased from Sigma Corporation (St. Louis, MO, USA) was freshly dissolved in phosphate-based saline
(PBS) buffer (0.14M NaC1,2.6mM KCI, 8mM Na2HP04, and 1.5mM KH2P04) and protected from light. Concentration-and time-depend- ent effects of SNP on osteoblasts were determined.
Quantijcation of lactate dehydrogenase
For evaluating the toxicity of SNP to osteoblasts, amounts of lactate dehydrogenase released in culture medium were determined. Osteoblasts (1 x 10’) were seeded in 24-well tissue culture plates (Corn- ing-Costar, Cambridge, MA, USA). After administration of SNP, the culture medium was collected and centrifuged. Levels of lactate dehydrogenase in supernatants were analyzed using a model 7450 automatic autoanalyzer system of Hitachi (Tokyo, Japan).
Assay of cell viability
A trypan blue exclusion method was carried out to determine the cytotoxicity of SNP to osteoblasts. Briefly, osteoblasts (2 x lo’) were cultured in 24-well tissue culture plates. After SNP administration, osteoblasts were trypsinized by 0.1% trypsine-EDTA (Gibco-BRL). Following centrifugation and washing, osteoblasts were suspended in
Ix PBS buffer and stained with an equal volume of trypan blue dye (Sigma). Fractions of dead cells with a blue signal were determined using a reverse-phase microscope.
QuantiJkation of DNA fragmentation
DNA fragmentation in osteoblasts was quantified to evaluate if SNP damaged nuclear DNA. The BrdU-labeled histone-associated DNA fragments in the cytoplasm of cell lysates were detected accord- ing to the instructions of the cellular DNA fragmentation enzyme- linked immunosorbent assay (ELISA) kit (Boehrin er Mannheim, Indianapolis, IN, USA). Briefly, osteoblasts (2 x 10 ) were sub-cul- tured in 24-well tissue culture plates and labeled with BrdU overnight. Cells were harvested and suspended in the culture medium. One hun- dred microliters of cell suspension was added to each well of 96-well tissue culture plates. Osteoblasts were cocultured with SNP for another 8 h at 37°C in a humidified atmosphere of 5% C02. Amounts of BrdU- labeled DNA in the cytoplasm were quantified using an Anthos 2010 microplate photometer (Anthos Labtec Instruments, Lagerhausstrasse, WalsISalzburg, Austria) at a wavelength of 450nm.
B
QuantiJcation of the mitochondria1 membrane potential
The mitochondrial membrane potential was determined following the method of Chen [5]. Briefly, osteoblasts (5 x 10’) were seeded in 12-well tissue culture plates overnight, and then treated with SNP for different time intervals. After SNP administration, osteoblasts were harvested and incubated with 3,3‘-dihexyloxacarbocyanine (DiOC6(3)), a positively charged dye, at 37°C for 30min in a humidi- fied atmosphere of 5% C02. After washing and centrifuging, cell pellets were suspended with Ix PBS buffer. Intracellular fluorescent intensities were analyzed using a flow cytometer (FACS Calibur, Becton Dickin- son, San Jose, CA, USA).
Assay of mitochondria1 NADH dehydrogenase activity
NADH dehydrogenase activity was determined using a colorimet- ric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay following the method of Wu et al. [37]. Briefly, osteoblasts (5 x 10’) were seeded in 96-well tissue culture plates overnight. After drug treat- ment, cells were cultured with new medium containing OSmg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-dipheny~tetrazo~ium bromide for another 3 h. A blue formazan product in cells was dissolved in dimethyl sulfoxide and spectrophotometrically measured at a wavelength of 570nm.
Quantification of cellular adenosine triphosphate ( A T P ) levels
Levels of cellular ATP in osteoblasts were determined with a bio- luminescence assay described previously [7]. This assay was based on luciferase’s requirement for ATP in producing emission light according to the protocol of Molecular Probes’ ATP determination kit (Molecu- lar Probes, Eugene, OR, USA). Luminent light (560nm) emitted by the
464 R.-M. Chen et al. I Journal of Orthopaedic Research 23 (2005) 462468
luciferase-mediated reaction of ATP and luciferin was detected using a WALLAC VICTOR&,, 1420 multilabel counter (Welch Allyn, Turku, Finland).
Table 1
Concentration-dependent effects of sodium nitroprusside on osteoblast viability
Determination of intracellular ROS
Levels of intracellular ROS were uantified following a method de- scribed previously [23]. Briefly, 5 x 10 osteoblasts were cultured in 12- well tissue culture plates overnight, and then cotreated with SNP and 2’,7’-dichlorofluorescin diacetate, an ROS sensitive dye. After drug treatment, osteoblasts were harvested and suspended in Ix PBS buffer. Relative fluorescence intensities of osteoblasts were quantified using a flow cytometer (FACS Calibur).
’3
Gel electrophoresis and immunoblotting analysis
After SNP treatment, osteoblasts were washed with lx PBS buffer. Cell lysates were prepared in ice-cold radioimmunoprecipitation assay (RIPA) buffer (25mM Tris-HCl pH 7.2, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 0.15M NaC1, and 1mM EDTA). To avoid protein degradation, a mixture of proteinase inhibitors, including 1 mM phenyl methyl sulfonyl fluoride, 1 mM sodium orthovanadate, and 5pg/ml leupeptin, was added to the RIPA buffer. Protein concen- trations were quantified by a bicinchonic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Cytosolic proteins (100 pg per well) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transfered to nitrocellulose membranes. Mem- branes were blocked with 5% non-fat milk at 37 “C for 1 h. Immunode- tection of Bcl-2 was carried out using a mouse monoclonal antibody against rat Bcl-2 (Transduction Laboratories, Lexington, KY, USA). Cellular p-actin protein was immunodetected using a mouse mono- clonal antibody against mouse p-actin (Sigma) as an internal standard. Intensities of the immunoreactive bands were determined using an UVIDOCMW version 99.03 digital imaging system (Uvtec, Cam- bridge, England, UK).
Statistical analysis
Statistical differences between the control and SNP-treated groups were considered significant when the
P
value of Duncan’s multiple- range test was less than 0.05. Statistical analysis between groups over time was carried out using two-way ANOVA.Results
Table 1 presents the concentration-dependent effects
of SNP on osteoblast viability. Administration of
0.5 and 1mM SNP in osteoblasts did not affect lactate
dehydrogenase release. SNP at 1.5 and 2mM
significantly increased amounts of lactate dehydroge-
nase by 85% and 154%, respectively. Analysis by a try-
pan blue exclusion method revealed that SNP at 0.5
and 1mM was not cytotoxic to osteoblasts (Table 1).
However, after administration of 1.5 and 2mM SNP,
viability of osteoblasts was decreased by 38% and
6270, respectively.
Table 2 shows the time-dependent effects of SNP
on
osteoblast viability. In 4-h-treated osteoblasts, SNP
did not affect lactate dehydrogenase release. After SNP
administration for
8
and 16h, amounts of lactate dehy-
drogenase were significantly augmented by 65% and
151%, respectively. Exposure to SNP for 4 h was not
cytotoxic to osteoblasts (Table 2). Viability of osteo-
SNP, mM Lactate dehydrogenase Cell viability
IUIL) (cell number x lo3)
0 41 f 11 1 8 8 f 44
0.5 3 8 f 10 187 f 38
1 35 f 8 179 f 45
1.5 76 f 8’ 116f 18‘
2 1 0 4 f 13’ 7 2 f 21’
Rat osteoblasts were exposed to 0.5, 1, 1.5, and 2mM sodium nitro- prusside (SNP) for 16h. Amounts of lactate dehydrogenase in the culture medium were determined by an autoanalyzer as described in “Materials and Methods”. Cell viability was assayed by the trypan blue exclusion method. Each value represents the mean f SEM for
n = 12 from four independent cell preparations.
Values significantly differ from the respective control,
P
< 0.05.Table 2
Time-dependent effects of sodium nitroprusside on osteoblast viability Time, h Lactate dehydrogenase Cell viability
( U W (cell number x
10”)
0 3 7 f 10 4 30 f 6 8 61 f 9’ 16 93 f 11’ 179 f 31 167 f 58 107 & 30‘ 81 f 19’Rat osteoblasts were exposed to 2mM sodium nitroprusside for 4, 8, and 16h. Amounts of lactate dehydrogenase in the culture medium were determined by an autoanalyzer as described in “Materials and Methods”. Cell viability was assayed by the trypan blue exclusion method. Each value represents the mean f SEM for n = 12 from four independent cell preparations.
* Values significantly differ from the respective control,
P
< 0.05.blasts was significantly reduced by 400/0 and 55%, respec-
tively, following SNP administration for
8
and 16h.
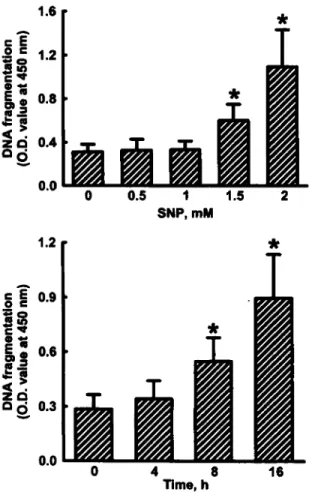
Fig. 1 presents the effects of SNP on DNA damage.
Exposure of osteoblasts to 0.5 and 1mM SNP for 16h
did not cause DNA injury (top panel). SNP at 1.5 and
2mM significantly increased levels of DNA fragments
by 92% and 267%, respectively. In 4-h-treated osteo-
blasts, SNP did not damage nuclear DNA
(bottom
panel).
After SNP administration for 8 and 16h, levels
of DNA fragments were enhanced by 65% and 205%,
respectively.
The mitochondria1 membrane potential of osteo-
blasts was determined and shown in Fig. 2. In l-h-trea-
ted osteoblasts, SNP did not change the membrane
potential of mitochondria. After administration of
SNP for 2 and 4h, the mitochondrial membrane
potential was significantly decreased by 16% and 36%,
respectively.
Table 3 shows the effects of SNP on NADH dehy-
drogenase activity and ATP synthesis. In 4-h-treated
osteoblasts, SNP did not affect NADH dehydrogenase
activity. Administration of SNP for 8 and 16 h signifi-
cantly reduced activities of NADH dehydrogenase by
R.-M. Chen et al. I Journal of Orthopaedic Research 23 (2005) 462-468 465
lS6
c
-
*
C E=
=
1.2
P
t P
nc
gg
2
0.82
6
0.40.0
0 0.51
1.5
2
SNP,mM
3
lime, h
Fig. 1. Effects of SNP on DNA fragmentation. Osteoblasts prepared from neonatal rat calvaria were exposed to 0.5, 1, 1.5, and 2mM SNP for 16 h ( f o p panel) or 2mM SNP for 4, 8, and 16h (bottom panel). DNA fragments in osteoblasts were quantified using a BrdU-labeled histone-associated DNA fragmentation ELISA kit as described in “Material and Methods”. Each value represents the mean ? SEM for
n = 9 from three independent cell preparations.
*
Values significantly differ from the respective control, P < 0.05.Table 3
Effects of sodium nitroprusside on NADH dehydrogenase activity and cellular adenosine triphosphate levels
Time, h NADH dehydrogenase ATP
(OD value at 550nm) (PmoU
0 1.03 f 0.21 4 0.98
f
0.17 8 0.74 f 0.18’ 16 0.62 ? 0.14’ 38 f 8 31 f 9 24 f 4’ 17f.5’ Rat osteoblasts were exposed to 2mM sodium nitroprusside for 0,4,8, and 16h. NADH dehydrogenase activity was assayed by a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide method as described in “Materials and Methods”. Cellular adenosine tri- phosphate (ATP) levels were determined by a bioluminescence assay. Each value represents the mean f SEM for n = 12 from four inde- pendent cell preparations.* Values significantly differ from the respective control, P < 0.05.
18%
and 58%, respectively. In 4-h-treated osteoblasts,
SNP did not change cellular ATP levels (Table 3). After
*
*
0
1
2
4
lime, h
Fig. 2. Effects of SNP on the mitochondrial membrane potential. Osteoblasts prepared from neonatal rat calvaria were exposed to 2 mM SNP for 1, 2, and 4h. The mitochondrial membrane potential of osteoblasts was analyzed using the fluorescent dye, DiOC6(3), and quantified by a flow cytometer. Each value represents the mean f SEM for n = 9 from three independent cell preparations.
*
Values signif- icantly differ from the respective control, P < 0.05.0
1
2
4
Time, h
Fig. 3. Effects of SNP on intracellular reactive oxygen species (ROS). Osteoblasts prepared from neonatal rat calvaria were exposed to 2 mM
SNP for 1, 2, and 4h. Levels of intracellular ROS were caught using the ROS-sensitive dye, 2’,7’-dichlorofluorescin diacetate, and quanti- fied by a flow cytometer. Each value represents the mean ? SEM for
n = 9 from three independent cell preparations.
*
Values significantly differ from the respective control, P < 0.05.administration of
S N P
for
8
and 16h, cellular ATP
levels were significantly decreased by 37% and 55%,
respectively
.
Intracellular levels of
ROS
were determined and are
shown in Fig.
3.
In 1-h-treated osteoblasts,
SNP
caused
a significant increase in
ROS
levels by 76%. Levels of
intracellular ROS in osteoblasts were respectively
466 R.-M. Chen et al. I Journal of Orthopaedic Research 23 (2005) 462468
1
2
Bcl-2
*
4 2 6 kD
f3-Actin
-26
kD
(A)
C
SNP
C
SNP
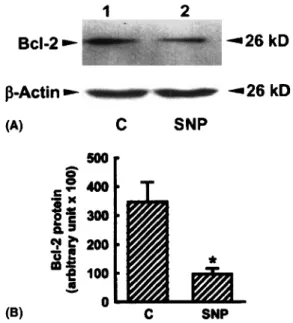
Fig. 4. Immunoblotting analysis of Bcl-2 protein. Osteoblasts pre- pared from neonatal rat calvaria were exposed to 2mM SNP. Cytosolic protein was prepared, separated by SDS-PAGE, and transferred to nitrocellulose membranes. Immunodetection of Bcl-2 protein was carried out using a monoclonal antibody against rat Bcl-2 protein (A, rop panel). P-Actin was immunodetected as an internal control (bottom panel). Intensities of protein bands were quantified by a digtal imaging system (B). Each value represents the mean f SEM for n = 6 from three independent cell preparations.
*
Values signif- icantly differ from the respective control,P
< 0.05.augmented by 3- and 5-fold following SNP administra-
tion for 2 and 4 h.
Fig. 4 shows the effects of SNP
on
Bcl-2 protein.
In
untreated osteoblasts, Bcl-2 protein was detectable
(Fig. 4A, fop panel, lane 1). Administration of SNP in
osteoblasts decreased Bcl-2 protein production (lane
2). p-actin was immunodetected as an internal control
(bottom panel). Quantificantion of these immunodetec-
ted protein bands revealed that levels of Bcl-2 protein
were significantly decreased by 82% following SNP
administration (Fig. 4B).
Table 4
Effects of lipopolysaccharide and IFN-y on nitrite production, cell viability, mitochondrial membrane Dotential. and ATP synthesis
Analysis Control LPS
+
IFN-yNitrite, pM 4 f 1 28 f 8'
Cell viability, cell number x lo3 58 ? 15'
Mitochondria1 membrane 100 61 ? 18'
ATP, pmol 4 6 f 13 2 4 2 6 '
Rat osteoblasts were exposed to a mixture of 1 pglml lipopolysaccha- ride (LPS) and IOOIUlml IFN-y for 16h. Amounts of nitrite in the culture medium were determined by the Griess reaction. Cell viability was assayed by the trypan blue exclusion method. The mitochondrial membrane potential was quantified by a flow cytometer. Levels of adenosine triphosphate (ATP) were assayed by a bioluminescence assay. Each value represents the mean f SEM for n = 6 from three independent cell preparations.
* Values significantly differ from the respective control,
P
< 0.05.137 f 39
potential
(YO
of control)Table 4 presents the effects of endogenous NO pro-
duction on cell viability, the mitochondrial membrane
potential, and ATP synthesis. Administration of lipo-
polysaccharide (LPS) and IFN-y significantly increased
nitrite production by 6.5-fold. Viability of osteoblasts
was decreased by 58% following administration of
LPS
and IFN-y. After administration of LPS and IFN-y,
the mitochondrial membrane potential and cellular
ATP levels were reduced by 39% and 48%, respectively.
Discussion
SNP can be decomposed to NO under light exposure
or
in
the presence of a biological reducing system [2,21].
As presented in our previous study, administration of
SNP significantly enhances the amounts of nitrite, which
corresponds to an increase in NO [6]. In parallel with the
increase in NO, the amounts of lactate dehydrogenase
released from osteoblasts into the culture medium were
significantly augmented. Analysis by the trypan blue
exclusion method showed that the membrane permeabi-
lity
of osteoblasts was disturbed following SNP admin-
istration. The breakage of cell membranes increased
the release
of
lactate dehydrogenase. Thus, NO decom-
posed from SNP broke down plasma membranes,
increasing levels of lactate dehydrogenase in the cul-
ture medium, and leading to osteoblast insults or even
death.
Our previous study provided several lines of evidence
which identified that NO-induced osteoblast death
mainly occurs via an apoptotic pathway [6]. This study
used a cellular DNA fragmentation ELISA to further
demonstrate that the nuclear DNA of osteoblasts was
fragmented following SNP administration. Fragmented
breakage of chromosome DNA is a critical characteris-
tic which indicates that cells are undergoing apoptosis
[
15,341. Our data reveal that NO can damage osteoblast
DNA and induce cell apoptosis. Dypbukt et al. reported
that high concentrations of NO donors increased the re-
lease of lactate dehydrogenase and caused cell necrosis
[13]. This does not rule out the possibility that SNP
partially induces osteoblast necrosis.
SNP significantly reduced cellular ATP levels. Mito-
chondria are critical ATP-synthesizing organelles. This
study shows that SNP can decrease the mitochondrial
membrane potential of osteoblasts. Previous studies re-
ported that disruption
of
the mitochondrial membrane
potential results in mitochondrial depolarization and
blocks the respiratory chain reaction [30,38]. Thus, one
of the possible mechanisms involved in the NO-induced
depletion of ATP levels in osteoblasts is through the
suppression of the mitochondrial membrane potential.
Administration of SNP significantly decreased mitoch-
ondrial complex
I
NADH dehydrogenase activity.
NADH dehydrogenase contributes to the respiratory
R.-M. Chen et al. I Journal of Orthopuedic Re.wurch 23 (2005) 462468 467
chain reaction and ATP synthesis [34]. A decrease in
NADH dehydrogenase activity is another possible
mechanism involved in the NO-induced ATP depletion
in osteoblasts. Reduction of ATP synthesis has been
shown to induce cell apoptosis [3]. Therefore, NO can
decrease cellular ATP levels through suppression of
the mitochondrial membrane potential and complex
I
enzyme activity in osteoblasts and thus induce cell
apoptosis.
This study shows that levels
of
intracellular
ROS
were significantly augmented following SNP administra-
tion.
ROS
is one of the mitochondrial apoptotic factors
[24]. Previous studies reported that depolarization of the
mitochondrial membrane potential enhances the release
of apoptotic factors, including ROS and cytochrome c,
from mitochondria to the cytoplasm and drives cells
undergoing apoptosis [21,32,37]. Li et al. showed that
rotenone, an inhibitor of NADH dehydrogenase, en-
hanced mitochondrial
ROS
and induced cell apoptosis
[22]. Therefore, SNP can increase intracellular
ROS
through suppression
of
the mitochondrial membrane
potential and NADH dehydrogenase activity. However,
the NO radical is one ROS. DCFH-DA was used in this
study to catch ROS. Previous studies demonstrated that
NO and peroxynitrite ( N 0 0 0 - ) can also directly react
with DCFH-DA [17,33]. Therefore, the elevation of
intracellular ROS in SNP-treated osteoblasts is partially
due to the enhancement of intracellular NO.
SNP decreased Bcl-2 protein production. Bcl-2, an
anti-apoptotic protein, can determine if cells undergo
apoptosis [35,36]. A decrease in Bcl-2 protein levels will
increase the risk that cells will undergo apoptosis.
Bax
is
a pro-apoptotic protein [25]. Immunocytochemical and
immunoblotting analyses were carried out in our previ-
ous study to validate that NO increased Bax protein lev-
els in osteoblasts [6]. Mitochondria1 apoptotic factors,
including ROS and cytochrome c, can regulate the cellu-
lar Bcl-2/Bax ratio [9,10]. Therefore, SNP can modulate
mitochondrial functions and increase the release of mit-
ochondrial apoptotic factors. The SNP-caused suppres-
sion of Bcl-2/Bax proteins can induce osteoblasts
apoptosis.
Osteoblasts were exposed to a mixture of LPS and
IFN-y to determine if endogenous NO has similar effects
as those of SNP. After administration of LPS and IFN-
y,levels of nitrite in osteoblsts were significantly
augmented. In parallel with the increase in NO, admin-
istration of LPS and IFN-y disrupted the mitochondrial
membrane potential, reduced cellular ATP levels, and
ultimately induced cell death. This study has shown that
the exogenous and endogenous forms of NO have the
same effects on the induction of mitochondrial dysfunc-
tion and cell death.
In summary, NO decomposed from SNP can cause
DNA fragmentation, thus the NO-induced death mech-
anism mainly occurs via an apoptotic pathway. SNP can
modulate mitochondrial functions through inhibition of
the mitochondrial membrane potential, NADH dehy-
drogenase activity and cellular ATP levels. Levels of
intracellular
ROS
and Bcl-2 protein are also regulated
by SNP. The modulating effects
of
NO decomposed
from SNP are also observed with endogenous NO.
Therefore, this study presents further data to validate
our hypothesis that NO can modulate mitochondrial
functions, intracellular oxidative stress, and Bcl-2 pro-
tein production to induce osteoblast apoptosis. The
roles of caspases in NO-induced osteoblast apoptosis
will be a further study in our laboratory.
Acknowledgements
This study was supported by grants NSC91-2314-B-
038-031 and NSC92-23 14-B-038-010 from the National
Science Council, Taipei, Taiwan, ROC.
References
[l] Armour KE, Van? Hof RJ, Grabowski PS, Reid DM, Ralston SH. Evidence for a pathogenic role of nitric oxide in inflammation-induced osteoporosis. J Bone Miner Res 1999; 1 4 21 3742.
[2] Bates JN, Baker MT, Guerra Jr R, Harrison DG. Nitric oxide generation from nitroprusside by vascular tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochem Pharmacol 1991;42:s15745.
[3] Blom WM, de Bont HJ, Nagelkerke JF. Regional loss of the mitochondrial membrane potential in the hepatocyte is rapidly followed by externalization of phosphatidylserines at that specific site during apoptosis. J Biol Chem 2003;278: 12467-74.
[4] Chang H, Tsai SY, Chang Y, Chen TL, Chen RM. Therapeutic concentrations of propofol protects mouse macrophages from nitric oxide-induced cell death and apoptosis. Can J Anaesth
[S] Chen LB. Mitochondria membrane potential in living cells. Ann Rev Cell Biol 1988;4155-81.
[6] Chen RM, Liu HC, Lin YL, Jean WC, Chen JS, Wang JH. Nitric oxide induces osteoblast apoptosis through the de novo synthesis of Bax protein. J Orthop Res 2002;20:295-302.
[7] Chen RM, Wu CH, Chang HC, Lin YL, Sheu JR, Chen TL. Propofol suppresses macrophage functions through modulating mitochondrial membrane potential and cellular adenosine tri- phosphate levels. Anesthesiology 2003;98: 1 178-85.
[8] Chow JW, Fox SW, Lean JM, Chambers TJ. Role of nitric oxide and prostaglandins in mechanically induced bone formation. J
Bone Miner Res 1998;13:1039-44.
[9] Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun 2001;282: 1075-9.
[lo] Chung KC, Park JH, Kim CH, Ahn YS. Tumor necrosis factor- alpha and phorbol 12-myristate 13-acetate differentially modulate cytotoxic effect of nitric oxide generated by serum deprivation in neuronal PC12 cells. J Neurochem 1999;72: 1482-8.
[ l l ] Collin-Osdoby P, Nickols GA, Osdoby P. Bone cell function,
regulation, and communication: a role for nitric oxide. J Cell Biochem 1995;57:399408.
468 R - M . Chen et al. I Journal of Orthopaedic Research 23 (2005) 462668
[I21 Damoulis PD, Hauschka PV. Nitric oxide acts in conjunction with proinflammatory cytokines to promote cell death in osteo- blasts. J Bone Miner Res 1997;12:412-22.
[I31 Dypbukt JM, Ankarcrona M, Burkitt M, Sjoholm A, Strom K, Orrenius S, et al. Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting RINm5F cells: the role of intracellular polyamines. J Biol Chem 1994;269:3055340.
[I41 Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 1999;18:7719-30.
[I51 Goyal L. Cell death inhibition: keeping caspases in check. Cell 2001; 1W80S8.
[I61 Hay E, Lemonnier J, Fromigue 0, Marie PJ. Bone morphogenetic protein-2 promotes osteoblast apoptosis through a Smad-inde- pendent, protein kinase C-dependent signaling pathway. J Biol Chem 200 1 ;276:29028-36.
[I71 Hikiji H, Shin WS, Koizumi T, Takato T, Susami T, Koizumi Y,
et al. Peroxynitrite production by TNF-a and IL-ID: implication
for suppression of osteoblastic differentiation. Am J Physiol [IS] Hock JM, Krishnan V, Onyia JE, Bidwell JP, Milas J, Stanislaus D. Osteoblast apoptosis and bone turnover. J Bone Miner Res 2001; 16:975-84.
[19] Hortelano S, Alvarez AM, Bosca L. Nitric oxide induces tyrosine nitration and release of cytochrome c preceding an increase of mitochondrial transmembrane potential in macrophages. FASEB J 1999;13:2311-7.
[20] Hortelano S, Bosca L. 6-Mercaptopurine decreases the Bcl-2IBax ratio and induces apoptosis in activated splenic B lymphocytes. Mol Pharmacol 1997;51:41&21.
[21] Kowaluk EA, Seth P, Fung HL. Metabolic activation of sodium nitroprusside to nitric oxide in vascular smooth muscle. J Pharmacol Exp Ther 1992;262:9 16-22.
[22] Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, et al. Mitochondrial complex I inhibitor rotenone induces apop- tosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 2003;278:85 16-25.
[23] Liu HC, Chen RM, Jian WC, Lin YL. Cytotoxic and antioxidant effects of the water extract of traditional Chinese herb gusuibu
(Drynaria fortuner] on rat osteoblasts. J Formos Med Assoc 200 1 ; 100:383-8.
[24] Mancini L, Moradi-Bidhendi N, Becherini L, Martineti V, MacIntyre I. The biphasic effects of nitric oxide in primary rat osteoblasts are cGMP dependent. Biochem Biophys Res Commun 2000;274:477-8 I.
[25] Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Mijake S, et al. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J Neurochem 1999;73:203746. 2OO0;278:E103 1-7.
[26] Mogi M, Kinpara K, Kondo A, Togari A. Involvement of nitric oxide and biopterin in proinflammatory cytokine-induced a p o p totic cell death in mouse osteoblastic cell line MC3T3-El. Biochem Pharmacol 1999;58:649-54.
[27] Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev 1991;43: 10942.
[28] Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci 1997;22:299-306.
[29] OShaughnessy MC, Polak JM, Afzal F, Hukkanen MV, Huang P, MacIntyre I, et al. Nitric oxide mediates 17P-estradiol-stimu- lated human and rodent osteoblast proliferation and differentia- tion. Biochem Biophys Res Commun 2000;277:60&10.
[30] Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A, et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem 2003;278: [3 I] Partridge NC, Alcorn D, Michelangeli VP, Kemp BE, Ryan GB, Martin TJ. Functional properties of hormonally responsive cultured normal and malignant rat osteoblastic cells. Endocrino- [32] Pearce LL, Epperly MW, Greenberger JS, Pitt BR, Peterson J. Identification of respiratory complexes I and 111 as mitochondrial sites of damage following exposure to ionizing radiation and nitric oxide. Nitric Oxide 2001;5:128-36.
[33] Rao KM, Padmanabhan J, Kilby DL, Cohen HJ, Currie MS, Weinberg JB. Flow cytometric analysis of nitric oxide production in human neutrophils using dichlorofluorescein diacetate in the presence of a calmodulin inhibitor. J Leukocyte Biol 1992;51: 496500.
[34] Rathmell JC, Thompson CB. Pathways of apoptosis in lympho- cyte development, homeostasis, and disease. Cell 2002; 109: [35] Srivastava RK, Sollott SJ, Khan L, Hansford R, Lakatta EG, Longo DL. Bcl-2 and Bcl-X (L) block thapsigargin-induced nitric oxide generation, c-Jun NH (2)-terminal kinase activity, and apoptosis. Mol Cell Biol 1999; 195659-74.
[36] Umansky V, Ushmorov A, Ratter F, Chlichlia K, Bucur M, Lichtenauer A, et al. Nitric oxide-mediated apoptosis in human breast cancer cells requires changes in mitochondrial functions and is independent of CD95 (APO-1IFas). Int J Oncol 2000;16
1 09-1 7.
[37] Wu CH, Chen TL, Chen TG, Ho WP, Chiu WT, Chen RM. Nitric oxide modulates pro- and anti-inflammatory cytokines in lipopolysaccharide-activated macrophages. J Trauma 2003;55: 540-5.
[38] Yu XH, Perdue TD, Heimer YM, Jones AM. Mitochondria1 involvement in tracheary element programmed cell death. Cell Death Differ 2002;9: 189-98.
2 8220-8.
logy 198 1 ;I 08~213-9.