R
ESEARCHA
RTICLEProteomic identification of specific glycosyltransferases
functionally implicated for the biosynthesis of a targeted
glyco-epitope
Chi-Hung Lin
1, 2, 3, Chia-Wei Lin
4and Kay-Hooi Khoo
1, 3, 41Chemical Biology and Molecular Biophysics Program, Taiwan International Graduate Program,
Academia Sinica, Taipei, Taiwan
2
Institute of Bioinformatics and Structural Biology, National Tsing-Hua University, Hsin-Chu, Taiwan
3Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan
4Institute of Biochemical Science, National Taiwan University, Taipei, Taiwan
Functional glycomic and glycoproteomic analyses often entail correlating the mapped glycosyla-tion pattern of a cell against the activities of specific glycosyltransferases it expresses. While the mRNA transcripts can be readily mapped, the expression of a functional glycosyltransferase at protein level has defied most current proteomic approaches. To enable identification of these low abundant Golgi residing membrane bound proteins, we have developed a novel semigel-based shotgun proteomic workflow incorporating subcellular fractionation and one-step affinity enrichment of the detergent solubilized Golgi preparation on resins derivatized with nucleotide diphosphates. Applying the strategy to a colonic adenocarcinoma, Colo205, which is known to aberrantly synthesize abundant fucosylated extended type 1 chain, we first validated that b3-galactosyltransferase 5 (b3GalT5) is indeed the overexpressed b3GalT. This and b4GalT1 are the two galactosyltrasferases which were positively identified by proteomic analysis of the eluted fractions from uridine diphosphate (UDP)-affinity column. Substituting UDP with a guanidine diphosphate (GDP)-affinity column and monitoring the eluted fractions for enriched a3/4-fuco-syltransferase (FucT) activities, we then identified FucT3 and FucT6 as the two major a3/4FucTs expressed in Colo205 at the protein level. Our proteomic analysis demonstrated that not all GDP-utilizing glycosyltransferases bind and are retained similarly by the GDP-affinity column and that specific activity assay along with optimization of binding and elution conditions is critical for successful identification of a particular subset of the targeted glycosyltransferases. Only OFUT1, a protein O-FucT, was additionally identified to coelute with the a3/4FucT activity, and not other GDP-fucose utilizing FucTs.
Received: July 19, 2007 Revised: November 7, 2007 Accepted: November 9, 2007
Keywords:
Affinity capture / Glycosyltransferases / Subcellular fractionation
1
Introduction
Glycan chains are known to modulate many biological func-tions such as signaling, immune response, and develop-ment, in part by mediating cell–cell and protein–protein interaction through terminal glyco-epitopes. The develop-mental stage and tissue specific expression of a particular glyco-epitope is mostly effected through concerted actions of glycosyltransferases residing in the Golgi apparatus. The most direct biochemical approach to understand and manipulate the functions of glycosylation is therefore to
Correspondence: Dr. Kay-Hooi Khoo, Institute of Biological Chemistry, Academia Sinica, 128, Academia Rd Sec 2, Nankang Taipei 11529, Taiwan - Republic of China
E-mail: [email protected] Fax: +886-2-27889759
Abbreviations: FucT, fucosyltransferase; GalT, galactosyltransfer-ase; GDP, guanidine diphosphate; Leb, Lewis b,
Fuca1-2Galb1-(Fuca1-3)4GlcNAcb-; (s)Lex, (Sialyl) Lewis x,
(sialyl)Galb1-(Fuca1-3)4GlcNAcb-; LNnT, neolactotetraose, Galb1-4GlcNAcb1-3Galb1-3Glc; PA, 2-aminopyridine; TX-100, Triton X-100; UDP, uridine diphosphate
determine the regulated expression of each of the implicated endogenous glycosyltransferases, followed by the studies of substrate specificities and kinetics of individually purified enzymes. A major difficulty, however, is that these enzymes are often unstable and very low in abundance [1]. Purification of glycosyltransferase usually requires large quantity of ani-mal tissue and multiple chromatography steps. Moreover, since most of these enzymes are ER and Golgi resident type II membrane proteins [2], the requisite use of detergent for solubilization and prevention of aggregation often compli-cates the purification procedure.
The first mammalian glycosyltransferase gene was cloned by using the partial sequence of purified enzyme [3]. The progress of gene cloning was slow then because purifi-cation is difficult and time consuming. In the early 90s, expression cloning and PCR cloning were extensively used to identify novel glycosyltransferase genes. This led to the cur-rent picture of mammalian glycosyltransferase families, each with its distinctive motifs, as well as common char-acteristics. More recently, in silico analysis of glycosyl-transferase gene sequences together with advances in hu-man genome projects has brought forth a comprehensive identification and cloning of human glycosyltransferase genes including those of unknown specificities and/or func-tions [4, 5]. This development provides a relatively easy way to map the expression of mRNA transcript, which is con-veniently taken as an indicator of the expressed enzyme ac-tivity.
A fundamental scientific rationale for proteomics is that expression of a gene transcript is often a good indicator but not equivalent to functional protein expression or activity. There are growing examples, which collectively show that the mRNA transcript level may not accurately reflect the true expression level of a functional protein [6] that is subjected to further translational and post-translational regulation. In the case of glycosyltransferases, the in vivo activity and specificity of the enzymes are determined not only at the transcription level but also by their subcellular localization, proteolytic processing, dimerization/oligomerization, and PTMs [7, 8], which therefore necessitate the identification and biochem-ical studies of endogenous enzymes. Furthermore, while the overall activity of a particular class of glycosyltransferases can be readily demonstrated by in vitro assay of cell lysates using appropriate acceptor, it remains difficult to directly attribute the observed activities to any one of the many family mem-bers without a facile way to identify the expressed enzymes at protein level.
Recent technical advances in MS-based proteomics have made it possible to identify proteins with greater ease and sensitivity than previously possible. Subcellular organellar proteomic studies have further provided many detailed mapping of the constituent proteins of various organelles [9]. Among these, large scale Golgi proteomic studies using rat liver as source material have led to the identification of an impressive list of Golgi proteins including many novel ones [10–13] but only a limited
number of glycosyltransferases [12, 13]. The most com-monly found glycosyltransferases are polypeptide N-acet-ylgalactosaminyltransferases for initiation of mucin type
O-glycosylation, N-acetylglucosaminyltransferases (GnTs)
for conversion of high mannose type N-glycans to com-plex type, the ubiquitous b1,4-galactosyltransferases (GalTs), and some of the sialyltransferases, which collec-tively represent only a tiny fraction of all glycosyl-transferases implicated to be functionally active for a gly-come under investigation. The only fucosyltransferase (FucT) identified in large scale Golgi proteomics is FucT8 [13], which catalyzes core fucosylation of N-glycan. Fur-thermore, very few Golgi proteomic studies actually used cultured human cells [14] and thus far no proteomic identification of human glycosyltransferases from this sample source has ever been reported. These suggest that specifically tailored isolation and enrichment step would be required to identify glycosyltransferases by MS-based proteomic approach.
We have initiated studies along this line to develop analytical strategy which would allow proteomic identifi-cation of low abundant glycosyltransferases inferred to be responsible for the synthesis of glyco-epitopes character-istic of a cell line, as defined by initial glycomic mapping. Colo205, a colonic adenocarcinoma cell line, was reported to have high level of fucosylation presented as terminal Lewis a(Lea) and the tumor specific dimeric Leb/a-Lea
gly-cotopes on their lacto-series glycosphingolipids [15]. The preference for a backbone sequence based on type 1 chain, -Galb1-3GlcNAcb1-, instead of the more common type 2 chain, -Galb1-4GlcNAcb1-, has been attributed to abnormally high activity of b3GalT [16]. Using this as our model system, we show here that proteomic identification of glycosyltransferases from microsomal or Golgi prepa-ration of cell culture is possible after one-step enrichment with uridine diphosphate (UDP)/guanidine diphosphate (GDP)-affinity column, coupled with careful monitoring of the targeted enzyme activity. The latter is critical and required since not all glycosyltransferases that utilize UDP- or GDP-based nucleotide sugar diphosphates as donors would bind equally well to the respective affinity column and eluted similarly. We identified b3GalT5 as the predominantly expressed b3GalT, along with FucT3 and FucT6 which contributed to the expressed a3/4FucT activities.
2
Materials and methods
2.1 Materials
All chemicals were purchased from Sigma unless otherwise noted. GDP-affinity resin, UDP-affinity resins, and pyr-idylaminated sugar chains 2-aminopyridine (PA-sugar) were purchased from Calbiochem.
2.2 Glycosyltransferase assay
Glycosyltransferase activity assays were based on detecting the enzymatic products by HPLC, using PA-glycans as acceptors. For b3GalT and b4GalT activities, GlcNAcb1-Lac-PA was used as acceptor substrate (where Lac stands for Galb1-4Glc). For activity assay, 5 mL of the enzyme source was added to give a final volume of 20 mL reaction solution containing 50 mM HEPES buffer, pH 7.0, containing 1% Triton X-100 (TX-100), 20 mM MnCl2, 3.3 mM UDP-Gal, and
5 mM PA-glycan acceptors. Reaction was conducted at 377C for 4 h. For FucT activity assay, 10 mM GDP-fucose and 10 mM neolactotetraose (LNnT)-PA were used as donor and acceptor substrates, respectively. After boiling at 1007C for 2 min and centrifugation at 13 000 rpm for 3 min, super-natant from the reaction mixture was applied to a TSK-gel ODS-80™column (4.66250 mm; Tosoh, Japan) fitted on an Agilent HP1100 HPLC system coupled with an HP1100 fluo-rescence detector. Products and nonreacted acceptors were eluted isocratically with 20 mM ammonia acetate at pH 4.0 and identified by elution time in reference to authentic PA-glycan standards. Further confirmation of the structures was afforded by MALDI-MS and MS/MS analysis of the collected peaks as permethyl derivatives, using an MALDI-TOF/TOF mass spectrometer (4700 Proteomics Analyzer, Applied Bio-systems, Framingham, MA), as described in ref. [17].
2.3 Cell culture and preparation of microsome and Golgi membrane fractions
Colo205 cells were cultured in RPMI 1640 medium contain-ing 10% FCS in 75 cm2culture flasks. After harvest, cells
(,16108) were washed three times in homogenization HES
buffer (20 mM HEPES, pH 7.5, containing 1 mM EDTA and 0.25 M sucrose) and cell lysate was prepared by 20 strokes of a Dounce device. Cell debris was removed by centrifugation at 20006g for 10 min. Postnuclear fraction was then sub-jected to ultrancentrifugation at 105 0006g in an SW 41 Ti rotor for 16 h. The resulting pellet was collected as micro-some fraction for activity assay and proteomic analysis. To obtain the Golgi membrane, the postnuclear fraction was subjected to ultracentrifugation at 105 0006g for 16 h in an SW 41 Ti rotor with step sucrose gradient from 15 to 55%. Aliquots of 0.5 mL were taken from the top of the cen-trifugation tube. The collected fractions were then assayed for bGalT activity to define the Golgi membrane enriched fractions, which were pooled, diluted by HES buffer, and pelleted by 105 0006g for 1 h.
2.4 Extraction of target glycosyltransferases and affinity chromatography
Enriched microsome or Golgi membrane pellet was resus-pended and extracted by 50 mM HEPES buffer, pH 7.0, con-taining 20 mM MnCl2, 150 mM NaCl, 1 mM DTT, 20%
glycerol, and 1% TX-100, for 16 h. Insoluble material was
removed by ultracentrifugation at 105 0006g for 1 h and the supernatant was collected as TX-100 extraction for direct ac-tivity assays or further affinity enrichment. For GalT, 1.5 mL UDP-affinity (10 mmol/mL) resin was packed and equili-brated with binding buffer (50 mM HEPES, pH 7.0, 20 mM MnCl2, 150 mM NaCl, 1 mM DTT, 20% glycerol, 1%
TX-100). For FucT, 1.5 mL GDP-affinity (10 mmol/mL) resin was packed and equilibrated with the same binding buffer except for the use of 20 mM MgCl2, instead of MnCl2. TX-100
extraction was loaded into column and washed by 36volume of the binding buffer. Bound fraction was eluted sequentially by 0.5 M NaCl, 20 mM EDTA, and 2 mM UDP for GalT, or 0.5 M NaCl and 2 mM GDP for FucT.
2.5 SDS-PAGE and in-gel digestion
Active fractions by enzymatic assay were pooled and sub-jected to trichloroacetic acid (TCA) precipitation. The result-ing pellets were redissolved in 20 mL SDS-PAGE sample buffer and boiled for 5 min. SDS-PAGE 1-D gel was run until the dye front passed the stacking gel and then stained by EZblue protein staining kit (Sigma). Stained region was excised for standard in-gel digestion. Briefly, the gel pieces were subjected to reduction with 55 mM dithioerythreitol in 25 mM ammonium bicarbonate, pH 8.5, at 377C for 1 h, and subsequently alkylated with 100 mM iodoacetamide in 25 mM ammonium bicarbonate, pH 8.5, at room tempera-ture for 1 h. The gel pieces were then washed twice with 50% ACN in 25 mM ammonium bicarbonate, pH 8.5 for 15 min each time, dehydrated with ACN for 5 min, dried, and finally rehydrated with 25 mM ammonium bicarbonate, pH 8.5, containing a total of 0.1 mg of sequencing grade, modified trypsin (Promega) for 16 h at 377C. Following digestion, tryptic peptides were extracted twice by 50% ACN containing 5% TFA for 3 min with 10 s interval sonication. The extract-ed solutions were poolextract-ed and driextract-ed.
2.6 LC-MS/MS and protein ID
LC-nano-ESI-MS/MS analysis were performed on an inte-grated nano-LC-MS/MS system (Micromass) with an auto-sampler, a stream select module configured for precolumn plus analytical capillary column, and a Micromass Q-Tof Ultima API mass spectrometer fitted with nano-LC sprayer, operated under MassLynx 4.2 control. Injected samples were first trapped and desalted isocratically on a homemade pre-column (Nuclesil C18, 5 mm particle size, 100 Å pore size) for 4 min with 0.1% formic acid delivered at 15 mL/min after which the peptides were eluted off from the precolumn and separated on an analytical C18 capillary column (packed with the same material as the precolumn) connected in-line to the mass spectrometer, for a 60 min gradient of 0–80%ACN in 0.1% formic acid, at ,300 nL/min.
After data acquisition, the individual MS/MS spectra within a single LC run were combined, smoothed, deiso-toped using the Micromass ProteinLynx™ Global Server
(PGS) 2.0 and output as a single peak list (.pkl) file. The peak list files were used to query the Swiss-Prot database for hu-man proteins using the MASCOT program (Version: 1.9.05). Enzyme digestion was set to trypsin allowing one miss cleavage. Carboxyamidomethylation on cysteine and oxida-tion on methionine were set as variable modificaoxida-tion. The precursor ion and fragment ion tolerance were set to 0.25 Da. For each sample, an exclusion list, based on protein ID result, was generated for a second LC-MS/MS run to exclude previous found peptides. The two separate peak list files from the same sample thus obtained were manually combined into one single.pkl file for database searching. To minimize false positive, each individual peptide ion with a MASCOT score lower than the significant level was removed (scores of 31 for p,0.05).
3
Results and discussion
The unusual occurrence of extended type 1 chain on the GSLs of colo205 has been attributed to an abnormally high expression level of b3GalT5 among the candidate b3GalTs, as demonstrated at the mRNA transcript level [18]. Conse-quently, we would expect b3GalT5 to be readily identified among the glycosyltransferases in a corresponding prote-omic analysis. This would serve as a positive control against
detecting any other targeted Golgi glycosyltransferases of interest. A major aim of current investigation was to estab-lish which of the a3/4FucTs among FucT3-7, and FucT9, can be identified at protein expression level, in comparison against their reported transcript level [19, 20]. However, initial attempt using either microsome or Golgi preparation failed to identify any of these glycosyltransferases through standard shotgun proteomic analysis. This is consistent with current limitations in the sensitivity and selectivity of detec-tion without target enrichment. Thus, short of readily avail-able mAb, a robust enzymatic assays which can resolve specifically the targeted b3GalT and a3/4FucT activities were first developed and applied to monitor the subfractionation procedures for a more targeted approach.
Instead of conventional radioisotope-based assay which is simple but lack of structure specific information, fluores-cence tagged PA-sugar chains were used as acceptor sub-strates for nonradioactivity-based enzymatic activity assays. This method confers the advantage of easy monitoring and separation of products by HPLC [21], which can be further subjected to facile MS and MS/MS analysis to ascertain the identity of the synthesized products. In particular, since both b3GalT and b4GalT compete for the same substrate and, likewise, all a3/4FucTs share the same lacto-series acceptor substrates with a2FucT, resolving the structural isomers of the enzymatic products are required. As shown in Fig. 1, the
Figure 1. GalT and FucT enrichment and activity assay based on PA-glycans as acceptor substrates. Enzyme reaction products were sepa-rated by RP-HPLC and identified by fluorescent detector, in reference to the retention times of authentic standards. Quantitative assess-ment of individual products relative to unreacted substrates was afforded by integration of peak area. bGalT activity is shown to be effi-ciently solubilized from the pelleted microsome fraction by 1% TX-100, by comparing the product yield afforded by the microsome (lower trace) relative to that of the TX-100 extracts (upper trace). Likewise, a3/4FucT activity was efficiently solubilized by 1% TX-100 (upper trace) from the Golgi membrane preparation (lower trace). The retention time of the a3-fucosylated product, Lewis x (Lex)-Lac-PA, was
deter-mined by using recombinant human FucT5 (rhFucT5, Calbiochem) as the enzyme source (see inset). The peak denoted by # indicates contaminant substance also found in the original PA-sugar substrate stock.
Figure 2. MALDI TOF/TOF CID-MS/MS sequencing of the GalT and FucT reaction products to distinguish between LNT-PA (A) and LNnT-PA (B), and to confirm the a3-fucosylation position at the GlcNAc of LNnT-PA, corresponding to Lex-Lac-PA (C). Critical fragment ions afforded
by the permethyl derivatives [17] are the D ions at m/z 268 and 472, which is specific to type 1 and 2 chain, respectively, as illustrated. For Lex-Lac-PA, the type 2 chain specific D ion at m/z 472 confirms the a3-fucosylation at GlcNAc, whereas the absence of a2-FucT product was
indicated by lack of both C and D ions at m/z 433 (Fuc-Gal-OH) and 646 (Fuc-Gal-DGlcNAc-OH, where D indicates an unsaturated bond due to elimination of 3-substituent). Symbol keys used and nomenclature of cleavage ions as annotated are further illustrated schematically (inset) and have been described in detail before [17].
unreacted GlcNAcb1-Lac-PA substrate, 3GlcNAcb1-Lac-PA (type 1 chain, lactotetraose (LNT)-PA) and Galb1-4GlcNAcb1-Lac-PA (type 2 chain, LNnT-PA) could be sepa-rated by RP-HPLC chromatography, identified first by refer-ring to the elution time of available standard PA-glycan chains and then confirmed by MS/MS analysis (Fig. 2). For FucT assay, since all FucTs known to show a4-fucosylation activity against type 1 chain can also a3-fucosylate type 2 chain, the acceptor LNnT-PA was used as a general substrate to monitor all a3/4FucT activities. The fucosylated product can be resolved from the precursor and the exact fucosylated position can be similarly distinguished based on both elution time and MS/MS analysis (Figs. 1 and 2).
Based on these PA-glycan assays, significant GalT and FucT activities were detected for the total lysates, as well as the microsome or Golgi fractions. As expected for Colo205, b3GalT activity which is normally very weak in other cell lines (data not shown) was prominently detected along with b4GalT activity. More importantly, we showed that the en-zyme activities could be efficiently extracted from the mem-brane by 1% TX-100 (Fig. 1). This is critical since the targeted glycosyltransferases are type II membrane proteins, which often require detergent solubilization to improve recovery
and prevent aggregation. Monitoring against the activities of b3/4GalTs, a significant portion of the solubilized activities was shown to be retained on a UDP-affinity column and eluted by 2 mM UDP, after extensive wash (Fig. 3), which therefore suggested that it may constitute an effective affinity enrichment step. The continued use of detergent and glyc-erol in the eluting buffer served to stabilize the membrane proteins but nevertheless rendered the sample not conducive to the following in solution digestion and direct LC-MS/MS analysis. To get around this problem, and to prepare the en-zyme active fractions for proteomic identification, the eluted sample was desalted and concentrated by TCA precipitation. The resulting pellet was then subjected to 1-D SDS-PAGE but the electrophoretic running was stopped immediately after the dye front migrated through the stacking gel. This method allows total proteins to be stacked as a band in the 1-D gel for subsequent in-gel digestion and LC-MS/MS analysis.
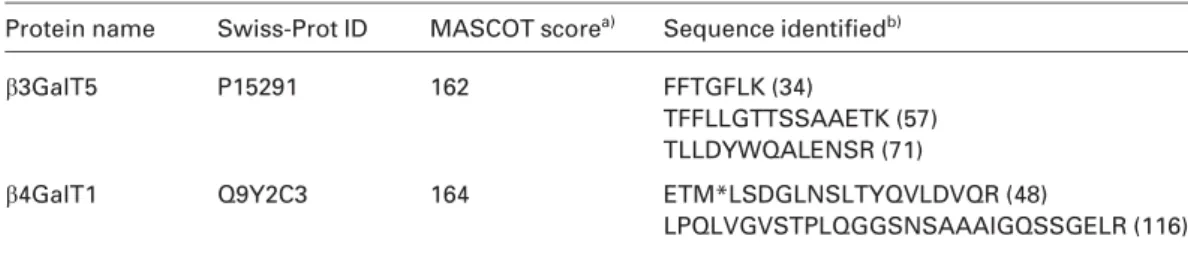
As a result, both b3GalT5 and b4GalT1 were positively identified by three and two matched peptides, respectively (Table 1). However, from a total of 95 proteins identified, only seven proteins may be classified as ER/Golgi proteins (data not shown). Despite the use of microsome preparation and
Table 1. GalTs identified
Protein name Swiss-Prot ID MASCOT scorea) Sequence identifiedb)
b3GalT5 P15291 162 FFTGFLK (34)
TFFLLGTTSSAAETK (57) TLLDYWQALENSR (71)
b4GalT1 Q9Y2C3 164 ETM*LSDGLNSLTYQVLDVQR (48)
LPQLVGVSTPLQGGSNSAAAIGQSSGELR (116)
a) MASCOT score of individual peptide is listed in parenthesis after the assigned peptide sequence. b) M* indicates oxidation at methionine.
Figure 3. UDP-affinity enrichment of b3GalT and b4GalT activities extracted from colo205 microsome preparation. Collected frac-tions were subjected to bGalT assay to monitor the distribution of b3GalT and b4GalT activities. Two-thirds of b3GalT was retained whereas b4GalT activity was fully retained by the UDP-affinity column. The bound fractions (#30–38) were subjected to SDS-PAGE and proteomic identification. Although b3GalT activity can be found in the flow through (#12–20), it cannot be efficiently identified in the presence of many other copurifying proteins.
the affinity step, it remains difficult to minimize the copur-ified cytosolic or nucleus proteins, including many DNA/ RNA binding proteins that presumably exhibited certain degree of affinity against UDP. To further improve the effi-ciency of enrichment, a Golgi preparation step was further incorporated into this overall protocol for subsequent prote-omic analysis of the FucTs, substituting UDP with a GDP-affinity column. Thus, Golgi membrane was separated from postnuclear fraction by zonal ultracentrifugation with step sucrose gradient. The activities of the targeted a3/4FucTs were shown to colocalize with the trans-Golgi marker, b4GalT activity (Fig. 4), and the enriched Golgi fractions were pelleted by ultracentrifugation and subjected to similar TX-100 extraction.
Before sample loading onto the GDP-affinity column, the TX-100 content was adjusted to the condition for maximum binding ability, which is often also the condition for best
Figure 4. Subcellular fractionation of Colo205 membrane was conducted by a step sucrose gradient from 15 to 55%. Fractions were collected from top to bottom of gradient. b4GalT activity assay was conducted to locate the distribution of Golgi mem-brane. Distribution of b3GalT and a3FucT was also evaluated and found to colocalize with the b4GalT activity. Left Y-axis indicates the relative activity of b3GalT whereas right Y-axis indicates the relative activities of b4GalT and a3FucT.
enzyme activity. To remove nonspecific binding, 36volume of binding buffer and 0.5 M NaCl in binding buffer were used consecutively to wash the GDP-affinity resin. GDP-af-finity bound fraction was then eluted by 2 mM GDP and subjected to a3/4FucT assay to evaluate the distribution of a3/4FucT activity. As shown in Fig. 5, the a3/4FucT activity was well retained by the GDP-affinity resin. As in the treat-ment of bGalTs, the detergent carrying eluates were TCA precipitated, similarly run into 1-D gel as a single focused band, followed by in-gel digestion and LC-MS/MS shotgun proteomic analysis of the extracted peptides.
Three FucTs, FucT3, FucT6, and OFUT1, were positively identified from the a3/4FucT active fraction (Table 2). In addition to three common peptide matches, the highly ho-mologous FucT3 and FucT6 [22] are distinguished respec-tively by one and two additional unique peptide matches of sufficiently good MASCOT scores. In vitro studies have demonstrated that only FucT3 and FucT5 possess a1,4FucT activity to fucosylate type 1 chain. Since the expression of FucT5 is undetectable at both mRNA transcript level [19, 20] and by current proteomic analysis, it could therefore be con-cluded that FucT3 is the only enzyme responsible for
Table 2. FucTs identified
Protein name Swiss-Prot ID MASCOT scorea) Sequence identifiedb)
FucT6 P51993 231 SFSWALAFC*K (41) EVMYNPSAQLPR (57) YLQELDKDHAR (51) NALEAWAVPVVLGPSR (48) FLPPDAFIHVDDFQSPK (34) FucT3 P21217 187 SFSWALDFC*K (54) YLQELDKDHAR (51) NALEAWAVPVVLGPSR (48) FLPPDAFIHVDDFQSPK (34) OFUT1 Q9H488 71 VISLEDFMEK (71)
a) MASCOT score of individual peptide is listed in parenthesis after the assigned peptide sequence.
b) Sequences underlined are common to FucT3 and FucT6; C* indicates carboxyamidomethylation at cysteine.
Figure 5. GDP-affinity enrichment of a3/4FucT activity extracted from Colo205 Golgi preparation. Collected fractions were sub-jected to a3FucT assay to locate the distribution of a3FucT activity. a3/4FucT activity was well retained by the GDP-affinity column. The bound fractions (#34–38) were subjected to SDS-PAGE and proteomic ID.
synthesizing Leaand (sialyl) Lewis a,
(sialyl)Galb1-(Fuca1-4)3GlcNAcb- (s)Lea in Colo205. On the other hand, FucT3 and FucT6 could both account for a3-fucosylation of type 2 chain in Colo205, among the many known a3FucTs. The proteomic results are in fact in good correlation with pre-vious findings, which demonstrated that Colo205 cells express high levels of FucT3 and FucT6 transcripts but rela-tively low level of other a3/4FucTs [23], like most other cells of epithelial origin [24].
An interesting result is the identification of OFUT1 in the a3/4FucT active fractions, which suggests an equally high binding affinity to GDP resin. Recent studies have implicated OFUT1 as an ER lumenal protein instead of Golgi resident membrane protein [25]. Our proteomic studies using microsome fraction for GDP-affinity enrichment has always led to identification of OFUT1 with higher sequence coverage (data not shown), instead of only one matched
peptide when using the Golgi preparation, whereas the reverse is true for FucT3 and FucT6. This not only suggests a better enrichment efficiency of a3/4FucTs by Golgi prepara-tion but also consistent with OFUT1 being primarily an ER protein. The better known biological function of OFUT1 is to catalyze protein O-fucosylation of the conserved EGF domain in a number of proteins involved in Notch signaling [26–29]. OFUT1 was shown to be highly expressed only in heart, brain, placenta, lung, liver, skeletal muscle, kidney, and pan-creas [28]. Our proteomic identification of OFUT1 suggests a relatively high protein expression level in Colo205 cells but the biological implication remains to be elucidated.
Aside from keratins, a total of 52 proteins were con-fidently identified in the a3/4FucT active fraction (see Table in Supporting Information) according to the criteria de-scribed in Section 2. Among these, those formally classified as ER/Golgi proteins according to Swiss-Prot annotation account for 31% of total, suggesting a good enrichment of ER/Golgi membrane comparable to that reported in the lit-erature [12, 14]. However, only five proteins have been shown to interact with GDP/GTP and 16 proteins with ATP/UDP, based on the Swiss-Prot annotation. It thus appears that a significant portion of coeluting proteins was not specifically enriched by affinity to GDP. More extensive wash and/or preadsorption with the resins would conceivably reduce fur-ther the nonspecific interactions but unlikely to have yielded only the desirable GDP-binding proteins.
4
Concluding remarks
Identification of glycosyltransferases represents one of the most daunting undertakings in current proteomics. The gly-cosyltransferases are not only of low abundance but are also Golgi resident membrane proteins. A range of technical issues therefore need to be considered including extraction, subcellular fractionation, detergent solubilization, affinity
enrichment, and efficient digestion, all of which must lead to final compatibility with MS analysis without incurring lengthy buffer exchange steps of poor recovery. In the absence of readily available antibodies against the endoge-nous enzymes or activity-based probe of sufficient reactivity and specificity, we have shown that both Golgi and GDP-af-finity enrichment, coupled with semigel-based approach, are the necessary minimal workflow for successful identification by shotgun proteomics. Our proteomic results, as exempli-fied here with applications to Colo205 cells, are largely con-sistent with mapping at the mRNA transcript level in so far as revealing the major b3GalT and a3/4FucT activities. How-ever, only at protein level, through further optimization and scale up, would it be possible to contemplate tackling poten-tial PTMs.
We are grateful to Professor Naoyuki Taniguchi and Dr. Tomohiko Taguchi (Department of Biochemistry, Osaka Uni-versity Graduate School of Medicine) for their technical advice to CHL in the biochemical handling of glycosyltransferases and PA-sugar activity assay. This work is supported by an Academia Sinica Program Project Grant. Proteomic analysis was performed at the National Core Facilities for Proteomics (NSC grant 95-3112-B-001-014), located at the Institute of Biological Chemistry, Academia Sinica.
The authors have declared no conflict of interest.
5
References
[1] Sadler, J. E., Beyer, T. A., Oppenheimer, C. L., Paulson, J. C. et al., Purification of mammalian glycosyltransferases. Meth. Enzymol. 1982, 83, 458–514.
[2] Paulson, J. C., Colley, K. J., Glycosyltransferases. Structure, localization, and control of cell type-specific glycosylation. J. Biol. Chem. 1989, 264, 17615–17618.
[3] Narimatsu, H., Sinha, S., Brew, K., Okayama, H., Qasba, P. K., Cloning and sequencing of cDNA of bovine N-acet-ylglucosamine (beta 1-4)galactosyltransferase. Proc. Natl. Acad. Sci. USA 1986, 83, 4720–4724.
[4] Narimatsu, H., Human glycogene cloning: Focus on beta 3-glycosyltransferase and beta 4-3-glycosyltransferase families. Curr. Opin. Struct. Biol. 2006, 16, 567–575.
[5] Narimatsu, H., Construction of a human glycogene library and comprehensive functional analysis. Glycoconj. J. 2004, 21, 17–24.
[6] Stults, J. T., Arnott, D., Proteomics. Meth. Enzymol. 2005, 402, 245–289.
[7] Hathaway, H. J., Evans, S. C., Dubois, D. H., Foote, C. I. et al., Mutational analysis of the cytoplasmic domain of beta1,4-galactosyltransferase I: Influence of phosphorylation on cell surface expression. J. Cell Sci. 2003, 116, 4319–4330. [8] de Graffenried, C. L., Bertozzi, C. R., The roles of enzyme
localisation and complex formation in glycan assembly within the Golgi apparatus. Curr. Opin. Cell Biol. 2004, 16, 356–363.
[9] Yates, J. R., III, Gilchrist, A., Howell, K. E., Bergeron, J. J., Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 2005, 6, 702–714.
[10] Mogelsvang, S., Howell, K. E., Global approaches to study Golgi function. Curr. Opin. Cell Biol. 2006, 18, 438–443. [11] Bell, A. W., Ward, M. A., Blackstock, W. P., Freeman, H. N. et
al., Proteomics characterization of abundant Golgi mem-brane proteins. J. Biol. Chem. 2001, 276, 5152–5165. [12] Wu, C. C., MacCoss, M. J., Mardones, G., Finnigan, C. et al.,
Organellar proteomics reveals Golgi arginine dimethylation. Mol. Biol. Cell 2004, 15, 2907–2919.
[13] Takatalo, M. S., Kouvonen, P., Corthals, G., Nyman, T. A., Ronnholm, R. H., Identification of new Golgi complex spe-cific proteins by direct organelle proteomic analysis. Prote-omics 2006, 6, 3502–3508.
[14] Wu, C. C., Yates, J. R., III, Neville, M. C., Howell, K. E., Prote-omic analysis of two functional states of the Golgi complex in mammary epithelial cells. Traffic 2000, 1, 769–782. [15] Stroud, M. R., Levery, S. B., Nudelman, E. D., Salyan, M. E. et
al., Extended type 1 chain glycosphingolipids: Dimeric Lea (III4V4Fuc2Lc6) as human tumor-associated antigen. J. Biol. Chem. 1991, 266, 8439–8446.
[16] Holmes, E. H., Characterization and membrane organization of beta 1-3- and beta 1-4-galactosyltransferases from human colonic adenocarcinoma cell lines Colo 205 and SW403: Basis for preferential synthesis of type 1 chain lacto-series carbohydrate structures. Arch. Biochem. Biophys. 1989, 270, 630–646.
[17] Yu, S. Y., Wu, S. W., Khoo, K. H., Distinctive characteristics of MALDI-Q/TOF and TOF/TOF tandem mass spectrometry for sequencing of permethylated complex type N-glycans. Gly-coconj. J. 2006, 23, 355–369.
[18] Isshiki, S., Togayachi, A., K, T., Nishihara, S. et al., Cloning udo, expression, and characterization of a novel UDP-galac-tose: Beta-N-acetylglucosamine beta1,3-galactosyltransfer-ase (beta3Gal-T5) responsible for synthesis of type 1 chain in colorectal and pancreatic epithelia and tumor cells derived therefrom. J. Biol. Chem. 1999, 274, 12499–12507. [19] Sasaki, K., Kurata, K., Funayama, K., Nagata, M. et al.,
Expression cloning of a novel alpha 1,3-fucosyltransferase that is involved in biosynthesis of the sialyl Lewis x carbo-hydrate determinants in leukocytes. J. Biol. Chem. 1994, 269, 14730–14737.
[20] Kaneko, M., Kudo, T., Iwasaki, H., Ikehara, Y. et al., Alpha1,3-fucosyltransferase IX (Fuc-TIX) is very highly conserved be-tween human and mouse; molecular cloning, characteriza-tion and tissue distribucharacteriza-tion of human Fuc-TIX. FEBS Lett. 1999, 452, 237–242.
[21] Taniguchi, N., Nishikawa, A., Fujii, S., Gu, J. G., Glycosyl-transferase assays using pyridylaminated acceptors: N-acetylglucosaminyltransferase III, IV, and V. Meth. Enzymol. 1989, 179, 397–408.
[22] de Vries, T., Knegtel, R. M., Holmes, E. H., Macher, B. A., Fucosyltransferases: Structure/function studies. Glycobiol-ogy 2001, 11, 119R–128R.
[23] Hiller, K. M., Mayben, J. P., Bendt, K. M., Manousos, G. A. et al., Transfection of alpha(1,3)fucosyltransferase antisense sequences impairs the proliferative and tumorigenic ability of human colon carcinoma cells. Mol. Carcinog. 2000, 27, 280–288.
[24] Kannagi, R., Izawa, M., Koike, T., Miyazaki, K., Kimura, N., Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004, 95, 377–384.
[25] Luo, Y., Haltiwanger, R. S., O-fucosylation of notch occurs in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 11289– 11294.
[26] Wang, Y., Lee, G. F., Kelley, R. F., Spellman, M. W., Identifi-cation of a GDP-L-fucose: Polypeptide fucosyltransferase and enzymatic addition of O-linked fucose to EGF domains. Glycobiology 1996, 6, 837–842.
[27] Wang, Y., Spellman, M. W., Purification and characterization of a GDP-fucose: Polypeptide fucosyltransferase from Chi-nese hamster ovary cells. J. Biol. Chem. 1998, 273, 8112– 8118.
[28] Wang, Y., Shao, L., Shi, S., Harris, R. J. et al., Modification of epidermal growth factor-like repeats with O-fucose. Molec-ular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J. Biol. Chem. 2001, 276, 40338–40345. [29] Haines, N., Irvine, K. D., Glycosylation regulates Notch