行政院國家科學委員會專題研究計畫 成果報告
奈米半導體光催化難分解有機物之研究(3/3)
計畫類別: 個別型計畫 計畫編號: NSC93-2211-E-002-001- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立臺灣大學環境工程學研究所 計畫主持人: 林正芳 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 94 年 10 月 31 日
奈米半導體光催化難分解有機物之研究(3/3)
計畫編號:93-2211-E-002-001-研究報告-完整報告
計畫主持人:林正芳
執行機關:國立臺灣大學環境工程學研究所
計畫執行起迄:2004.08.01 至 2005.07.31
摘要
半導體光催化程序高級氧化程序之一環,其利用適當的光線照射半導體觸 媒,使其產生電子-電洞對進而產生氫氧自由基對有機物進行分解,為一有效的 處理方法,相較於其他處理方式可不用添加藥劑或需進一步處理如生物處理所產 生的污泥,若能進一步利用太陽光則為一節省能源,且更環保的污染處理技術。 然而光觸媒目前仍以 TiO2最為有效,但是 TiO2因有其缺點而還無法快速的 應用於實場操作。其主要缺點有懸浮態的 TiO2雖然接觸面積大,效率較佳,但 後續固液分離與回收困難,尤其是在使用奈米級的 TiO2時;固定形式 TiO2雖然 不用進行觸媒的分離與回收程序,但其表面積無法如懸浮態的大而受限於質傳條 件,光催化效率無法有效提升;TiO2需使用波長約 385 nm 以下的紫外光才能被 激發以進行光催化反應;TiO2 被激發所產生的電子電洞容易再結合而使光催化 效果不易提升。 基於上述這幾項 TiO2原有的缺點,本計畫以複合半導體的顆粒間電子傳送 程序特性為主軸,選擇不同的種類的半導體與不同的方法,製備複合半導體光觸 媒進行研究,並於第三年研究成果中,以顆粒狀活性碳作為載體使其固液分離容 易,利用活性碳的吸附能力以期增加光催化效果,並利用 WO3與 MoO3的特性 使光觸媒可利用可見光範圍的波長來進行光催化反應。目錄
頁次 摘要...i 目錄...ii 表目錄...iii 圖目錄...iii 第一章 研究計畫之背景及目標...1 1.1 研究背景...1 1.2 研究目標...11 第二章 第一、二年研究成果...12 2.1 第一年研究成果...12 2.1.1 催化劑物化性質測定...12 2.1.2 pH 值效應...12 2.1.3 結合半導體催化劑...13 2.2 第二年研究成果...14 2.2.1 觸媒之性質鑑定...14 2.2.2 pH 值對 4-氯酚光催化降解之影響 ...16 2.2.3 複合觸媒對 4-氯酚降解之影響 ...17 第三章 第三年之計畫研究背景說明...19 3.1 研究背景...19 3.2 研究目的...20 第四章 第三年計畫研究方法...21 4.1 光觸媒/活性碳製備方法 ...21 4.2 光觸媒/活性碳之背景吸附實驗 ...21 4.3 光觸媒/活性碳之光催化實驗 ...21 4.4 藥品與儀器設備...23 4.4.1 實驗用藥...23 4.4.2 儀器設備...23 第五章 第三年計畫研究成果...24 5.1 光觸媒/活性碳之比表面積、SEM、EDS 分析...24 5.2 光觸媒/活性碳之吸附與光催化實驗結果 ...28 5.2.1 TiO2/GAC 之實驗結果 ...28 5.2.2 複合半導體之實驗結果...29 5.2.3 使用針鐵礦為擔體之實驗結果...32 第六章 結論與建議...336.1 結論...33
6.2 建議...33
參考文獻...34
表目錄 表 2.1 TiO2、ZnO、TiO2與 ZnO 混合及 TiO2與 ZnO 混合燒結催化劑在 pH=7 及 pH=11 下,光催化降解 4-氯酚之擬一階速率常數 ...14 表 2.2 單一及複合型態觸媒之比表面積...15 表 2.3 觸媒之臨界波長及能隙...15 表 2.4 Cd、Ti 元素之 XPS 定量分析結果...16 表 2.5 波長 254 及 365 nm,觸媒在 pH=7 及 11 下之擬一階速率常數 k(min-1) ...16 表 5.1 活性碳與光觸媒/活性碳的比表面積 ...24 表 5.2 EDS 分析複合半導體光觸媒計算之 Ti、W 與 Mo 之組成 ...27 圖目錄 圖 1.1 光催化研究領域架構圖...3 圖 1.2 CdS/TiO2半導體電子與電洞傳遞圖 ...7 圖 2.1 燒結催化劑之雷射光粉末繞射...12 圖 2.2 混合燒結催化劑之顯微照片...13 圖 2.3 TiO2與 ZnO 在不同 pH 時光催化降解 4-氯酚之擬一階反應速率常數...13
圖 2.4 CdSe/TiO2之 XPS survey spectra ...15
圖 2.5 波長 254 nm 、pH=7 及 11 下之 TiO2及 CdSe/TiO2觸媒 4-CP 降解關係圖 ...16 圖 2.6 波長 254 nm、pH 7,TiO2及 CdSe/TiO2觸媒之光催化實驗比較圖...17 圖 2.7 波長 365 nm、pH=7,TiO2及 CdSe/TiO2觸媒之光催化實驗比較圖...18 圖 4.1 光催化反應設備圖...22 圖 5.1 未負載觸媒之 GAC 表面 SEM 觀察照片...24 圖 5.2 TiO2/GAC (1:2)之表面 SEM 觀察照片...25 圖 5.3 TiO2/GAC (1:1.5)之表面 SEM 觀察照片...25
圖 5.4 WO3-TiO2/GAC (1:2)與 MoO3-TiO2/GAC (1:2)之表面 SEM 觀察照片...26
圖 5.5 WO3-TiO2/GAC 之 EDS 分析結果 ...26
圖 5.6 MoO3-TiO2/GAC 之 EDS 分析結果...28
圖 5.7 無曝氣時 TiO2/GAC (1:4)與 TiO2/GAC (1:1.5)之光催化與吸附結果 ...29
圖 5.8 TiO2/GAC (1:1.5)之光催化分解 4-CP 與 GAC、TiO2/GAC (1:1.5)之 吸 附結果比較...29
圖 5.9 複合半導體之光催化分解與其吸附 4-CP 結果比較 ...29 圖 5.10 複合半導體於 365 波長之光催化分解礦化 4-CP 結果比較 ...30 圖 5.11 複合半導體使用不同光波長光催化分解 4-CP 結果比較 ...30 圖 5.12 複合半導體/活性碳於 435 波長之光催化分解礦化 4-CP 結果比較 ...30 圖 5.13 複合半導體/活性碳於 435 波長不同 pH 值之光催化分解 4-CP 結果比較 ...31 圖 5.14 TiO2/Goethite 之吸附與光催化試驗結果...32
第一章
研究計畫之背景及目標
1.1
研究背景
去除難分解之有機污染物是重要之環境污染消減課題,為了保障生活環境並 符合日益嚴格的環境法令,以減低毒性物質對人體健康及環境生態的危害,發展 一能對有機毒性污染物達完全降解或礦化作用的處理技術是必要的。 傳統廢水處理技術,在生物處理上活性污泥法是常被應用的,但其所需處理 時間較長,同時也面臨微生物受毒性物質的抑制、難分解有機物與污泥需後續處 理等問題(Jardim et al., 1997)。在物化處理上粒狀活性碳吸附或是氣提法程序 中都會面臨後續有害廢棄物處理的問題;在焚化處理有機廢棄物的效率並非穩定 且常會因燃燒不完全而將有毒物質排入大氣環境中。因此一個有效的處理系統應 該是能將污染物質在排放至環境之前完全的降解(Robertson , 1996)。 基 於 上 述 傳 統 方 法 處 理 難 分 解 有 機 污 染 物 的 缺 失 , 半 導 體 光 催 化 (semiconductor photocatalysis)程序是一值得研發的程序,此種程序是屬於高級氧 化程序(advanced oxidation process)之一環,利用紫外光加上半導體觸媒(如 UV/TiO2),其原理為利用高於能隙(band gap)的紫外光激發半導體產生具還 原能力的電子與具氧化能力電洞,以破壞目標污染物及其中間產物,達成對污染 物降解與礦化目的,半導體光催化程序在過去十年間已有多位學者應用在降解水 中污染物(Jardim et al., 1997;Kang et al., 1999;Herrmann , 1999;San et al., 2001; Sokmen et al., 2001)與空氣中污染物(Alberici et al., 1997;Yang et al., 2000;Zornet al., 1999, 2000)。 難分解有機污染物中含氯苯環有機物在水體環境造成嚴重之污染問題,常見 的氯酚類如 2-氯酚(2-CP)、4-氯酚(4-CP)主要為合成殺蟲劑之用,是水體中重要 的污染物種類,其對於哺乳動物和水生生物的毒性被歸類為中等毒性,但具有相 當之生物累積性,口服恕限值為 0.1 ppb (Doong et al., 2001)。水體中氯酚污染主 要來自工業生產之中間產物和工廠加氯於含酚類放流水過程中產生。另外,農藥 分解的副產物也會形成氯酚而殘留於環境中,飲用水加氯消毒亦可能因有機物的 存在之氯化作用(Chlorination)而形成氯酚。目前台灣放流水標準(1987)及自 來水水質標準(1990)中,放流水標準氯酚類為 1.0 ppm,而自來水水質標準則 定氯酚類為 0.001 ppm,五氯酚為 0.005 ppm。隨著公眾對於飲用水質之要求及環
境污染之關注,促使政府、民間致力各種污染處理科技之研究開發來分離除去水 中之污染物。
本研究計畫將以半導體為光催化劑進行異相光催化反應(heterogeneous photocatalytic reactions)研究,其基本原理一般可歸納如後(Litter , 1999):半 導體的特性是由電子能帶結構來決定,在半導體內存在一能隙(band gap)將共 價帶(valence band)與傳導帶(conduction band)隔開,此能隙是在半導體完整 結晶中的隱藏能量;當一光子能量高於或等於能帶能量會被半導體表面所吸附, 進而電子(e-)將由共價帶被激發至傳導帶,同時在傳導帶有一電洞(h+)形成; 但在極短時間(nanoseconds, ns)內,傳導帶電子會與共價帶電洞在半導體表面 或周圍再結合(recombination),或是被受困在表面而與吸附在半導體表面的供 給者(donor, D)或接受者(acceptor, A)進行氧化還原反應。而此說明了半導 體在經過光照後所可能進行的反應,其反應過程可以下式表示: Light Absorption SC + hυ→ e- + h+ Recombination e- + h+ → heat Oxidation D + h+ → D+ Reduction A + e- → A-目前在半導體光催化(semiconductor photocatalysis)研究主題大致可歸納為以 下兩大部份:其一為針對半導體光催化劑之製備及其特性改良(Serpone et al., 1995;Fuji et al., 1998;Kang et al., 1999;Shi et al., 2000;Piscopo et al., 2001);
另一為利用半導體光催化劑對不同污染物進行降解、礦化及其處理效率的改善 (Hoffmann et al., 1995; Jardim et al., 1997;Herrmann , 1999;Hsien et al., 2001)。
在半導體光催化劑的特性改良上,主要的方式是以現有的製備方法中加入不 同的改良方式,獲得不同物化特性之光催化劑成品,以提升光催化劑之反應性與 光活性。在對污染物降解的研究上,利用經由改良製造的光催化劑來對不同污染 物進行降解、礦化以比較反應性能之提昇狀況;並經由改變光催化劑的反應型態 以獲得較佳的實際應用條件。以下對於目前半導體光催化過去文獻的研究重點以 及發展流程整理歸納如圖 1.1,其詳細說明如後所述。
光催化研究 光觸媒製備 污染物之去除 溶膠-凝膠法 液相沈積法 氣相沈積法 粉末燒結法 陽極氧化法 製備方法 改良方式 添加金屬 添加電極 改變燒結溫度 改變粒徑大小 不同金屬結合 探討觸媒之物性 /化性 操作參數 觸媒劑量 光波長 光強度 污染物初始 濃度 溫度 溶氧量 pH值 檢測方法 SEM XRD Thermal analysis FT-IR spectroscopy BET UV-Vis 孔隙度 粒徑 反應途徑 中間產物 最終產物 反應動力式 反應機構 反應器設計 觸媒懸浮式 (批次式) 觸媒固定式 (連續式) 反應性效能提升 添加電子/電 洞再結合抑 制劑 觸媒再生處 理 不同半導體結合 材質之性質測定 能隙大小 量子產率 光活性大小 開發高活性 、高反應性 之光觸媒 反應產物評估 毒性測試 去除效率評 估 圖 1.1 光催化研究領域架構圖 1. 半導體光催化劑之製備及其特性改良 近來對於許多光催化反應降解污染物研究上,皆是以半導體化合物做 為光催化劑,此乃因光催化反應的程序中最重要之因素來自於半導體光催 化劑之基本物性(包括半導體本身能隙大小、量子產率與光活性大小)。 當半導體催化劑受到能量大於其能隙之光照時,會將催化劑的價帶電子提 升至傳導帶,而留下一帶正電之電洞於價帶中,若價帶正電位足以產生氫 氧自由基於催化劑表面(氧化反應)而傳導帶電位負到可以還原分子氧 時,會產生一連串之自由基反應。氫氧自由基是一強氧化物質,會攻擊催 化劑表面附近之污染物而造成其分解,最後完全碳化至 CO2;光激發之電
子、電洞促使光催化反應之發生,但其也有再結合的反應,且其反應速率 通常比標的反應大上幾個級數,光量子產率不高是光催化反應應用之最大 限制。
一般應用在光催化反應上的半導體催化劑有下列數種,如 TiO2(Mill et
al., 1996; Ku et al., 1996; Chen et al., 1999)、ZnS(Yin et al., 2001)、CdS(Yin
et al., 2001)、ZnO(Serpone et al., 1995),其中又以 TiO2的研究最多,原因
除了其具有高活性外,化學穩定性佳,容易取得又無毒性,不會造成二次 污染。TiO2有兩種具有光活性之晶相,分別為 anatase 與 rutile,其中又以 anatase 具有較佳之光活性。
根據 Bacsa et al. (1998)在其研究中指出,以四異丙基鈦酸酯為前驅
物,經溶膠凝膠法(sol-gel)製備與水熱處理後所得之奈米級晶狀 TiO2粉末
(25~50nm),含有 30%之 rutile 與 70% 之 anatase,其光活性比備製單純 anatase 、 rutile 相 之 TiO2 佳 , 且 比 商 業 粉 Degussa(20% rutile, 80% anatase)TiO2光活性良好(Bacsa et al., 1998)。此外提高鍛燒溫度亦可以改
善奈米級 TiO2粉末之結晶性,而提高光活性,但若溫度太高(>600℃),
顆粒成長使比表面積下降,反而使 TiO2 之光活性下降(Zhang et al.,
2000)。另外,在催化劑型態方面,粒徑大小、比表面積、孔隙度與表面 之酸、鹼基等,皆會影響催化劑之光活性,Bacsa et al. (1998)以各種鈦的 烷氧化物為前驅物所備製之奈米級 TiO2中,以表面孔隙口徑分佈在 6-25 Å 之催化劑對於污染物的吸附效率最高。通常當光催化劑粒徑在 5-25 nm 時,會展現出量子效應,使量子產率接近 1,且由於構成單顆粒的單體在 2000 個左右,而使得催化劑的能隙擴大,激發電子所需之光往短波長方 向移動,即所謂的藍位移(Blue Shift)。具有量子效應之光催化劑顯現較好 之光活性,但常常由於表面晶格缺陷與粒徑不一所抵銷(Mills et al., 1997)。 對於半導體光催化劑之一般特性可由以下因子來探討(Li , 1997): (1)半導體的能隙與能帶位置 半導體的特性通常是以共價帶與傳導帶間的能隙來決定,而能隙 大 小 是 決 定 於 能 引 起 半 導 體 激 發 或 電 子 分 離 的 波 長 (Serpone and Perlizzetti , 1989)。在半導體被激發後,其傳導帶的電子與共價帶的電 洞將與吸附在表面的物質進行電子傳遞反應或是電子電洞的再結合。
在電子傳遞反應的效率決定半導體作為光催化劑在氧化還原反應 的能力,半導體光催化效率是半導體的能帶位置與被吸附物質的氧化 還原能力有函數關係。對於一理想的電子傳遞反應而言,電子接受者 的能階應該要比傳導帶為低,相對地電子供給者的能階應該要比共價 帶為高。而在傳導帶能階底部的電子是具有還原能力,在共價帶能階 頂部的電洞是具有氧化能力,此二者能階的差距(能隙)是整個系統 氧化還原的能力(Schiavello , 1993)。 (2)半導體表面的競爭吸附 污 染 物 質 濃 度 對 光 催 化 動 力 學 上 的 影 響 可 以 由 Langmuir–Hinshelwood 方程式來說明(Kang et al., 1999)。在光催化反 應中,除了反應物外還有很多其他分子(溶劑、反應中間產物、反應 產物以及其他非反應之溶劑)會同時吸附在半導體表面。在大多數的 實驗濃度範圍中,來自中間產物與最終產物的競爭性是可以忽略的。 對於污染物降解而言,其主要目的是將初始物質完全轉變成水及二氧 化碳。 (3)電子與電洞的捕獲(scavenging) 為了抑止電子與電洞的再結合以及持續提供共價帶電洞來進行 氧化反應,可利用一犧牲性電子接受者來捕捉傳導帶的電子。氧分子 是一在光催化氧化過程中常使用的電子接受者,因為氧分子具有從大 多數半導體的傳導帶捕捉電子的能力。 (4)反應器設計 一般可區分為懸浮態與固定態兩種設計。在懸浮態半導體可提供 很大的表面積因而能有高光催化效率,但應用於懸浮態的半導體大多 是次微米(submicron)的顆粒,面對處理後的分離是一艱鉅的工作。而 固定態半導體針對懸浮態半導體顆粒回收之缺點來改善。 近年來,已有許多研究致力於解決光催化反應光量子產率不高的問 題,包括在催化劑表面嵌入貴重金屬,如 Pd 或 Pt (Sanchez et al., 1995; Sclafani et al., 1998),因為其具有捉住光激發電子的能力,可以降低電子 與電洞再結合(recombination)比率,但 Sclafani et al.(1998)在其研究中指
出,附著於 anatase 相 TiO2上之 Pt 會捕集電子,並吸引電洞而成為再結
合中心,而在 rutile 相 TiO2上之 Pt 則無這樣的現象發生。在溶液中加入
過渡金屬離子 Mn2+,會誘發類似 Photo-Fenton 的反應,而減少電子與電
洞再結合並加快污染物之降解速率(Chen et al., 1999)。此外,水中溶氧也
有捕集電子的功能,溶氧與光激發電子形成 O2-後與溶液中之 H+反應成
HO2‧,幫助破壞水中污染物(Sclafani et al., 1998)。其他研究如在 TiO2表 面附著染料,照射可見光激發染料之電子後,電子傳送至催化劑之傳導 帶,以此路徑達成破壞污染物之目的,而在反應過程中催化劑之價帶保持 不變,因此無電子與電洞再結合之問題(Cho et al., 2001)。 為了加強光催化反應的效率,改進光催化劑的活性的主要方法是改 變光催化劑之表面特性(Linsebigler et al., 1995)。一般增加光催化劑光 活性的方法有:添加重金屬,如 Pt、Pd 或 Au 等,添加重金屬的目的 是抑制已激發的電子電洞產生再結合,因為當電子經激發後會再經由電 位差而轉移至金屬上,能有效的分離電子及電洞。複合半導光催化劑, 當兩種光催化劑被激發後,電子因為電位差而轉移至傳導帶電位較低的 光催化劑上;而電洞則會轉移至價帶電位較高的另一光催化劑上,其目 的也是有效的分離電子電洞對,降低再結合反應的發生,以增進光催化 劑之活性 ( Linsebigler et al., 1995; Serpone et al., 1995)。根據 Linsebigler
et al.(1995)的研究歸納顯示在改變光催化劑表面特性的方法有下列幾
種:
(1)以金屬半導體修正(Metal semiconductor modification)
目前改善半導體催化效能的方法是阻止半導體內已形成的電子與 電洞的再結合(recombination),可採用添加重金屬如 Pt, Pd, Au 以及 其他半導體,來將半導體表面經光激發所產生之電子快速傳至金屬顆 粒 , 有 效 地 將 電 子 與 電 洞 分 離 , 以 加 強 光 催 化 反 應 的 降 解 效 率 (Linsebigler et al., 1995;Hirano et al., 1997;Wilke et al., 1999)。在 半導體中添加重金屬可以經由改變半導體表面特性而改變光催化的過 程,以及加強特定產物產率、光催化反應的速率,亦可改變反應的產
物。添加重金屬的作用機制是電子經激發後遷移至金屬上並捕捉住, 因而可抑止電子與電洞之再結合發生,而游離的電洞可以在半導體表 面自由擴散至有機物所在將其氧化。
(2)耦合半導體系統(Coupled semiconductor system)
結合兩種半導體的光催化劑系統的作用原理是將其中具有較低能 隙(band gap)半導體激發使其電子與電洞分離,再藉此將電子傳至 另一較高能隙半導體的傳導帶,可使電子與電洞的再結合機率降低以 提 高 半 導 體 之 光 活 性 , 在 對 提 昇 有 機 物 降 解 是 一 個 有 效 的 作 法 (Linsebigler et al., 1995;Serpone et al., 1995;Vinodgopal et al., 1995; Mills et al., 1997;Shi et al., 2000;Doong et al., 2001)。兩種半導體的 結合因各自有不同的共價帶與傳導帶而具有不同的氧化還原的能階, 因而提供一方法來達到更有效率的電子分離、增加電子攜帶者的存在 時間(lifetime)以及加強界面電子傳送到吸附物質的效率(Serpone et
al., 1995)。Serpone et al.(1995)利用 CdS/TiO2, ZnO/TiO2, TiO2/Fe2O3,
以及 ZnO/Fe2O3 等分散結合態的半導體,對 phenol, 2-chlorophenol 與 pentachlorophenol 進行光催化反應,發現 CdS/TiO2能提升對於污染物 的降解率,相對於 TiO2可達兩倍;同時發現 CdS 表面的電子會有方向 性移至未被光激發的 TiO2顆粒上,其電子移動的示意圖如圖 1.2。 ⊕ Θ Θ CB VB VB CB CdS TiO2 hν D+ D A-A 圖 1.2 CdS/TiO2半導體電子與電洞傳遞圖
在結合兩種半導體的光催化系統可分為兩型:一為分散懸浮態 (Linsebigler et al., 1995;Serpone et al., 1995;Doong et al., 2001)、另 一為結合附著態(Kang et al., 1999;Shi et al., 2000)。根據 Shi et al. (2000) 利用同相沈降法 (homogeneous precipitation method)來結 合兩種不同能帶(band gap)的半導體製備結合半導體(coupled semiconductors),其方法是以超細 TiO2為種體(seeds)再將 SnO2附 著在其上,形成 SnO2- TiO2之結合顆粒。研究發現在 SnO2添加量為 TiO2之 18.4%時對 Active red X-3B 具有最佳之光催化降解速率,相對
於單獨存在之光催化劑(SnO2、TiO2)其光催化降解速率均不如結合的 SnO2- TiO2催化劑的效果。 而在分散懸浮態半導體光催化的研究中,Doong et al. (2001)針對 2-chlorophenol (2-CP)的光催化降解採用 CdS 加入 TiO2懸浮液中進行 光催化降解,發現相對於單一半導體系統,將 CdS 與 TiO2之結合半導 體系統能提高對 2-CP 的去除速率。
另外在 Kang et al.(1999)的研究對於 4-chlorophenol (4-CP)在不同型
態(TiO2與 TiO2/CdS 之粉末狀、薄膜電極)的光催化系統中之光催化
降解過程,發現 4-CP 在半導體催化劑表面的吸附作用是異相光催化動 力學中一項重要的影響因子。該研究為了加強對 4-CP 的光降解,將較 低傳導帶能階之 CdS(作為 TiO2之 photosensitizer)附著在 TiO2 表面 形成結合態半導體(coupled semiconductors)來進行光催化反應,發 現結合態半導體的光降解效率遠大於單一半導體的光降解效率。 (3)表面敏化 (Surface sensitization) 利用染料分子(作為光敏劑)可將光催化劑之表面進行光敏化作用 (photosensitization),進而增加半導體表面電子激發的效率。光敏化 過程經由光敏劑(photosensitizer)的激發將電子傳至半導體表面能擴 大光催化劑被激發的波長範圍。如果染料分子被激發的氧化能階較半 導體傳導帶能階為高時,染料分子可將激發電子傳至半導體之傳導帶。 (4)過渡金屬之塗佈(Transition metal doping)
過渡金屬塗佈的好處是可以改善激發過程中電子的捕捉,進而抑止 電子與電洞的再結合發生。根據 Fujihira et al.(1982) 與 Butler et
al.(1990)的研究只有特定的過渡金屬(Fe+3、Cu+2 )能實質上抑制電子與 電洞的再結合。其他過渡金屬(如 Cr+3)的塗佈將產生位置而增加電 子電洞的再結合(Herrmann et al.,1984)。一般相信,過渡金屬所產生電 子接受及供給中心會直接發生再結合。相對於其他改善光催化劑表面 性質的方法中,針對以過渡金屬加入 TiO2系統處理不同有機物種的光 反應研究工作是很少的(Linsebigler et al., 1995)。 2. 光催化反應對污染物降解效率之研究 自從光催化反應被應用在污染物降解、礦化之後,被選來處理之污染 物大多是屬於難分解的有機毒性污染物,如多環碳氫化合物(Polycyclic aromatic hydrocarbons, PAH)、酚(Phenol)、氯酚(Chlorphenol)、多 氯聯苯(Ploychlorinated biphenyls, PCBs)、各種染料等,其中農藥是主 要的來源。 由圖一之研究領域架構可將污染物去除之研究概分為反應器設計、污 染物反應途徑與生成產物之探討以及實驗操作上參數之最佳條件。根據上 述之分類中的研究主要目的是提升整個光催化反應效率的提高與對於反 應產物破壞效率之評估。其中部份相關研究文獻如下頁表所示。 以下將針對圖 1.1 中污染物去除部分所提的研究領域分項說明。 (1)反應器設計 一般在光催化反應的設計上最主要的考量是確保光線可以均勻的分 佈在光催化劑之表面。因此在設計反應槽時有兩個重要的因子需要考量: 反應槽內的光線分佈與光催化劑所提供之表面積。 根據反應器的設計可區分為批次式與連續式反應設計。在批次式反 應槽主要是以懸浮態光催化劑為主,雖然懸浮態反應槽有較佳的量子產率 與容積效率,但面對光催化劑與反應環境的分離將是一困難的工作。而固 定態光催化劑反應槽則沒有分離上的問題,但卻面對反應效率較低的瓶 頸,其原因為固定態的設計會限制光催化劑與溶劑接觸的表面積,所以在 設計上必須能充分提高固定態光催化劑與溶劑、光線接觸的機會。
污染物 光催化劑 及其型態 操作條件 產物/中間產物 重要結果 文獻 Malathion, Phorate, Diazion, Methamidophos, EPN Direct photolysis UV-TiO2 UV-H2O2 UV-TiO2–H2O2 光波長: 253-578nm 光源: 100W、450W 沒有探討 添 加 H2O2 於 UV -TiO2 系 統 可 提 高 3-10 倍之量子產率。 發現有機磷農藥的降 解速率與其個別特性 有關。 Doong et al. (1997) parathion UV/H2O2 UV/Fe0/H2O2 UV/Fe+2/H2O2 UV/TiO2/H2O2 光波長: 253-578nm 光源 100W Diethylphosphoric acid, Diethylmonothiophosphoric acid,Oxalate, p-nitrophenol, O,O-ethyl p-nitrophenyl 添 加 (Fe0, Fe+2) 於 UV/H2O2 系 統 可 將 parathion 之降解速率 常數提升三倍。 Doong et al. (1998) Cyanotoxin cylindrospermopsin UV-TiO2 pH=4,7,9 temp=19-34℃ 沒有探討 發 現 污 染 物 不 會 在 TiO2表面吸附。 在鹼性環境下降解最 快。 Senogeles et al. (2001) Chlorinated paraffins(CPs) UV UV/H2O2 光波長 254,302,313, 366,405/408nm 長鏈含氯烷類可降解成短鏈 含氯 paraffins 產物之生物毒性測試 Koh et al. (2001) 2-methylthiophene UV/TiO2 Solvent:H2O, CH2Cl2/H2O 光源:sunlight, 可見光,UV 中間產物: thiophene-2-aldehyde, 2-methyl-5-(2-thienylmethyl) thiophene, 2-hydroxymethylthiophene, 5-methyl-(5H)thiophen-2-one 推出四個可能之污染 物降解機構。 Sokmen et al. (2001) Phenol, Guaiacol, 2-chlorophonel, catechol UV TiO2 UV/TiO2 pH=3, temp=25℃ TiO2=1.5g/dm3 利用 HPLC,HPLC/MS, GC/MS 鑑定各污染物之中 間產物。 求得各污染物之光催 化降解常數與平衡常 數。 Peiró et al. (2001) Benzamide UV/TiO2(P25) UV/TiO2(Sol-Gel ) λ=300-800 nm pH= 5-6, TiO2=1 g/L 4-hydroxybenzamide hydroquinone quinone 比較商用與 sol-gel 製
備之 TiO2 降解能力 Piscopo et al.
(2)操作參數的影響 一般光催化實驗常會採用之操作參數有:催化劑劑量、光波長與強 度、污染物初始濃度、溶氧含量以及 pH 值。調整各類操作參數的目的是 針對不同光催化劑特性而選取其適合的範圍來進行實驗,以求得操作參數 的最佳化。 (3)反應動力與反應途徑 在研究光催化反應過程,Langmuir–Hinshelwood 方程式是最常被使 用在評估反應動力平衡。
1.2
研究目標
針對前述研究提到的各種提升半導體光催化效率方法,本研究之三年計畫主 要方向將著重於結合半導體系統(Coupled semiconductor system)來探討,並依下 列三個年度目標來進行研究: 本研究計畫第一年目標,以 TiO2與 ZnO 之粉末為催化劑,將其混合、燒結 分別探討及比較對於鄰氯酚之光催化降解效率,並針對以燒結所結合之兩種半導 體的特性做一研究,來探討兩種不同能隙之半導體在光催化反應過程所提升的效 能,提供做為未來發展污染防治技術之基礎。 本研究計畫第二年目標,以複合奈米級半導材料二氧化鈦(TiO2)及硒化鎘 (CdSe)為光催化劑,其主要目的是利用自行製備之半導體具有較細小之粒徑以加 強光催化效能,並將之應用於 4-氯酚(4-Chlorophenol)之光催化降解反應與進行複 合光催化劑光活性的探討。 本研究計畫第三年目標,將複合半導體材料負載於具吸附能力之吸附劑形成 一混合光催化劑(hybrid photocatalysis),來進行低濃度環境污染物之光降解反 應,並探討其吸附動力平衡模式與 4-氯酚(4-Chlorophenol)降解效率之評估,進而 嘗試瞭解污染物在混合光催化劑表面的吸附與光降解反應的機構,以應用未來各 種可能之污染物型態。第二章
第一、二年研究成果
2.1
第一年研究成果
2.1.1 催化劑物化性質測定
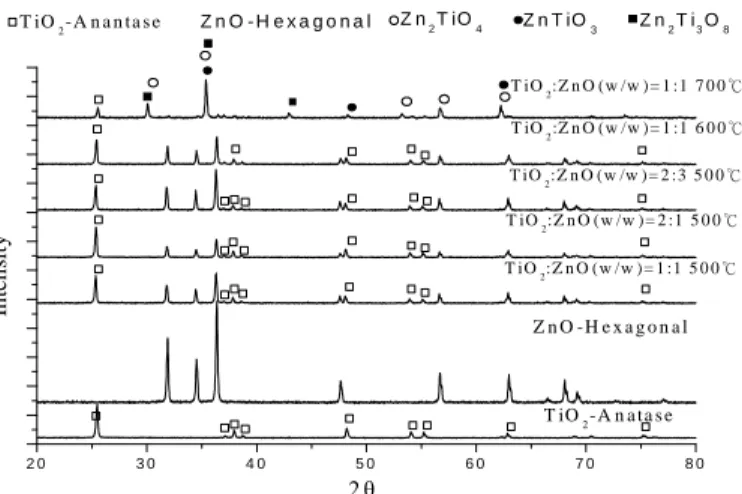
圖 2.1 為催化劑之雷射光粉末繞射圖。TiO2原始之晶相為 anatase 而 ZnO 為 ZnO-hexagonal,以 TiO2:ZnO (w/w) = 1:1、2:1 及 2:3 混合、500 ℃燒結後 之催化劑晶相無變化,其繞射圖是由 anatase 及 ZnO-hexagonal 晶相之繞射峰所 組成,而對於 TiO2:ZnO (w/w) = 1:1 混合、600 ℃燒結後之催化劑晶相仍無變 化,但當燒結溫度提高至 700 ℃時,繞射圖之繞射峰產生偏移的情形,代表燒 結產生其他晶相,經由繞射峰之比對,產生之晶相為 ZnTiO3、Zn2TiO4及 Zn2Ti3O8。 由於 700 ℃燒結催化劑產生其他晶相,而這些晶相也有其特定之能隙及價 帶與傳導帶能階,可能會使電子傳遞機制改變或消失,而為了驗證 TiO2與 ZnO 之電子間傳遞現象,所以後續光催化反應實驗採用 500 ℃及 600 ℃燒結之催化 劑。 圖 2.2 為 TiO2:ZnO (w/w) = 1:1 混合,500 ℃燒結後之掃瞄式電子顯微照 片。由圖可以發現,除了 ZnO 之稜角變得比較圓滑外,也可以看見幾團之催化 劑黏結在一起,與未燒結前之催化劑在形態上有明顯之變化。 2.1.2 pH 值效應 圖 2.3 為 TiO2與 ZnO 在不同 pH 時,光催化降解 4-氯酚之擬一階反應速率 常數。TiO2在 pH = 4、7、11 時之速率常數分別為 0.0055 min -1、0.0018 min- 1及 0.0019 min-1,在中、鹼性環境之速率常數小於在酸性環境之速率常數,酸 性環境之速率常數約為中、鹼性環境之速率常數的三倍。但對於 ZnO 而言,則 顯現與 TiO2相反之趨勢。由於 ZnO 在 pH = 4 有酸腐蝕之現象,故反應速率常數 相當低,為 0.0005 min-1,而在中性環境時,速率常數為 0.0096 min-1,隨著 pH 上升至 11,此時速率常數增加到 0.0123 min-1。 2 0 3 0 4 0 5 0 6 0 7 0 8 0 Z n2T i3O8 T iO2-A n a n ta s e Z n O - H e x a g o n a l Z n2T iO4 Z n T iO3 T iO2:Z n O ( w /w ) = 1 :1 7 0 0 ℃ T iO2:Z n O ( w /w ) = 1 :1 6 0 0 ℃ T iO2:Z n O ( w /w ) = 2 :3 5 0 0 ℃ T iO2:Z n O ( w /w ) = 2 :1 5 0 0 ℃ T iO2:Z n O ( w /w ) = 1 :1 5 0 0 ℃ Z n O -H e x a g o n a l T iO2-A n a ta s e In te n si ty 2θ 圖 2.1 燒結催化劑之雷射光粉末繞射
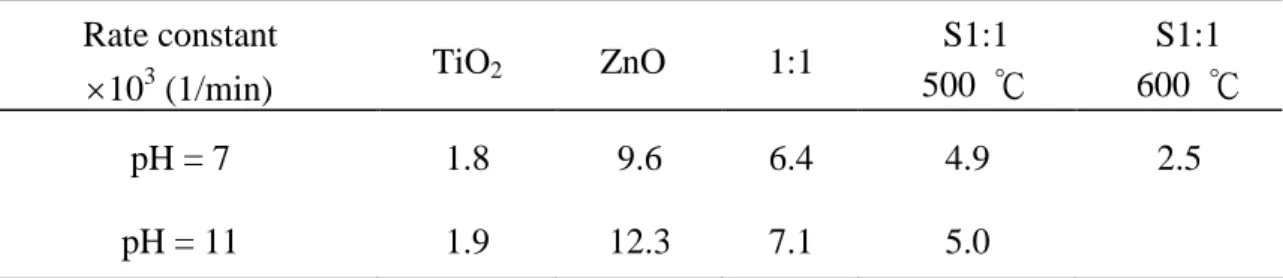
圖 2.2 混合燒結催化劑之顯微照片 0 2 4 6 8 10 12 14 0 2 4 6 8 10 12 14 pH k × 1 0 3 (1 /m in ) TiO2 ZnO 圖 2.3 TiO2與 ZnO 在不同 pH 時光催化降解 4-氯酚之擬一階反應速率常數 2.1.3 結合半導體催化劑 如表 2.1,在 pH=7 時,TiO2之速率常數為 0.0018 min -1,ZnO 之速率常數 為 0.0096 min-1,而混合催化劑 TiO 2:ZnO (w/w) = 1:1 之速率常數為 0.0064 min -1,500 ℃燒結催化劑 TiO 2:ZnO (w/w) = 1:1 之速率常數為 0.0049 min -1, 小於混合催化劑,推測之原因為:比表面積之收縮造成用於傳遞電子、電洞至污 染物之面積減少,因此,燒結催化劑之光催化活性降低。而提高燒結溫度造成催 化劑比表面積之收縮更嚴重,故 600 ℃燒結之催化劑光催化活性又小於 500 ℃ 燒結催化劑。對於 TiO2、ZnO、TiO2與 ZnO 混合及 TiO2與 ZnO 混合燒結催化 劑在 pH=11 下光催化降解 4-氯酚效率而言,也是呈現 ZnO 降解 4-氯酚之效率
表 2.1 TiO2、ZnO、TiO2與 ZnO 混合及 TiO2與 ZnO 混合燒結催化劑在 pH=7 及 pH=11 下,光催化降解 4-氯酚之擬一階速率常數
Rate constant 103
(1/min) TiO2 ZnO 1:1
S1:1 500 ℃ S1:1 600 ℃ pH = 7 1.8 9.6 6.4 4.9 2.5 pH = 11 1.9 12.3 7.1 5.0
Serpone et al.(1995)在 pH = 6.7,波長>355 nm 之光源,曝空氣下,以 ZnO、 TiO2及 ZnO/TiO2之系統光催化降解酚,ZnO 之擬一階速率常數為(614)10-3 min-1,TiO2之速率常數為(191)10-3min
-1,而 ZnO/TiO
2之系統為(712) 10-3
min-1,結合催化劑提升光催化速率。又以波長大於 320 nm 之光源在 pH = 12,曝空氣之狀況下,使用 CdS、TiO2及 CdS/TiO2光催化降解酚、2-氯酚及五 氯酚。對於酚之降解而言,CdS 之速率為 1.090.04 μM/min,而 TiO2與 CdS/TiO2 之系統之反應速率分別為 2.050.06 μM/min 和 2.0 0.2 μM/min;而對於 2-氯酚,CdS 之速率為 0.30.1 μM/min,TiO2與 CdS/TiO2之系統各為 5.10.1 μ M/min 和 4.10.5μM/min;CdS 對五氯酚之降解速率為 0.76 0.11 μM/min, 而對於 TiO2 和 CdS/TiO2之系統分別為 3.61 0.06 μM/min 及 4.9 0.2 μ
M/min,由上數知數據,可知除了以 CdS/ TiO2降解五氯酚顯現加強之光活性外, 對於降解酚及 2-氯酚而言,CdS/ TiO2並未明顯地展現出預期提升之反應速率。 對於本研究而言,標的污染物為 4-氯酚,並未得到預期提升之降解效率,經由上 述數據推論,可能為不同標的污染物之關係,而造成結果不一致。另外,在結合 催化劑之系統中,若其一催化劑之光活性低於另一個,可能會抵銷顆粒間電子傳 送造成電子、電洞分離之益處,而使結合催化劑系統整體反應速率降低。
2.2
第二年研究成果
2.2.1 觸媒之性質鑑定表 2.2 為 TiO2及 CdSe/TiO2觸媒之比表面積分析結果。複合態 CdSe/TiO2之
比表面積較 TiO2為小,可以證明 CdSe 之比表面積較 TiO2小。而觸媒之比表面
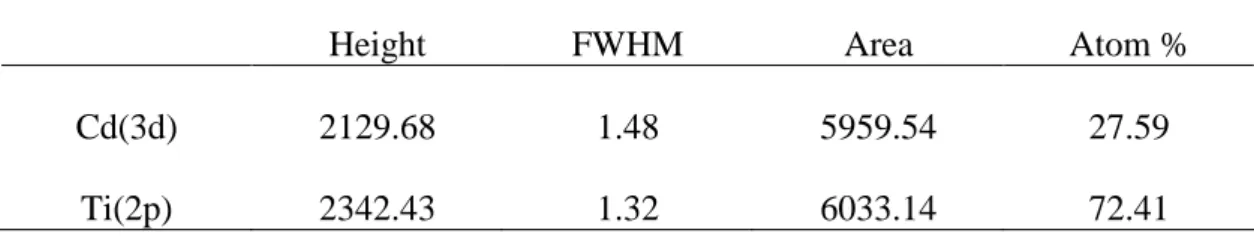
積大小會造成其光活性之差異。 經 UV-Vis 分析,可以得知,二氧化鈦在波長小於 400 nm 後,其吸收度驟 增,即紫外光能量被二氧化鈦分子大量吸收,此時電子獲得足夠能量由價帶激發 到傳導帶。而硒化鎘吸收度則在波長小於 620 nm 後驟增,表示硒化鎘的激發光 波長約在 620 nm 左右。表 2.3 為 TiO2 及 CdSe/TiO2 觸媒之臨界波長及能隙大小。 將觸媒進行 XPS(或稱 ESCA)分析,可以得知觸媒表面之化學環境及 CdSe 沈積於 TiO2 表面之定量分析。圖 2.4 為複合觸媒 CdSe/TiO2 之 XPS survey spectrum,由圖中可以確定 Ti(2p)的 binding energy 為 460 eV、Cd(3d) peak 位於
406 eV、O(1s) peak 位於 532 eV。表 2.4 為複合態 CdSe/TiO2觸媒進行 Cd 及 Ti 的元素定量分析結果,結果顯示以化學沈積法製備之 CdSe/TiO2中,其 Cd:Ti 比約為 1:2.62。 表 2.2 單一及複合型態觸媒之比表面積 觸媒 比表面積(m2 /g) TiO2 8.848 CdSe/TiO2 7.047 表 2.3 觸媒之臨界波長及能隙 樣品 臨界波長λ (nm) 能隙 E (eV) TiO2 396 3.13 CdSe 620 2.00 0 1000 2000 3000 4000 5000 6000 200 300 400 500 600
Binding Energy (eV)
In te n si ty (c p s)
圖 2.4 CdSe/TiO2之 XPS survey spectra O1s Ti2
表 2.4 Cd、Ti 元素之 XPS 定量分析結果
Height FWHM Area Atom %
Cd(3d) 2129.68 1.48 5959.54 27.59 Ti(2p) 2342.43 1.32 6033.14 72.41 2.2.2 pH 值對 4-氯酚光催化降解之影響 圖 2.5 為在波長 254 nm 下,分別添加 TiO2及 CdSe/TiO2兩種觸媒時,在 pH=7 及 11 之 4-CP 之降解圖。由圖中可發現,不管添加何種觸媒,在 pH=11 的環境 下,4-CP 降解效率皆比 pH=7 時好。波長 365 nm 下的結果亦同。另外,4-CP 的 降解關係遵循擬一階速率式,故可以擬一階速率常數值(k)來判斷 4-CP 之降解速 度,表 2.5 列出波長分別為 254 及 365 nm 時,兩種觸媒在不同 pH 值下,4-CP 降解之擬一階速率常數值。由表中發現,在 pH=11 之 k 值皆大於 pH=7 之 k 值。 0 0.2 0.4 0.6 0.8 1 1.2 0 50 100 150 200 250 300 Time(min) C /C0 TiO2(R-dH)-pH7 TiO2(R-dH)-pH11 CdSe/TiO2-pH7 CdSe/TiO2-pH11 圖 2.5 波長 254 nm 、pH=7 及 11 下之 TiO2及 CdSe/TiO2觸媒 4-CP 降解關係圖 表 2.5 波長 254 及 365 nm,觸媒在 pH=7 及 11 下之擬一階速率常數 k(min-1) 254 nm 365 nm pH 7 11 7 11 TiO2 0.0068 0.0116 0.0018 0.0019 CdSe/TiO2 0.0135 0.0433 0.00196 0.002
2.2.3 複合觸媒對 4-氯酚降解之影響 (1) 波長 254 nm
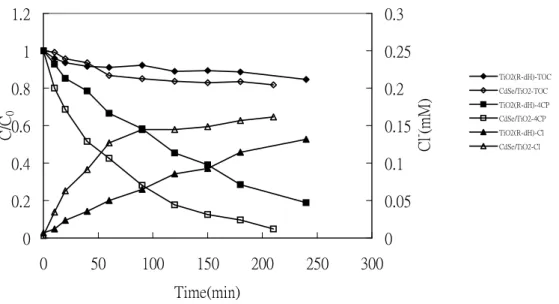
圖 2.6 為波長 254 nm、pH=7 的情形下,添加量為 1.2 g/L 之 TiO2及複合態
CdSe/TiO2觸媒光催化實驗之比較圖。由圖中得知,不論 4-CP 濃度、TOC、Cl
離子濃度趨勢,複合 CdSe/TiO2觸媒的效果都較單獨 TiO2(R-dH)為佳。TiO2於 180 min 內可去除約 80%之 4-CP;而複合態之觸媒於 180 min 內可去除 90%之 4-CP。由表 5 所示,TiO2(R-dH)觸媒之擬一階速率常數 k 值為 0.0068 min-1, CdSe/TiO2觸媒之 k 值為 0.0135 min-1,複合觸媒之效果約為單一觸媒之 2 倍。若 以 TOC 的結果來看,單獨 TiO2及複合態觸媒在 180 min 內之 TOC 的去除率分 別為 12%及 16.5%,再度顯示複合態觸媒對光催化效率的確有提升的效果。鹼性 環境 pH=11 的情形下,單一 TiO2與複合觸媒的擬一階速率常數 k 值則分別為 0.0116 及 0.0433 min-1,複合觸媒之效果約為單一觸媒之 3.7 倍,提升的效果較 中性環境更為顯著。 0 0.2 0.4 0.6 0.8 1 1.2 0 50 100 150 200 250 300 Time(min) C /C0 0 0.05 0.1 0.15 0.2 0.25 0.3 C l - (m M ) TiO2(R-dH)-TOC CdSe/TiO2-TOC TiO2(R-dH)-4CP CdSe/TiO2-4CP TiO2(R-dH)-Cl CdSe/TiO2-Cl 圖 2.6 波長 254 nm、pH 7,TiO2及 CdSe/TiO2觸媒之光催化實驗比較圖 (2) 波長 365 nm 圖 2.7 為波長 365 nm、pH=7 的情形下,添加量為 1.2 g/L 之 TiO2(R-dH) 及複合態 CdSe/TiO2(R-dH)觸媒進行光催化實驗之比較圖。由圖中的 4-CP、TOC 及 Cl 離子趨勢發現,複合 CdSe/TiO2的效果也較單獨 TiO2的效果為佳。以 4-CP 濃度來說,單獨 TiO2觸媒於 180 min 內可去除約 31%之 4-CP;而以 CdSe 結合 TiO2(R-dH)之觸媒於 180 min 內可去除 33%之 4-CP。由表 4 所示,TiO2(R-dH) 觸媒之擬一階速率常數 k 值為 0.0018 min-1,CdSe/TiO
2觸媒之 k 值為 0.00196
min-1,複合觸媒之效果約可提高 9%。若以 TOC 的結果來看,單獨 TiO2及複合
態 CdSe/TiO2在 180 min 內之 TOC 的去除率相差不大,因為在 365 nm 的波長及 中性環境下,TOC 的去除效果並不顯著。鹼性環境(pH=11)的情形下之光催化結
果與中性環境差不多,在 180 min 內,複合觸媒可去除 34%之 4-CP,單獨 TiO2 可去除 30%之 4-CP,兩者對 TOC 的去除效果較中性環境好。整體而言,複合觸 媒對 4-CP 的光催化效果仍較單一觸媒為佳。 0.5 0.6 0.7 0.8 0.9 1 1.1 0 50 100 150 200 250 300 Time(min) C /C0 0 0.05 0.1 0.15 0.2 0.25 0.3 C l - (m M ) TiO2(R-dH)-TOC CdSe/TiO2-TOC TiO2(R-dH)-4CP CdSe/TiO2-4CP TiO2(R-dH)-Cl CdSe/TiO2-Cl 圖 2.7 波長 365 nm、pH=7,TiO2及 CdSe/TiO2觸媒之光催化實驗比較圖
第三章
第三年之計畫研究背景說明
3.1
研究背景
本研究計畫第三年目標是將複合半導體材料負載於具吸附能力之吸附劑形 成一混合光催化劑(hybrid photocatalysis),來進行低濃度環境污染物之光降解反 應,並探討其吸附動力平衡模式與 4-氯酚(4-Chlorophenol)降解效率之評估,進而 嘗試瞭解污染物在混合光催化劑表面的吸附與光降解反應的機構,以應用未來各 種可能之污染物型態。 在前兩年的研究結果中,結合 TiO2及 ZnO 光催化降解 4-氯酚來探討顆粒間電子傳送程序(Interparticle Electron Transfer , IPET)對於光催化效率之影響,實
驗之結果顯示結合 TiO2及 ZnO 催化劑未能有效提升光催化效率。而使用複合半
導體 CdSe/TiO2在光波長 365 nm 的照射下,效果雖然有提升但仍不顯著,由半
導體能隙及能階圖發現,CdSe 雖然價帶與 TiO2有很大的電位差,但就傳導帶而
言,CdSe 的傳導帶電位只比 TiO2高一些,而這可能造成 CdSe 上的電子傳遞至
TiO2 的效應不強,導致被激發的電子電洞分離情形,不如我們所預期,也造成
複合態 CdSe/TiO2光催化效果的限制。而且利用 CdSe 作為光觸媒材料恐有 Cd
溶出形成環境二次污染的問題。
基於上述前兩年實驗結果考量,於本研究計畫第三年中,複合半導體的材料
則採用 WO3與 MoO3與 TiO2進行結合並負載於吸附劑擔體上。目前也已有許多
應用 WO3/TiO2與 MoO3/TiO2於光催化分解水中或氣相有機物之研究,添加 WO3 與 MoO3可提升 TiO2的光催化能力(Papp et al., 1994; Dube et al., 1996; Martin et
al., 1997; Song et al., 2001; Yang et al., 2004),另外由於 WO3與 MoO3的能隙約
為 2.6-2.8 eV,因此可利用波長大於 400 nm 作為其光催化之激發光源,Li et
al.(2001)使用可見光(光波長 400-800 nm)進行 WOx-TiO2觸媒分解水中甲基藍
進行研究。
而在光觸媒結合吸附劑的研究中包括有利用沸石或分子篩(Cheng et al., 2001; Reddy et al., 2003; Fukahori et al., 2003)、活性碳(Matos et al., 2001; Nazir
et al., 2003; Colon et al., 2004; El-Sheikh et al., 2004),其中以 TiO2結合活性碳的
研究較為眾多,主要是因為活性碳本身就是一個吸附能力強的吸附劑,尤其是對 於水中或氣相中有機物吸附效果,可以克服光觸媒本身受限於其與污染物質的碰
撞機率,使得對於低濃度環境下會使光催化劑的反應速率降低的缺點。至目前文 獻的研究結果中發現,光觸媒結合活性碳進行光催化分解有機物反應時可減少溶 液中的中間產物的量,並縮短有機物礦化時間。但
3.2
研究目的
基於上述之研究背景說明,第三年的研究使用 WO3與 MoO3與 TiO2結合作 為本研究之複合的半導體光觸媒,並將其覆載於粒狀活性碳上,至目前為止光觸 媒結合活性碳的研究中除 Lu et al.(1999)是使用粒狀活性碳外,大多仍然主要以TiO2與粉末狀活性碳(PAC)進行研究,使用 PAC 則還是有後續固液分離與觸
媒/活性碳回收的問題產生。
另外,目前亦未有利用可見光激發複合半導體光觸媒結合活性碳之研究,因 此本計畫第三年的研究可作為此方向研究之後續研究可行性參考。
第四章
第三年計畫研究方法
4.1
光觸媒/活性碳製備方法
本研究以採用 Sol-Gel 方法製備 TiO2、WO3/TiO2與 MoO3/TiO2之溶液並加 入適量之 GAC,Sol-Gel 溶液配製參考 Arabatzis et al.(2002)之方法說明如下:
TiO2的 Sol-Gel 溶液配製方法:於充滿惰性氣體(氮氣或氬氣)且相對濕度
小於 10%的手套箱內進行配製,將 4.3 克的 titanium butoxide(相當於 1 克的 TiO2) 慢慢滴入 2.5 M 的 Acetylacetone Acetylacton 溶液(溶於 n-butanol)中,使最後 體積為 100 mL,此混合物於室溫下攪拌 15 分鐘,準備一含有 0.19 克之 p-toluene sulfonic acid 之水溶液 1 mL,加入前述之溶液中於室溫下攪拌 30 分鐘。
WO3/TiO2與 MoO3/TiO2的 Sol-Gel 配製方法:本研究
將 2 克的 GAC(Merk,使用粒徑為 0.5-1 mm)加入上述配製好之 Sol-Gel 溶液中攪拌均勻後(此為 TiO2:GAC=1:2 比例)入 150oC 烘箱使其乾燥後,以 450oC 溫度鍛燒 2 小時,為避免活性碳於高溫時與氧氣接觸而燃燒消失,觸媒/ 活性碳樣品置入加蓋的坩鍋內進行鍛燒,以隔絕外界的氧氣。 待光觸媒/活性碳樣品鍛燒完成冷卻至室溫後,以去離子水清洗以去除附著 不佳之光觸媒顆粒,清洗後置入 110o C 烘箱乾燥以待使用。
4.2
光觸媒/活性碳之背景吸附實驗
配製 2×10-4 M 之 4-CP 溶液,活性碳或光觸媒/活性碳之添加量為 0.5 g/L, 攪拌器的轉速定為 400rpm,進行吸附實驗時,於容器外包覆鋁箔紙防止光線照 射光觸媒之影響,於 0.5、1、1.5、2、3、4、5、6 小時進行採樣分析,樣品以 HPLC 進行分析。4.3
光觸媒/活性碳之光催化實驗
可分為背景實驗與操作參數兩部分,實驗條件如實驗架構圖所示。背景實驗 中之吸附實驗與操作參數中之催化劑量、pH、溶氧及濃度效應實驗分別以 TiO2 和 ZnO 為催化劑,其加量以催化劑加量實驗中所得之最佳加量為準。 主要器材為光反應槽,分內部燈光套管及外部反應容器兩部分。 內部燈光套為石英夾層(玻璃會吸收紫外光,降低光能量),其內徑約 57 mm 外徑 36 mm,光反應槽中軸向位置放入內部登套管,內部燈光套中放置光源,內 部燈光套管有一循環水夾層,主要光源波長使用 365 及 435 nm。使用波長 365 nm時測量反應器外壁之光強度約為 0.25 mW/cm2,而使用波長 435 波長時外壁之光 強度約為 148 LUX。 外部反應容器為玻璃夾層,其內徑約 10 ㎝,外徑 12 ㎝,高 23 ㎝(Iwaki, 訂製),其為放置待處理之水樣。光反應槽之內外夾層均連接恆溫循環水槽,外 部反應容器上方設活動孔,使 pH 控制電極、酸鹼加藥管線及取樣管線通入光反 應槽中,其他週邊設備見下圖(圖 4.1): 8 4 5 9 1 2 3 7 10 6 1.玻璃光反應槽 6.溫度控制系統 2.石英燈套管 7.攪拌器 3.紫外線光 8.電源供應 4.pH 電極 9.採樣口 5.pH 控制系統 10.反應箱 圖 4.1 光催化反應設備圖 光催化反應實驗說明如下: (1)加入適量之 4-氯酚儲存溶液於反應槽中,稀釋至 3 L,加入光觸媒/活 性碳(使用量為 0.5 g/L)並以鹼控制器控制反應 pH,打開循環水控 制器,開啟攪拌器攪拌約 30 min,使催化劑表面氫氧基均勻。 (2)打開光源開始光催化反應。 (3)依時序取出 20 mL 樣品,經過濾後作 HPLC 分析,此為光反應後濃度。
(4)部分水樣上 IC 測其氯離子濃度。
(5)部分水樣以總有機碳(TOC)分析儀分析溶液中殘留之有機碳濃度。
4.4
藥品與儀器設備
4.4.1 實驗用藥
活性碳:Merk, Charcoal activated granular(about 2.5 mm)
Titanium(IV) n-butoxide:Fluca, 99%。
Tungsten(VI) ethoxide:Alfa Aesar, 5% w/v in ethanol, 99.8%。
Molybdenum(V) isopropoxide:Alfa Aesar, 5% w/v in isopropanol, 99.6%。 1-Butanol, anhydrous:Sigma Aldrich, 99.8%。
Acetylacetone Acetylacton:Fluca, 98%。
Toluenesulfonic acid monohydrate:Riedel-deHaen, 99%。
HClO4: Fisher Scientific, 70%。 NaOH:NACALAI TESQUE, >96%。
4.4.2 儀器設備
HPLC 分析儀器:Viscotek model 500 variable UV/Vis detector。 TOC 分析儀器:OI Analytical 1010。
IC 分析儀器:Metrohm 790 Personal IC。 手套箱:Plas-Labs LP Compact Glove Box。
燈管:365 nm,Philips PL-L 36 W。
435 nm,訂製,50 W。
光強度測定:UVP Multi-Sense Ms-100 Optical Radiometer。
比表面積測定儀:Micrometrtics Instrument Corpration ASAP 2000。
台大貴儀 SEM:Hitachi S-800 Field Emission Scanning Electron Microscope
Hitachi S-2400 Scanning Electron Microscope(附 Kevex Delta class 80000 EDS)
第五章
第三年計畫研究成果
5.1
光觸媒/活性碳之比表面積、SEM、EDS 分析
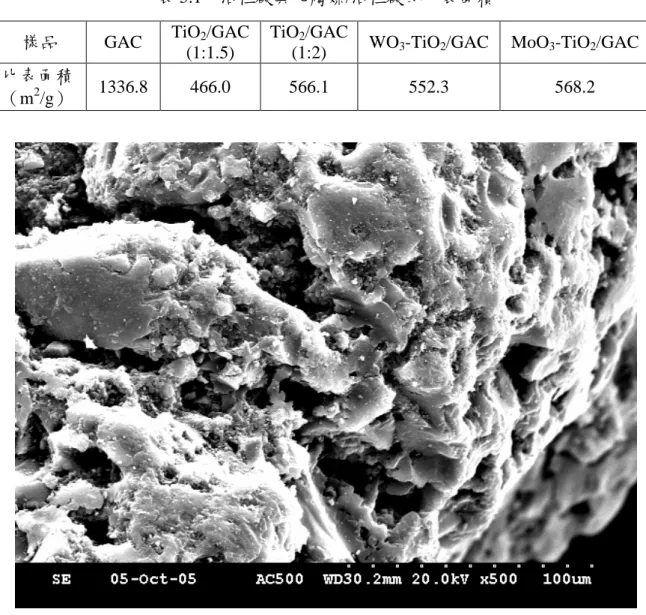
表 5.1 為活性碳與光觸媒/活性碳的 BET 比表面積結果,未負載光觸媒之活 性碳比表面積高達 1336.8 m2 /g,在負載光觸媒後之比表面積則明顯減少至原來 的 35 至 43%,其中光觸媒負載量最多的 TiO2/GAC(1:1.5)的如預期中比表面積最 小,在 TiO2與 GAC 相同比例為 1:2 的情況下,各添加 W/Ti 與 Mo/Ti 之比皆為 5%的複合半導體光觸媒/活性碳,其比表面積幾無變化。圖 5.1 至 5.4 為 GAC 及 TiO2/GAC 的 SEM 觀察照片,在未負載觸媒的活性 碳上可以明顯的看出其除了原本多孔性的結構外,其最外層的表面仍然光滑,在
負載光觸媒後,其表面已被光觸媒覆蓋,尤其是在負載量最多的 TiO2/GAC (1:1.5)
表 5.1 活性碳與光觸媒/活性碳的比表面積
樣品 GAC TiO2/GAC
(1:1.5)
TiO2/GAC
(1:2) WO3-TiO2/GAC MoO3-TiO2/GAC 比表面積
(m2
/g) 1336.8 466.0 566.1 552.3 568.2
圖 5.2 TiO2/GAC (1:2)之表面 SEM 觀察照片
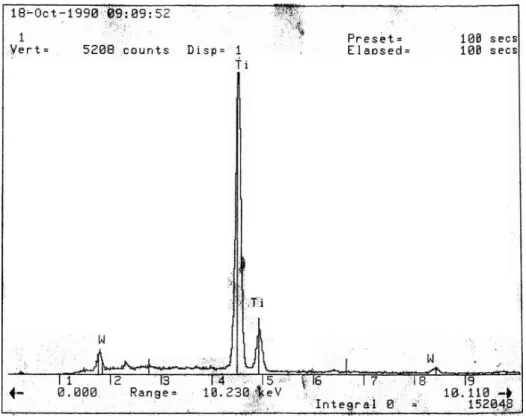
圖 5.4 WO3-TiO2/GAC (1:2)與 MoO3-TiO2/GAC (1:2)之表面 SEM 觀察照片 樣品(圖 5.3)原本的多孔結構與平滑表面幾被覆蓋,且外觀更為粗糙。 圖 5.4 則是不同時段預約另一學校的 SEM 貴儀所觀察的照片,是有添加 W/Ti 與 Mo/Ti 為 5%的樣品,在 TiO2的表面上可以明顯看少量顏色較深且不同的結晶 物,是添加 WO3和 MoO3的物質。經由 EDS 進行表面元素分析結果之圖譜如圖 5.5 與 5.6 所示。表面大部分除了 Ti 及添加的 W 或 Mo 外為有其他元素。
圖 5.5 WO3-TiO2/GAC 之 EDS 分析結果
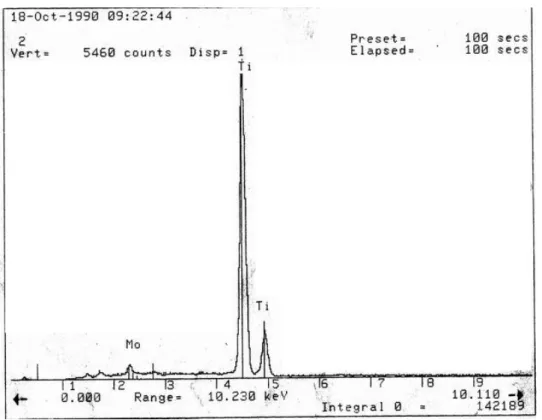
圖 5.6 MoO3-TiO2/GAC 之 EDS 分析結果
表 5.2 則為 EDS 分析計算之 Ti、W 與 Mo 之組成結果。由於 EDS 是分析一 選取之範圍進行表面元素的分析,故每一複合半導體光觸媒之樣品選取三個不同 範圍作 EDS 分析,其結果可看出 W 與 Mo 的分佈不均勻,其中 W 所含有的量 比原本配置之 W/Ti 為 5%要少。應是有些 WO3或 MoO3被 TiO2所覆蓋或進入了 活性碳的孔洞內。
表 5.2 EDS 分析複合半導體光觸媒計算之 Ti、W 與 Mo 之組成
Element Weight Percent Atomic Percent
Ti W 93.09 6.91 98.1 1.9 Ti W 90.83 9.17 97.44 2.56 Ti W 91.39 8.61 97.6 2.4 Ti Mo 95.84 4.16 97.88 2.12 Ti Mo 89.44 10.56 94.43 5.57 Ti Mo 93.11 6.89 96.44 3.56
5.2
光觸媒/活性碳之吸附與光催化實驗結果
5.2.1
TiO
2/GAC 之實驗結果
圖 5.7 為本計畫初期實驗時,先以 Sol-Gel 溶液配置時,TiO2:GAC 重量比 為 1:4 與 1:1.5 之 TiO2/GAC 進行吸附與光催化效果比較,TiO2/GAC 用量為
0.5 g/L,發現實驗結果中的 TiO2/GAC 光催化部分沒有光催化分解效果,只有原 本的吸附結果,其原因主要是因為在這次實驗中,未進行曝氣的結果所致,因為 溶液中的氧氣為光催化時的電子補集者(scavenger),可把光激發半導體所產生 的電子-電洞對中的電子消耗掉,以避免電子與電洞的再結合,讓電洞可以有效 的去氧化分解有機物或與水分子形成氫氧自由基去破壞有機物。因此在後續的實 驗中,光催化的反應都必須持續的曝氣以提供足夠的氧作為電子補集者。
圖 5.7 無曝氣時 TiO2/GAC (1:4)與 TiO2/GAC (1:1.5)之光催化與吸附結果
圖 5.8 為 TiO2/GAC (1:1.5)樣品進行不同 pH 值之光催化與活性碳吸附實驗結 果比較,由於活性碳本身即是一個良好的吸附劑,在十小時的吸附後已將原本為 2×10-4 M 之 4-CP 吸附降至 10%以下,而在負載光觸媒後的 TiO2/GAC 則因其孔 洞被 TiO2所覆蓋,比表面積減少,使得其吸附能力降低許多,在使用 365 nm 波 長的紫外光同時進行光催化反應的實驗中,可使溶液中的 4-CP 濃度隨反應時間 增加而降低,其不同的 pH 值下前 6 個小時的擬一階反應速率 k 值為 pH 4:0.0027 0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.14 0.16 0.18 0 100 200 300 400 500 600 Time (min) 4 -C P C o n c . (m M ) TiO2/GAC(1:4) Adsorption TiO2/GAC (1:1.5) Adsorption TiO2/GAC (1:4) 365 nm pH 7 TiO2/GAC (1:1.5) 365 nm pH 7
min-1、pH 7:0.0045 min-1、pH 10:0.0038 min-1。 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 200 400 600 800 1000 1200 1400 1600 Time (min) 4-C P (C /C 0) TiO2/GAC (1:1.5) 365 nm pH 10 TiO2/GAC (1:1.5) 365 nm pH 7 TiO2/GAC (1:1.5) 365 nm pH 4 GAC Adsorption TiO2/GAC (1:1.5) Adsorption
圖 5.8 TiO2/GAC (1:1.5) 之光催化分解 4-CP 與 GAC、TiO2/GAC (1:1.5)之吸附 結果比較
5.2.2
複合半導體之實驗結果
圖 5.9 為使用複合半導體/活性碳於 365 nm 波長光源照射與其吸附之實驗結
果,複合半導體與 TiO2/GAC 一樣的除了原本的吸附能力外,有其光催化的效果,
其中 WO3-TiO2/GAC(1:2)與 MoO3-TiO2/GAC(1:2)前 6 個小時的的擬一階反應速 率 k 值分別為 0.0025 min-1與 0.0023 min-1。圖 5.10 則為其分解礦化 4-CP 之分析 結果,TOC 的減少與 4-CP 的降解情況差距不大,可推斷大部分降解的 4-CP 也 被礦化。 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 200 400 600 800 1000 1200 1400 1600 Time (min) 4 -C P (C /C 0) WO3-TiO2/GAC (1:2) Adsorption MoO3-TiO2/GAC (1:2) Adsorption WO3-TiO2/GAC (1:2) 365 nm pH 7 4-CP MoO3-TiO2/GAC (1:2) 365 nm pH 7 4-CP 圖 5.9 複合半導體之光催化分解與其吸附 4-CP 結果比較
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 200 400 600 800 1000 1200 1400 1600 Time (min) 4-C P an d T O C (C /C 0) or C l-(p pm ) WO3-TiO2/GAC (1:2) 365 nm pH 7 4-CP MoO3-TiO2/GAC (1:2) 365 nm pH 7 4-CP WO3-TiO2/GAC (1:2) 365 nm pH 7 TOC MoO3-TiO2/GAC (1:2) 365 nm pH 7 TOC WO3-TiO2/GAC (1:2) 365 nm pH 7 MoO3-TiO2/GAC (1:2) 365 nm pH 7 Cl-圖 5.10 複合半導體於 365 波長之光催化分解礦化 4-CP 結果比較 由於本研究中所選擇使用的 WO3與 MoO3的添加半導體,除了探討顆粒間
電子傳送程序(Interparticle Electron Transfer , IPET)對於光催化效率之影響外, 另一目的是希望可用可見光來進行光催化反應,圖 5.11 則為紫外光與可見光波 長的光催化實驗比較結果。結果可看出用可見光範圍的 435 nm 波長同樣可達到 光催化降解 4-CP 的能力,且其降解效果稍比使用波長 365 nm 的好一些,使用波 長 435 nm 的 WO3-TiO2/GAC(1:2)與 MoO3-TiO2/GAC(1:2)前 6 個小時的的擬一階 反應速率 k 值分別為 0.0032 min-1與 0.0028 min-1。圖 5.12 為其礦化分解結果。 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 200 400 600 800 1000 1200 1400 1600 Time (min) 4-C P (C /C 0 ) WO3-TiO2/GAC (1:2) 365 nm pH 7 MoO3-TiO2/GAC (1:2) 365 nm pH 7 WO3-TiO2/GAC (1:2) 435 nm pH7 MoO3-TiO2/GAC (1:2) 435 nm pH 7 圖 5.11 複合半導體使用不同光波長光催化分解 4-CP 結果比較
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 200 400 600 800 1000 1200 1400 1600 Time (min) 4-C P an d T O C (C /C 0) or C l-(p pm ) WO3-TiO2/GAC (1:2) 435 nm pH 7 4-CP MoO3-TiO2/GAC (1:2) 435 nm pH 7 4-CP WO3-TiO2/GAC (1:2) 435 nm pH 7 TOC MoO3-TiO2/GAC (1:2) 435 nm pH 7 TOC WO3-TiO2/GAC (1:2) 435 nm pH 7 MoO3-TiO2/GAC (1:2) 435 nm pH 7 Cl-圖 5.12 複合半導體/活性碳於 435 波長之光催化分解礦化 4-CP 結果比較 圖 5.13 則為複合半導體/活性碳於 435 波長於 pH 值 7 與 10 條件下之光催化 分解 4-CP 結果比較,結果顯示 pH 10 的光催化能力明顯降低至只有原本的吸附 效果。但由於計畫時程已不足以多做此情況的研究,且尚無類似本研究內容相關 的研究文獻,未來本研究室將繼續進行此相關實驗與研究。 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 200 400 600 800 1000 1200 1400 1600 Time (min) 4 -C P (C /C 0 ) WO3-TiO2/AC 435 nm pH7 MoO3-TiO2/AC 435 nm pH 7 WO3-TiO2/AC 435 nm pH 10 MoO3-TiO2/AC 435 nm pH 10 圖 5.13 複合半導體/活性碳於 435 波長不同 pH 值之光催化分解 4-CP 結果比較
5.2.3
使用針鐵礦為擔體之實驗結果
最後本計畫亦同時選用針鐵礦(Goethite)作為另一光觸媒擔體的材料,但 由於原本針鐵礦並非是用於吸附有機物的材料,其主要是應用於吸附還原水中金 屬之用,因此在進行負載 TiO2後進行吸附與光催化實驗後如圖 5.14 所示,無法 有效吸附 4-CP 或進行光催化反應。 0 0.2 0.4 0.6 0.8 1 1.2 0 200 400 600 800 1000 1200 1400 1600 Time (min) 4 -C P (C /C 0 ) TiO2/Goethite (1:2) 365 nm pH7 TiO2/Goethite (1:2) Adsorption 圖 5.14 TiO2/Goethite 之吸附與光催化試驗結果第六章 結論與建議
6.1
結論
本計畫經由第一年以 TiO2與 ZnO 之粉末混合、燒結方式,第二年以複合奈 米級半導材料合成 CdSe/TiO2,到第三年以活性碳為載體製備複合半導體材料, 已慢慢地克服原本單獨使用 TiO2作為光催化觸媒的固液分困離與受限質傳條件 的缺點。另外,亦利用複合半導體的顆粒間電子傳送程序特性,並選用可見光(波 長 435 nm)可激發的複合半導體 WO3/TiO2與 MoO3/TiO2進行研究,克服 TiO2 電子-電洞對容易再結合而降低光催化效果,以 4-chlorophenol 為探究其反應效果 之參考污染物質,顯示有與單獨使用 TiO2之同等效果,大大提升半導體光催化 之應用層面,以及克服 TiO2受限於使用紫外光為光源的限制。6.2
建議
至目前為止結合光觸媒與活性碳的研究中,大多以 TiO2觸媒為主,仍少有 以複合行是的光觸媒與活性碳結合進行研究,且結合光觸媒與活性碳處理水中有 機物時,吸附與光催化分解間的反應機制至目前尚未有一明確的研究說明,大部 分的研究文獻中多是由反應結果,推論被活性碳吸附的化合物是經由擴散移動至 光觸媒附近在進而光催化分解。因此本研究建議有下列幾點: 可繼續設計一實驗流程,瞭解光觸媒結合活性碳的反應機制 嘗試於活性碳上合成奈米級光觸媒研究其可行性 應用於其他難分解之水中有機物之研究 設計一連續式反應槽,評估光觸媒/活性碳的應用性 利用可見光或是太陽光進行水處理的研究參考文獻
Alivisatos A.P. (1996) Perspectives on the physical chemistry of semiconductor nanocrystals. J.Phys.Chem. 100, 13226-13239.
Arabatzis, M.I.; Antonaraki, S.; Stergiopoulos, T.; Hiskia, A.; Papaconstantinou, E.; Bernard, M.C.; Falaras, P. (2002) Preparation, characterization and photocatalytic activity of nanocrystalline thin film TiO2catalysts towards 3,5-dichlorophenol degradation, J. Photoch. Photobio. A. 149, 237-245.
Bacsa R.R., Kiwi J. (1998) Effect of rutile phase on the photocatalytic properties of nanocrystalline titania during the degradation of p-coumaric acid. Appl. Catal.B 16,19-29
Cachet H., Essaaidi H., Froment M. and Maurin G. (1995) Chemical bath deposition of CdSe layers from Cd(Ⅱ)-selenosulfite solutions. J. Electro. Chem. 396, 175-182. Chang H.T., Wu N.M. and Zhu F. (2000) A kinetic model for photocatalytic
degradation of organic contaminants in a thin-film TiO2 catalyst. Wat.Res. Vol.34, No.2, 407-416.
Chen J.N., Chan Y.C., Lu M.C. (1999) Photocatalytic oxidation of chlorophenols in the presence of manganese ions. Wat. Sci. Tech. 39,225-230.
Cheng S.F., Tsai S.J. and Lee Y.F. (1995) Photocatalytic decomposition of phenol over titanium oxide of various structures. Catal. Today. 26, 87-96.
Cho Y.M., Choi W.Y, Lee C.H., Hyeon T.H, Lee H.I. (2001) Visible light-induced degradation of carbon tetrachloride on dye-sensitized TiO2. Environ. Sci. Technol. 35,966-970
Doong R.A., Chen C.H., Maithreepala R.A. and Chang S.M. (2001) The influence of pH and cadmiun sulfide on the photocatalytic degradation of 2-chlorophenol in titanium dioxide suspensions. Wat. Res. Vol.35, No.12, 2873-2880.
Doong R.A. and Chang W.H. (1998) Photodegradation of parathion in aqueous titanium dioxide and zero valent iron solutions in the presence of hydrogen peroxide. J. Photochem. Photobiol. A. 116,221-228.
Doong R.A. and Chang W.H. (1997) Photoassisted titanium dioxide mediated degradation of organophosphorus pesticides by hydrogen peroxide. J. Photochem. Photobiol. A. 107,239-244.
El-Sheikh A.H., Newman A.P., Al-Daffaee H., Phull S., Cresswell N., York S. (2004) Deposition of anatase on the surface of activated carbon. Surf. Coat. Tech. 187, 284-292.
Fox M.A. and Dulay M.T. (1993) Heterogenous photocatalysis. Chem.Rev. 93, 341-357.
Fujii H., Ohtaki M., Eguchi K. and Arai H. (1998) Preparation and photocatalytic activities of a semiconductor composite of CdS embedded in a TiO2 gel as a stable oxide semiconducting matrix. J. Molecular Catalysis A 129,61-68.
Fujishima A., Rao T.N. and Tryk D.A. (2000) Titanium dioxide photocatalysis. J. Photochem. Photobiol. C: Photochem. Reviews. 1 ,1-21.
Fu X., Clark L.A., Yang Q., Anderson M.A. (1996) Enhanced photocatalytic performance of titania-based binary metal oxides: TiO2/SiO2 and TiO2/ZrO2. Environ. Sci. Tech. 30,647-653
Fukahori S., Ichiura H., Kitaoka T., Tanaka H. (2003) Photocatalytic decomposition of bisphenol A in water using composite TiO2-zeolite sheets prepared by a
papermaking technique. Environ. Sci. Technol. 37, 1048-1051.
Gorer S. and Hodes G. (1994) Quantum size effects in the study of chemical solution deposition mechanisms of semiconductor films. J. Phys. Chem. 98, 5338-5346. Gorer S., Hodes G., Sorek Y. and Reisfeld R. (1997) Crystal phase transformation in sol-gel films of nanocrystalline CdSe and CdS. Meter. Lett. 31, 209-214.
Herrmann J.M. (1995) Heterogeneous photocatalysis: an emerging discipline involving multiphase systems. Catalsis Today 24,157-164
Herrmann J.M. (1999) Hetergeneous photocatalysis: fundamentals and applications to the removal of various types of aqueous pollutants. Catalysis Today 53, 115-129. Hirano K., Asayama H., Hoshino A. Wakatsuki H. (1997) Metal powder addition effect on the photocatalytic reactions and the photo-generated electric charge
collected at an inert electrode in aqueous TiO2 suspensions. J. Photochem. Photobiol. A. 110, 307-311.
Hoffmann M.R., Martin S.T., Choi W. and Bahnemann D.W. (1995) Environmental application of semiconductor photocatalysis. Chem. Rev. 95, 69-96.
Houas A., Lachheb H., Ksibi M., Elaloui E., Guillard C. and Herrmann J.M. (2001) Photocatalytic degradation pathway of methylene blue in water. Appl.Catal.B 31, 145-157.
Hsien Y.H., Chang C.F., Chen Y.H. and Cheng S. (2001) Photodegradation of aromatic pollutants in water over TiO2 supported on molecular sieves. Appl.Catal.B 31, 241-249.
Jardim W.F., Moraes S.G. and Takiyama M.K. (1997) Photocatalytic degradation of aromatic chlorinated compounds using TiO2 : toxicity of intermediates. Wat.Res. Vol.31, No.7, 1728-1732.
Kang M.G.,Han H.E. and Kim K.J. (1999) Enchanced photodecomposition of 4-chlorophenol in aqueous solution by deposition of CdS on TiO2. J. Photochem. Photobiol. A. 125, 119-125.
Kingery W.D., Bowen H.K. and Uhlmann D.R. (1976) Introduction to Ceramics, 2nd ed., John Wiley&Sons, New York, pp.187,476.
Kishimoto H., Takahama K., Hashimoto N., Aoi Y., Deki S. (1998) Photocatalytic activity of titanium oxide prepared by liquid phase deposition (LPD). J. Mater. Chem. 8(9),2019-2024.
chlorinated paraffins and identification of degradation products. J. Photochem. Photobiol. A. 139, 205-215.
Kormann C., Bahnemann D.W., Hoffmann M.R. (1991) Photolysis of chloroform and other organic molecules in aqueous TiO2 suspansions. Environ. Sci. Tech.
25,494-500
Ku Y., Leu R.M., Lee K.C. (1996) Decomposition of 2-chlorophenol in aqueous solution by UV irradiation with the presence of titanium dioxide. Wat. Res. 30,2569-2578
Kumar A. and Jain A.K. (2001) Photophysics and photochemistry of colloidal
CdS-TiO2 coupled semiconductors—photocatalytic oxidation of indole. J. Molecular Catalysis A 165, 265-273.
Lee G.D., Tuan V.A. and Falconer J.L. (2001) Photocatalytic oxidation and decomposition of acetic acid on titantium silicalite. Environ. Sci. Technol.35, 1252-1258.
Li X.Z., Li F.B., Yang C.L., Ge W.K. (2001) Photocatalytic activity of WOx-TiO2 under visible light irradiation. J. Photoch. Photobio. A. 141, 209-217.
Li Y. (1997) Organic Photochemistry-Photochemical reactions on semiconductor particles for organic synthesis. pp 295-303.
Linsebigler A.L., Lu G. and Yates Jr.J.T. (1995) Photocatalysis on TiO2 surface: principles, mechanisms, and selected results. Chem.Rev. 95, 735-758.
Litter M.I. (1999) Heterogeneous photocatalysis transistion metal ions in photocatalytic systems. Appl.Catal.B 23, 89-114.
Lu M.C., Chen J.N, Chang K.T. (1999) Effect of Adsorbents coated with titanium dioxide on the photocatalytic degradation of propoxur. Chemosphere. 38. 617-627. Mandal K. C. and Savadogo O. (1991) High-efficiency chemically deposited CdSe photoelectrochemical solar cells: effect of WiW12O404- incorporation. J. Mater. Sci. Lett. 10, 1446-1448.
Martin C., Solana G., Rives V., Marci G., Palmisano L., Sclafani A. (1997) Physico-chemical properties of WO3/TiO2 systems employed for 4-nitrophenol photodegradation in aqueous medium. Catal. Lett. 49, 235-243.
Matos J., Laine J. and Herrmann J.M. (2001) Effect of the type of activated carbons on the photocatalytic degradation of aqueous organic pollutants by UV-irradiated titania. J. Catal. 200, 10-20.
Milla A., Davies R.H. and Worsley D. (1993a) Water purification by semiconductor photocatalysis. Chem.Soc.Rev. 22, 417-425.
Mills A., Belghazi A., Rodman D. (1996) Bromate removal from drinking water by semiconductor photocatalysis. Wat. Res. 30,1973-1978
Mills A. and Le Hunte S. (1997) An overview of semiconductor photocatalysis. J. Photochem. Photobiol. A. 108, 1-35.
coupled SnO2/CdSe nanocrystalline semiconductor films. J. Electroanalytical Chem. 420, 201-207.
Nazir M., Takasaki J. and Kumazawa H. (2003) Photocatalytic degradation of gaseous ammonia and trichloroethylene over TiO2ultrafine powers deposited on activated carbon particles. Chem. Eng. Comm. 190, 322-333.
Nemec P., Mikes D., Rohovec J., Uhlirova E., Trojanek F. and Maly P. (2000) Light-controlled growth of CdSe nanocrystalline films prepared by chemical deposition. Meter. Sci. Eng. B69-70, 500-504.
Nozik A.J. and Memming R. (1996) Physical chemistry of semiconductor -Liquid interfaces. J. Phys. Chem. 100, 13061-13078.
O’Regan B.,MoserJ.,Anderson M.and GrätzelM.(1990)Vectorialelectron injection into transparent semiconductor membranes and electric field effects on the dynamics of light-induced charge separation. J.Phys.Chem. 94, 8720-8726.
Ormad M.P., Ovelleiro J.L. and Kiwi J. (2001) Photocatalytic degradation of
concentrated solutions of 2,4-dichlorophenol using low energy light identification of intermediates. Appl.Catal.B 32,157-166.
Patil A.J., Shinde M.H., Potdar H.S., Deshpande S.B., Sainkar S.R., Mayadevi S. and Date S.K. (2001) Chemical synthesis of titania (TiO2) power via mixed precursor route for membrane applications. Materl.Chem.Phys. 68,7-16.
Peiró A.M., Ayllón J.A., Peral J. and Doménech (2001) TiO2-Photocatalyzed degradation of phenol and ortho-substituted phenolic compounds. Appl.Catal.B 30,359-373.
Piscopo A., Robert D. and Weber J.V. (2001) Comparison between the reactivity of commercial and synthetic TiO2 photocatalysts. J. Photochem. Photobiol. A.
139,253-256.
Ranjit K.T., Willner I., Bossmann S.H. and Braun A.M.(2001) Lanthanide
oxide-doped titanium dioxide photocatalysts: Novel photocatalysts for the enhanced degradation of p-Chlorophenoxyacetic Acid. Environ. Sci. Techol. 35 , 1544-1549. Reddy E.P., Davydov L. and Smirniotis P. (2003) TiO2-loaded zeolites and
mesoporous materials in the sonophotocatalytic decomposition of aqueous organic pollutants: the role of the support. Appl. Catal. B-Environ. 42, 1-11.
Robertson P.K.J. (1996) Semiconductor photocatalysis: an environmentally
acceptable alternative production technique and effluent treatment process. J.Cleaner Prod. Vol.4, No.3-4, 203-212.
Ruan S., Wu F., Zhang T., Gao W., Xu B. and Zhao M. (2001) Surface state studies of TiO2 nanoparticles and photocatalytic degradation of methyl orange in aqueous TiO2 dispersions. Materl.Chem.Phys. 69, 7-9.
San N.,Hatipoğlu A.,Koçtürk G.and ÇinarZ.(2001)Prediction ofprimary intermediates and photodegradation kinetics of 3-aminophenol in aqueous TiO2 suspensions. J. Photochem. Photobiol. A. 139,225-232.
titanian and platinum-titania sol-gel materials. Mater. Lett. 25,271-275
Schiavello M. (1993) Some working principles of heterogeneous photocatalysis by semiconductors. Electrochimica Acta 38, No.1,11-14.
Sclafani A., Herrmann J.M. (1998) Influence of metallic silver and platinum-silver bimetallic deposits on the photocatalytic activity of titania (anatase and rutile) in organic and aqueous media. J. Photochem. Photobiol. A 113,181-188
Senogles P.J., Scott J.A., Shaw G. and Stratton H. (2001) Photocatalytic degradation of the cyanotoxin cylindrospermopsin, using titanium dioxide and UV irradiation. Wat.Res. Vol.35, No.5, 1245-1255.
Serpone N., Pichat P., Herrmann J.M. and Perlizzetti E. (1987) Inter-particle electron transfer in semiconductor dispersions : A new strategy in photocatalysis.
V.Balzani(ed.), Supramolecular Photochemistry, 415-434.
Serpone N. and Perlizzetti E. (1989) Photocatalysis-Fundamentals and applications., John Wiley & Sons : New York.
Serpone N., Maruthamuthu P., Pichat P., Perlizzetti E. and Hidaka H. (1995)
Exploiting the interparticle electron transfer process in the photocatalysed oxidation of phenol, 2-chlorophenol and pentachlorophenol: chemical evidence for electron and hole transfer between coupled semiconductors. J. Photochem. Photobiol. A. 85,24 7-255.
Shi L., Li C., Gu H. and Fang D. (2000) Morphology and properties of ultrafine SnO2-TiO2 coupled semiconductor particles. Materl.Chem.Phys. 62,62-67. Sokmen M., Allen D.W., Hewson A.T. and Clench M.R. (2001) Photocatalytic oxidation degradation of 2-methylthiophene in suspensions of TiO2: identification of intermediates and degradation pathways. J. Photochem. Photobiol. A. 141,63-67. Stafford U., Gray K.A. and Kamat P.V. (1994) Radiolytic and TiO2-assisted
photocatalytic degradation of 4-chlorophenol. A comparative study. J.Phys.Chem. 98, 6343-6351.
Sun L. and Bolton J.R. (1996) Determination of the quantum yield for the
photochemical generation of hydroxyl radicals in TiO2 suspensions. J.Phys.Chem. 100, 4127-4134.
Takeda N.,Iwata N., Torimoto T. and Yoneyama H. (1998) Influence of carbon black as an adsorbent used in TiO2 photocatalyst films on photodegradation behaviors of Propyamide. J.Catalysis 177 , 240-246.
Tennakone K., Tilakaratne C.T.K., Kottegoda I.R.M. (1997) Photomineralization of carbofuran by TiO2-supported catalyst. Wat. Res. 31,1909-1912
Topalov A., Molnár-gáborD.,KosanićM.and AbramovićB.(2000)
Photomineralization of the herbicide mecoprop dissolved in water sensitized by TiO2. Wat.Res. Vol.34, No.5, 1473-1478.
Torimoto T., Ito S., Kuwabata S. and Yoneyama H. (1996) Effects of adsorbents used as supports for Titanium Dioxide loading on photocatalytic degradation of