Synthesis of (Vinylidene)- and (Cyclopropenyl)ruthenium Complexes Containing
a Tris(pyrazolyl)borato (Tp) Ligand
Yih-Hsing Lo,
[a]Ying-Chih Lin,*
[a]Gene-Hsiang Lee,
[a]and Yu Wang
[a]Keywords: Cyclopropenyl / N ligands / Ruthenium / Vinylidene ligands
A convenient high-yield route to [Ru(C⬅C−Ph)(Tp)(PPh3)2]
[2; Tp = HB(pz)3, pz = pyrazolyl] has been found through
the intermediacy of [RuCl2(Hpz)2(PPh3)2] (1). This complex is
readily obtained on treatment of [RuCl2(PPh3)3] with 2 equiv.
of pyrazole in boiling THF. The molecular structures of com-plexes 1 and 2 have been confirmed by single-crystal X-ray diffraction analysis. A number of new cationic vinylidene complexes [Ru{=C=C(Ph)CH2R}(Tp)(PPh3)2]+[3a, R = CN; 3b,
R = HC=CH2; 3c, R = CH=C(CH3)2; 3d, R = Ph; 3e, R =
C(O)OMe] have been prepared by electrophilic addition of organic halides to complex 2. The deprotonation reaction of
3ayields the cyclopropenyl complex 4a. One phosphane
li-Introduction
The hydrotris(pyrazol-1-yl)borate (Tp) ligand has been
used to stabilize a wide variety of transition-metal
com-plexes since its discovery by Trofimenko,
[1]and the
develop-ment of Tp chemistry within group VIII, in particular, has
accelerated recently.
[1c,1d]Tp is often compared with Cp (η
5-C
5H
5) due to the same charge and number of electrons
do-nated, although their differences in size and electronic
properties are obvious. Thus, the cone angle of Tp (close to
180
°) is well above the value of 100° calculated for Cp. The
steric bulkiness of the Tp ligand appears to disfavor higher
coordination numbers or bulky metal fragment. Much of
the chemistry of the [CpRu(PPh
3)
2]
⫹fragment can be
traced to the strongly π-basic nature of the ruthenium
center. Replacing Cp with Tp increases the basicity of the
metal center further, and it has been claimed that it also
leads to more ideally octahedral hybridization.
[2]The
chemistry of (vinylidene)transition-metal complexes has
attracted increasing attention in recent years especially
be-cause of their application as key intermediates in
stoichio-metric and catalytic transformations of organic molecules.
[3]Representative examples of ruthenium-based catalysis
involving vinylidene complexes have been reported for the
cyclization
of
dienylalkynes,
[4a]the
dimerization
of
HC
⬅CtBu,
[4b]and the tandem cyclization/reconstructive
addition of propargyl alcohols with allyl alcohols.
[4c]A key
characteristic of vinylidene complexes appears to be the
[a] Department of Chemistry, National Taiwan University,Taipei, Taiwan 106, Republic of China
gand of 4a is remarkably labile, being replaced by donor li-gands L to yield diastereomeric mixtures of the cycloprope-nyl complexes 5a−5d mostly in an approximate 4:1 ratio. The cyclopropenyl rings in 4a and 5a are susceptible to ring ope-ning by I2. In addition, treatment of 4a with nBuNC in the
presence of MeOH results in substitution of a phosphane li-gand by nBuNC followed by protonation of the three-mem-bered ring by MeOH. This is then followed by addition of methoxide to give the vinyl ether complex [Ru{C(OMe)= C(Ph)CH2CN}(Tp)(PPh3)(nBuNC)] (8a).
( Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
electrophilicity of the α-carbon atom, which adds, often
easily, amines,
[5]alcohols,
[6,7]phosphanes,
[8]and even
fluoride.
[6a]The reactions of metal complexes with cyclopropenes
often generate interesting chemistry, mainly due to the large
amount of strain energy (ca. 50 kcal/mol) in the
three-mem-bered ring.
[9]This molecule has played a crucial role in the
development of the important concept of aromaticity, and
its chemical reactivity has been extensively explored.
[10]The
syntheses of cyclopropenyl-containing metal derivatives in
which the metal atom is bonded to the sp
3-hybridized
car-bon atom of the cyclopropene ring have been reported in
the literature.
[11]However, only a few examples of such
de-rivatives in which the metal atom is bonded to the sp
2-car-bon atom of the three-membered ring have been
re-ported.
[12]A few structurally different
(cyclopropenylidene)-transition-metal complexes, mostly prepared from
dichloro-cyclopropene,
[13]and
a
number
of
π-cyclopropene
complexes,
[14]are also known.
During the course of our investigations into
(vinyl-idene)ruthenium chemistry, we previously studied the
for-mation of several interesting neutral cyclopropenyl
com-plexes.
[6]For example, the cationic complex [RuCp{
⫽C⫽
C(Ph)CH
2CN}(PPh
3)
2]
⫹was found to undergo
depro-tonation to afford the yellow cyclopropenyl complex. The
cationic nature of the vinylidene complex, along with the
presence of an electron-withdrawing functionality such as a
CN group at C-γ of the vinylidene ligand, play a role in
enhancing the acidity of the proton next to the CN group.
Thus, addition of a base successfully brings about an
intra-molecular cycloaddition leading to the formation of a
neu-tral cyclopropenyl complex. To elaborate the breadth of
such a system, we set out to study analogous ruthenium
complexes containing a Tp ligand. Herein, we report the
synthesis of a series of new cationic vinylideneruthenium
complexes and new neutral cyclopropenyl complexes in
high yields. The reaction of the cyclopropenyl complex with
I
2to give a new vinylidene complex is also reported.
Results and Discussion
Preparation of (Tp)metal Acetylide Complexes
We have previously reported
[6b]that [RuCl(Tp)(PPh
3)
2]
reacts with phenylacetylene in the presence of NaOMe to
afford the acetylide complex [Ru(C
⬅C⫺Ph)(Tp)(PPh
3)
2]
(2). However, complex 2, prepared using this method, is
persistently contaminated with the starting material
be-cause of incomplete conversion. Therefore, a new synthetic
approach to prepare 2 has been developed (Scheme 1). This
new, convenient, high-yield route to 2 proceeds through the
intermediacy of [RuCl
2(Hpz)
2(PPh
3)
2] (1) (Hpz
⫽
pyr-azole). This compound is readily obtained as an
air-sensi-tive yellow solid in 97% yield upon treatment of
[RuCl
2(PPh
3)
3] with 2 equiv. of pyrazole in boiling THF for
1 h. Complex 1 is soluble in CHCl
3, CH
2Cl
2, MeOH,
CH
3CN and THF but insoluble in hexane, and was
charac-terized by a combination of elemental analysis and
1H and
13C{
1H} NMR spectroscopy. Characteristic
1H NMR
spec-troscopic data for 1 include a singlet at δ
⫽ 11.67 ppm
as-signable to the proton of the pyrazole ligand. In addition,
the solid-state structure was determined by a single-crystal
X-ray diffraction study. An ORTEP diagram is shown in
Figure 1 (with selected bond lengths and angles). The
coor-dination geometry of 1 is approximately octahedral with
both pyrazole ligands N-bonded. The ruthenium atom is at
the center, with the nitrogen atoms of the two pyrazole
li-gands and two PPh
3ligands occupying the equatorial
posi-tions and the axial posiposi-tions being occupied by two
trans-chlorine atoms. The Ru
⫺N(1) and Ru⫺N(3) bond lengths
are 2.133(5) and 2.127(5) A
˚ , respectively. The two Ru⫺Cl
Scheme 1
bonds of 2.428(2) and 2.426(2) A
˚ are similar to those found
in other (pyrazole)ruthenium complexes, such as 2.427(2)
and 2.392(2) A
˚ in [RuCl
2(HPz)(DMSO)
3] and 2.427(1) and
2.397(1) A
˚ in [RuCl
2(HPz)
2(DMSO)
2].
[15]Pyrazoles and
their deprotonated form (pyrazolate anions) are attractive
ligands that exhibit a rich coordination chemistry.
[16]Pyr-azoles and substituted pyrPyr-azoles usually behave as
mono-dentate ligands
[17]and these monodentate pyrazoles may
give rise to interesting processes such as prototropic
equilib-rium or reversible metal
⫺ligand binding, which are relevant
to biological systems.
[18]Figure 1. ORTEP drawing of complex 1 (30% probability ellip-soids); selected bond lengths [A˚ ] and angles [°]: Ru⫺P(1) 2.3423(20), Ru⫺P(2) 2.3421(20), Ru⫺N(1) 2.133(5), Ru⫺N(3) 2.127(5), Ru⫺Cl(1) 2.4283(19), Ru⫺Cl(2) 2.4261(19); N(1)⫺Ru⫺N(3) 83.24(20)
The one-pot reaction of 1 with NaTp and
phenylacety-lene in the presence of NaOMe gives the ruthenium
acetyl-ide complex [Ru(C
⬅C⫺Ph)(Tp)(PPh
3)
2] (2). The yield of
this one-pot reaction was higher than the yield obtained
from the previously reported reaction that required several
steps.
[6b]The ν(B
⫺H) vibration of 2 is found at 2489 cm
⫺1,
which is characteristic of Tp bound to a metal center in a
terdentate (N,N,N) manner. Yellow crystals of 2 were
ob-tained by slow diffusion of hexane into a CHCl
3solution
of 2 at room temperature for 3 d. The molecular structure
of 2 was determined by an X-ray diffraction study. An
OR-TEP diagram is shown in Figure 2 (with selected bond
lengths and angles). The coordination geometry of complex
2 is approximately octahedral. The Ru⫺C(1) bond length
of 2.006(6) A
˚ is typical of an Ru⫺C single bond. The
Ru
⫺C(1)⫺C(2) bond angle of 173.9(5)° and C(1)⫺C(2)
bond length of 1.205(8) A
˚ are characteristic of a ruthenium
acetylide complex.
Cationic (Tp)(vinylidene)metal Complexes
Treatment of 2 with ICH
2CN affords the cationic
vinyl-idene complex [Ru{
⫽C⫽C(Ph)CH
2CN}(Tp)(PPh
3)
2]I (3a)
in 90% yield (Scheme 1).
[9b]In the presence of excess
NH
4PF
6the counteranion is replaced by PF
6⫺. Similarly,
Figure 2. ORTEP drawing of complex 2 (30% probability ellip-soids); the carbon atoms of the phenyl groups (except the ipso-carbon atoms) on the triphenylphosphane have been eliminated for clarity; selected bond lengths [A˚ ] and angles [°]: Ru⫺C(1) 2.006(6), C(1)⫺C(2) 1.205(8), C(2)⫺C(3) 1.439(9); Ru⫺C(1)⫺C(2) 173.9(5), C(1)⫺C(2)⫺C(3) 167.8(6)
the
preparation
of
other
vinylidene
complexes
[Tp(PPh
3)
2Ru
⫽C⫽C(Ph)CH
2R]
⫹[3b, R
⫽ CH⫽CH
2; 3c,
R
⫽ CH⫽C(CH
3)
2; 3d, R
⫽ Ph; 3e, R ⫽ C(O)OMe] was
accomplished by treating 2 with the corresponding halides;
they were all isolated in high yields. With the exception of
3e, all the vinylidene complexes mentioned above were
pre-pared in CH
2Cl
2at room temperature; mild heating was
required for the synthesis of 3e, and a mixture of CH
2Cl
2/
CHCl
3(3:1, v/v) was used as solvent in order to achieve a
slightly higher reaction temperature. Complexes 3a
⫺3e are
all soluble in polar solvents such as CHCl
3, CH
2Cl
2, MeOH
and CH
3CN but insoluble in acetone, diethyl ether and
hex-ane. These complexes are green in the solid state.
Character-istic spectroscopic data of theses vinylidene complexes
con-sist of a strongly deshielded C-α resonance as a triplet at
δ
⫽ 370 ⫾ 5 ppm in the
13C NMR spectrum and a singlet
31P NMR resonance at δ
⫽ 36 ⫾ 1 ppm in CDCl
3
at room
temperature; this is due to the fluxional behavior of the
vinylidene ligand.
[19]The spectroscopic data of the Cp
ana-logues are similar; the triplet C-α resonance appears at δ
⫽
340
⫾ 5 ppm in the
13C NMR spectrum and a singlet
31P
NMR resonance is observed at δ
⫽ 42 ⫾ 1 ppm. The newly
formed carbon
⫺carbon bond of these vinylidene complexes
is easily cleaved in the presence of acid. Complexes 3a
⫺3d
are all stable at room temperature for a period of 3 d, after
which they decomposed to give unidentified products.
(Cyclopropenyl)(Tp)metal Complexes
The
neutral
complex
[Ru{C
⫽C(Ph)CH(CN)}(Tp)-(PPh
3)
2] (4a) can be readily prepared in high yield by
depro-tonation of 3a at 0
°C in CH
2Cl
2(Scheme 2). However, no
deprotonation was observed for the other vinylidene
com-plexes 3b
⫺3d, even in the presence of NaOMe, nBu
4NF (1
in THF), DBU (1,8-diazabicyclo[5.4.0]undecene) or KOH
(dissolved in a minimum amount of H
2O) at room
tempera-ture for 24 h. We have previously prepared neutral
cyclopro-penyl complexes containing a Cp ligand by deprotonation
of the cationic vinylidene precursors.
[19]Cyclopropenyl
complexes with substituents such as vinyl, dimethylvinyl,
phenyl and methyl ester groups are all achievable. The
chemistry of the [CpRu(PPh
3)
2]
⫹fragment can be traced to
the strongly π-basic nature of the ruthenium center.
Replac-ing Cp with Tp increases the basicity of the metal center
and reduces the cationic character of the vinylidene
com-plexes. Complexes 3b
⫺3e all lack enough cationic character
to lead to intramolecular cycloaddition. We thus believe
that the facile deprotonation of cationic vinylidene
com-plexes may be ascribed to the combined effect of the
cat-ionic character and the presence of the
electron-with-drawing substituent of the vinylidene complex.
Scheme 2
The analogous Cp complex of 4a is stable with respect to
ligand substitution — the phosphane ligand binds strongly
to the ruthenium center, making the coordination site
un-available for an incoming substrate. In contrast, the Tp
complex 4a is susceptible to ligand substitution under
rela-tively mild conditions. This may be attributed to the
in-creased steric bulkiness of the Tp ligand relative to Cp. For
example, when 2 equiv. of PhCN, p-CF
3C
6H
4CN, nBuNC
or tBuNC were added at room temperature to a CH
2Cl
2solution of 4a a smooth reaction ensued over 1 h which
afforded good yields of the bright-yellow
(cyclopropenyl)ru-thenium complexes [Ru{C
⫽C(Ph)CH(CN)}(Tp)(PPh
3)(L)]
(5a, L
⫽ nBuNC; 5b, L ⫽ tBuNC; 5c, L ⫽ p-CF
3C
6H
4CN;
5d, L
⫽ PhCN), respectively (Scheme 2). Significantly,
when these reactions were repeated using only 1 equiv. of L
much lower yields (ca. 8%) were obtained.
Complexes 5a
⫺5d all contain two diastereomers in a 4:1
ratio. The
1H NMR resonances of 5a attributed to the
CHCN moiety of the three-membered rings of the major
and the minor isomers appear at δ
⫽ 0.93 and 1.16 ppm,
respectively. In the
13C{
1H} NMR spectrum, singlet
13C
resonances at δ
⫽ 3.8 and 3.5 ppm and doublet resonances
at δ
⫽ 128.7 and 128.8 ppm, with
2J
P,C
coupling constants
of 11.6 and 11.5 Hz, are assigned to the CHN and the
ru-thenium-bonded C-α carbon atoms of the major and the
minor isomers, respectively. Interestingly, in the cases of 5c
and 5d the major isomer is more stable than the minor
iso-mer; the minor isomer decomposes within about 3 h.
[6b]In
the
13C{
1H} NMR spectrum of 5c, the singlet resonance at
δ
⫽ 4.10 ppm and the doublet resonance at δ ⫽ 132.6 ppm,
with a
2J
P,C
coupling of 12.3 Hz, are assigned to the CHN
and the ruthenium-bonded C-α carbon atoms of the major
isomer; the
13C NMR spectrum of the minor isomer was
not obtained because of its lower stability. Complexes 5a
and 5b are stable in diethyl ether and THF, but in CHCl
3compounds 5b, 5c, 5d are less stable than 5a. Furthermore,
5b decomposes in CHCl
3producing [RuCl(Tp)(PPh
3)-(tBuNC)] and some unidentified organic products.
De-composition of 5c and 5d produces complicated mixtures.
The stability of the substituted cyclopropenyl complexes
was found to decrease in the order nBuNC
⬎ tBuNC ⬎
p-CF
3C
6H
4CN
⬎ PhCN.
Opening of the Three-Membered Ring by Electrophiles
Treatment of 4a with I
2at 0
°C afforded the cationic
vinylidene
complex
[Ru{
⫽C⫽C(Ph)CH(I)(CN)}(Tp)-(PPh
3)
2]I (6) in 79% yield (Scheme 2). Complex 6 is a green
solid and its spectroscopic data display the features of a
vinylidene complex. The pattern of two-doublet resonances
at δ
⫽ 34.3 and 33.6 ppm, with a J
P,Pcoupling of 26.9 Hz,
in the
31P NMR spectrum arises from the stereogenic C-γ
center. In the
1H NMR spectrum of 6 the resonance at δ
⫽
3.23 ppm is assigned to the CHICN group, and in the
13C{
1H} spectrum the triplet resonance at δ
⫽ 374.5 ppm,
with a
2J
P,C
coupling of 15.1 Hz, is assigned to the
vinyl-idene C-α atom. Similarly, treatment of 5a with I
2affords
the addition product [Ru{
⫽C⫽C(Ph)CH(I)(CN)}(Tp)-(PPh
3)(nBuNC)]I (7) in high yield. Interestingly, only one
diastereoisomer is observed for 7. The
1H NMR spectrum
of 7 displays one singlet resonance at δ
⫽ 3.56 ppm,
as-signed to the CHICN group; the doublet resonance at δ
⫽
367.3 ppm, with a
2J
P,C
coupling of 16.4 Hz, in the
13C
NMR spectrum is assigned to the vinylidene C-α atom.
Formation of these vinylidene complexes occurs by selective
cleavage of the cyclopropenyl single bond near the metal
center. No alkylation is observed when 4a is treated with
CH
3I, CH
3CH
2I, CH
2⫽CHCH
2Br, CH
⬅CHCH
2Br or
ICH
2CN.
Reaction of 4a with nBuNC in the Presence of MeOH
Reaction of 4a with nBuNC in the presence of MeOH
causes substitution of a phosphane ligand by nBuNC.
This is followed by protonation of the three-membered
ring to give a vinylidene intermediate and addition
of MeO
⫺to give the vinyl ether complex [Ru{C(OMe)
⫽
C(Ph)CH
2CN}(Tp)(PPh
3)(nBuNC)]
(8a).
Similarly,
the
reaction
of
4a
with
tBuNC
in
the
presence
of
MeOH gives the vinyl ether product [Ru{C(OMe)
⫽
C(Ph)CH
2CN}(Tp)(PPh
3)(tBuNC)] (8b) in lower yield,
which may be due to steric effects. The
1H NMR spectrum
of 8a displays an AB pattern for the CH
2CN moiety, with
two doublets centered at δ
⫽ 3.86 and 2.79 ppm with a
coupling constant of 16.4 Hz. The characteristic
13C{
1H}
NMR spectroscopic features of 8a and 8b comprise
down-field resonances at δ
⫽ 179.1 and 182.2 ppm, respectively,
assignable to the vinyl C-α atom. In the absence of MeOH
these reactions give the simple substitution products 5a and
5b, respectively. The reaction proceeds with substitution as
the first step, followed by opening of the cyclopropenyl ring
by MeOH to give the vinylidene ligand. This was confirmed
by a separate experiment where 5 was allowed to react with
MeOH, resulting in protonation followed by a nucleophilic
attack of methoxide at C-α of the vinylidene ligand to give
the final product. In our previous paper we reported that
MeOH is able to open three-membered rings in some
cases.
[6]Concluding Remark
The (cyclopropenyl)ruthenium complex 4a, containing a
Tp ligand, has been prepared by deprotonation of the
vinyl-idene precursor 3a. No deprotonation was observed in the
reaction of 3b
⫺3e with NaOMe or nBu
4NOH which may
be attributed to the reduced cationic character of vinylidene
complexes caused by the presence of the Tp ligand. Unlike
its Cp analogue, complex 4a undergoes a facile phosphane
substitution reaction with several two-electron donor
mol-ecules. This property was taken advantage of to prepare
no-vel complexes of Ru. For example, nBuNC readily displaces
one of the phosphane ligands of 4a to give a mixture of
diastereomers 5 in a 4:1 ratio. Reaction of 4a with nBuNC
in the presence of MeOH gives the vinyl ether product 8a,
which results from a displacement reaction followed by
nu-cleophilic addition of the MeO
⫺group at C-α.
Experiment Section
General Procedures: All manipulations were performed under
nitro-gen using vacuum-line, drybox, and standard Schlenk techniques. CH3CN and CH2Cl2were distilled from CaH2and diethyl ether
and THF from Na/benzophenone ketyl. All other solvents and reagents were of reagent grade and were used without further puri-fication. NMR spectra were recorded with Bruker AC-200 and AM-300WB FT NMR spectrometers at room temperature (unless stated otherwise) and are reported in δ units with residual protons in the solvent as an internal standard [CDCl3: δ ⫽ 7.24 ppm;
CD3CN: δ⫽ 1.93 ppm; CD3C(O)CD3: δ⫽ 2.04 ppm]. FAB mass
spectra were recorded with a JEOL SX-102A mass spectrometer. Elemental analyses and X-ray diffraction studies were carried out at the Regional Center of Analytical Instrument at the National Taiwan University. The complexes [RuCl2(PPh3)3][20]and 4a[6]were
prepared according to literature methods.
Synthesis of [RuCl2(C3H3NNH)2(PPh3)2] (1): Pyrazole (0.38 g, 5.60 mmol) was added to a solution of [RuCl2(PPh3)3] (2.45 g,
2.80 mmol) in 20 mL of THF, and the reaction mixture was heated to 60°C for 1 h. After cooling and removal of the solvent under reduced pressure, the residue was dissolved in CH2Cl2. Addition of
hexane afforded a bright yellow precipitate, which was filtered off, washed with hexane and dried under vacuum to give 1 (2.12 g, 91% yield).1H NMR (CDCl 3): δ ⫽ 5.91 (t, JH,H⫽ 2.3 Hz, 3 H, Tp), 7.32⫺6.91 (m, Ph, Tp), 8.09 (d, JH,H⫽ 2.1 Hz, 3 H, Tp), 11.67 (s, 2 H, NH) ppm. 31P NMR (CDCl 3): δ⫽ 40.8 ppm. MS (FAB): m/z⫽ 832.1 [M⫹], 764.2 [M⫹⫺ HPz], 502.2 [M⫹⫺ HPz ⫺ PPh3]. C42H38BCl2N4P2Ru (832.68): calcd. C 60.58, H 4.60, N 6.73; found C 60.46, H 4.54, N 6.65.
Preparation of [Ru(C⬅CⴚPh)(Tp)(PPh3)2] (2): An excess of phen-ylacetylene (4.17 mL, 36.1 mmol) and NEt3 (6.3 mL, 36.1 mmol)
were added to 50 mL of an MeOH solution of 1 (3.00 g, 3.61 mmol), and the solution was heated to reflux for 90 min. The yellow precipitate thus formed was filtered off and washed with MeOH and hexane. The product was dried under vacuum and was subsequently identified as compound 2 (3.43 g, 97% yield). Yellow crystals of 2 were obtained by slow diffusion of hexane into a CHCl3solution of 2 at room temperature.1H NMR (CDCl3): δ⫽
5.20 (d, JH,H⫽ 1.8 Hz, 1 H, Tp), 5.31 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 5.54 (t, JH,H⫽ 2.3 Hz, 2 H, Tp), 7.24⫺6.91 (m, Ph, Tp), 7.40 (d, JH,H⫽ 2.0 Hz, 2 H, Tp), 7.58 (d, JH,H⫽ 2.1 Hz, 1 H, Tp) ppm.13C NMR [CD3C(O)CD3]: δ⫽ 122.7 (C-β), 135.8 (t, JC,P⫽ 12.3 Hz, C-α), 145.7⫺127.2 (Ph, Tp) ppm.31P NMR (CDCl 3): δ⫽ 48.6 ppm. MS (FAB): m/z⫽ 940.1 [M⫹], 678.1 [M⫹⫺ PPh3], 577.1 [M⫹⫺ PPh3⫺ C2Ph], 363.0 [M⫹⫺ PPh3⫺ C2Ph⫺ Tp]. C53H44BN6P2Ru (938.75): calcd. C 67.81, H 4.72, N 8.95; found C 67.94, H 4.59, N 8.91. Synthesis of [Ru{ⴝCⴝC(Ph)CH2(CHⴝCH2)}(Tp)(PPh3)2]I (3b): ICH2⫽CH2 (0.46 mL, 3.5 mmol) was added to a Schlenk flask
charged with complex 2 (1.41 g, 1.50 mmol) in 50 mL of CH2Cl2.
The clear solution was stirred for 16 h and then the volume of solvent was reduced to about 5 mL. This mixture was slowly added to 90 mL of vigorously stirred diethyl ether. The green precipitate thus formed was filtered off and washed with diethyl ether and hexane to give compound 3b (1.33 g, 77%).1H NMR (CDCl
3): δ⫽ 3.05 (d, JH,H⫽ 5.5 Hz, 2 H, CH2), 4.95 (dd, JH,H⫽ 15.3, 2.7 Hz, 1 H, ⫽CH), 5.05 (dd, JH,H⫽ 10.3, 2.7 Hz, 1 H, ⫽CH), 5.38 (d, JH,H⫽ 1.9 Hz, 1 H, Tp), 5.53 (m, 1 H, CH⫽), 5.56 (t, JH,H ⫽ 1.9 Hz 1 H, Tp), 5.61 (t, JH,H⫽ 2.1 Hz, 2 H, Tp), 6.59⫺7.42 (m, Ph), 7.57 (d, JH,H⫽ 2.2 Hz, 2 H, Tp), 7.84 (d, JH,H⫽ 1.9 Hz 1 H, Tp) ppm.13C NMR (CDCl 3): δ⫽ 14.0 (CH2), 105.8 (⫽CH2), 118.4 (C-β), 105.9⫺146.2 (m, Ph, Tp), 153.8 (⫽CH), 377.5 (t, JC,P ⫽ 16.3 Hz, C-α) ppm.31P NMR (CDCl 3): δ⫽ 37.8 ppm. MS (FAB): m/z⫽ 981.3 [M⫹⫺ I], 719.3 [M⫹⫺ I ⫺ PPh3], 577.1 [M⫹⫺ I ⫺ PPh3⫺ C2PhCH2CH⫽CH2]. C56H50BIN6P2Ru (1107.7): calcd. C 60.71, H 4.55, N 7.59; found C 60.62, H 4.43, N 7.63.
[Ru{ⴝCⴝC(Ph)CH2CHⴝC(Me)2}(Tp)(PPh3)2]Br (3c): Yield 1.36 g, in 83%; prepared in a similar manner from 1.43 g (1.52 mmol) of 2 and excess BrCH2CH⫽C(Me)2 (3.7 mmol) at room
temperature.1H NMR (CDCl 3): δ⫽ 1.18 (s, 3 H, Me), 1.62 (s, 3 H, Me), 3.12 (d, JH,H⫽ 5.1 Hz, 2 H, CH2), 4.90 (m, 1 H, CH⫽), 5.42 (t, JH,H⫽ 1.9 Hz, 1 H, Tp), 5.54 (t, JH,H⫽ 2.2 Hz, 2 H, Tp), 6.41 (d, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.45 (d, JH,H⫽ 2.0 Hz, 2 H, Tp), 6.96⫺7.42 (m, Ph) 7.66 (d, JH,H⫽ 2.0 Hz, 2 H, Tp), 7.83 (d, JH,H⫽ 2.1 Hz, 1 H, Tp) ppm.13C NMR (CDCl 3): δ⫽ 18.1 (CH2), 21.2 (Me), 25.6 (Me), 118.3 (C-β), 104.3⫺146.5 (Ph, Tp), 379.8 (t, JC,P⫽ 15.8 Hz, C-α) ppm.31P NMR (CDCl3): δ⫽ 37.9 ppm. MS (FAB): m/z⫽ 1009.3 [M⫹⫺ Br], 747.3 [M⫹⫺ Br ⫺ PPh3], 577.1 [M⫹ ⫺ Br ⫺ PPh3⫺ C2PhCH2CH⫽CMe2]. C58H54BBrN6P2Ru (1088.8): calcd. C 63.98, H 5.00, N 7.72; found C 63.87, H 4.91, N 7.81. [Ru{ⴝCⴝC(Ph)CH2Ph}(Tp)(PPh3)2]I (3d): Yield 1.45 g, in 87%; prepared in a similar manner from 1.50 g (1.59 mmol) of 2 and excess BrCH2Ph (0.40 mL, 3.0 mmol) at room temperature. 1H
NMR (CDCl3): δ⫽ 3.90 (s, 2 H, CH2), 5.29 (br, 1 H, Tp), 5.34 (br, 1 H, Tp), 5.39 (br, 1 H, Tp), 5.54 (t, JH,H⫽ 2.0 Hz, 2 H, Tp), 7.73⫺6.59 (m, Ph, Tp), 7.86 (d, JH,H⫽ 2.1 Hz, 1 H, Tp) ppm.13C NMR (CDCl3): δ⫽ 16.0 (CH2), 147.3⫺108.4 (Ph, Tp), 378.5 (t, JC,P⫽ 15.8 Hz, C-α) ppm.31P NMR (CDCl3): δ⫽ 37.4 ppm. MS (FAB): m/z⫽ 1031.4 [M⫹⫺ Br], 769.5 [M⫹⫺ Br ⫺ PPh3], 577.1 [M⫹⫺ Br ⫺ PPh3⫺ C2PhCH2Ph]. C60H52BBrN6P2Ru (1110.8): calcd. C 64.87, H 4.72, N 7.57; found C 64.91, H 4.67, N 7.41.
Preparation of [Tp(PPh3)2RuⴝCⴝC(Ph)CH2COOMe]Br (3e): A mixture of complex 2 (2.80 g, 3.10 mmol) and BrCH2COOMe
(0.5 mL, 5.10 mmol) in 40 mL of CH2Cl2/CH3Cl (3:1) was heated
to reflux for 6 h. The workup procedure was the same as that for
3d. Purification by recrystallization from CH2Cl2/hexane (1:5) gave
3e (1.45 g, 87% yield).1H NMR (CDCl 3): δ⫽ 3.10 (s, 2 H, CH2), 3.59 (s, 3 H, OMe), 5.45 (br, 1 H, Tp), 5.53 (br, 1 H, Tp), 5.61 (br, 1 H, Tp), 5.77 (t, JH,H⫽ 2.0 Hz, 2 H, Tp), 6.32 (br, 2 H, Tp), 7.57⫺6.59 (m, Ph, Tp), 7.65 (d, JH,H⫽ 1.9 Hz, 2 H, Tp), 7.79 (d, JH,H⫽ 2.0 Hz, 1 H, Tp) ppm.13C NMR (CDCl3): δ⫽ 19.1 (CH2), 57.1 (CH3), 106.4⫺145.6 (Ph, Tp), 171.4 (COO), 378.5 (t, JC,P⫽ 15.6 Hz, C-α) ppm.31P NMR (CDCl 3): δ⫽ 38.4 ppm. MS (FAB): m/z⫽ 1013.3 [M⫹⫺ Br], 751.2 [M⫹⫺ Br ⫺ PPh3], 577.4 [M⫹⫺ Br ⫺ PPh3⫺ C2PhCH2COOMe]. C56H50BBrN6O2P2Ru (1092.7): calcd. C 61.55, H 4.61, N 7.69; found C 61.61, H 4.51, N 7.82.
Synthesis of 5a: Complex 4a (0.50 g, 0.51 mmol) was dissolved in
CH2Cl2 (20 mL), and nBuNC (0.05 mL, 0.51 mmol) was added.
The mixture was stirred for 50 min to afford a bright-yellow solu-tion. The solvent was then removed under vacuum and the solid residue was extracted with 20 mL of diethyl ether. The extract was filtered through Celite, and the filtrate was dried to give a bright-yellow solid, which was washed with hexane (2⫻ 10 mL), and dried under vacuum to give a mixture of diastereoisomers of 5a (0.327 g, 80.3% yield).1H NMR [CD
3C(O)CD3]: major isomer: δ⫽ 0.75 (t,
JH,H⫽ 7.5 Hz, 3 H, CH3), 0.93 (s, 1 H, CH), 1.23 (m, 2 H, CH2), 1.53 (m, 2 H, CH2), 3.72 (t, JH,H ⫽ 6.6 Hz, 2 H, CH2), 5.88 (t, JH,H⫽ 2.2 Hz, 1 H, Tp), 5.96 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.01 (t, JH,H⫽ 1.8 Hz, 1 H, Tp), 6.76 (br, 1 H, Tp), 6.78 (br, 1 H, Tp), 6.86 (br, 1 H, Tp), 7.69 (d, JH,H⫽ 2.0 Hz, 1 H, Tp), 7.07⫺7.40 (m, Ph), 7.81 (d, JH,H⫽ 2.0 Hz, 1 H, Tp), 7.88 (d, JH,H⫽ 2.1 Hz, 1 H, Tp) ppm; minor isomer: δ⫽ 0.77 (t, JH,H⫽ 7.4 Hz, 3 H, CH3), 1.16 (s, 1 H, CHCN), 1.26 (m, 2 H, CH2), 1.55 (m, 2 H, CH2), 3.84 (t, JH,H⫽ 6.7 Hz, 2 H, CH2), 6.14 (t, JH,H⫽ 2.2 Hz, 1 H, Tp), 6.21 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.34 (t, JH,H⫽ 2.0 Hz, 1 H, Tp), 6.76 (br, 1 H, Tp), 6.94 (br, 2 H, Tp), 7.40⫺7.07 (m, Ph), 7.71 (d, JH,H⫽ 2.1 Hz, 1 H, Tp), 7.92 (d, JH,H⫽ 1.9 Hz, 1 H, Tp), 8.12 (d, JH,H⫽ 2.0 Hz, 1 H, Tp) ppm.13C NMR [CD3C(O)CD3]: major isomer: δ ⫽ 3.8 (CH), 13.5 (CH3), 20.2 (CH2), 32.3 (CH2), 44.8 (CH2), 116.3 (CN), 128.7 (d, JC,P⫽ 11.6 Hz, C-α), 146.9 ⫺129.3 (Ph, Tp), 160.1 (d, JC,P⫽ 22.5 Hz, CN) ppm; minor isomer: δ ⫽ 3.5 (CH), 13.5 (CH3), 20.2 (CH2), 32.2 (CH2), 44.7 (CH2), 115.2 (CN), 128.8 (d, JC,P⫽ 11.6 Hz, C-α), 129.3⫺146.9 (Ph, Tp), 163.2 (d, JC,P⫽ 21.3 Hz, CN) ppm.31P NMR [CD3C(O)CD3]: δ⫽ 54.1, 52.7 (4:1) ppm. MS (FAB): m/z ⫽ 800.3 [M⫹], 660.3 [M⫹ ⫺ C2PhCHCN], 577.2 [M⫹ ⫺ C2PhCHCN ⫺ nBuNC].
C42H40BN8PRu (799.65): calcd. C 63.08, H 5.04, N 14.01; found C
62.98, H 4.96, N 13.89.
Synthesis of 5b: Complex 4a (1.01 g, 1.03 mmol) was dissolved in
The mixture was stirred at room temperature to afford a bright-yellow solution. The solvent was then removed under vacuum, and the solid residue was extracted with 20 mL of diethyl ether. The extract was filtered, and the filtrate was dried to give a bright-yellow solid, which was washed with hexane (2⫻ 10 mL) and dried under vacuum to give a diastereomeric mixture of 5b (0.62 g, 79.1% yield).1H NMR [CD
3C(O)CD3]: major isomer: δ⫽ 0.96 (s, 1 H,
CH), 1.47 (s, 9 H, Me), 5.81 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 5.97 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.11 (t, JH,H⫽ 1.9 Hz, 1 H, Tp), 6.71 (br, 1 H, Tp), 6.74 (br, 1 H, Tp), 6.83 (br, 1 H, Tp), 7.46⫺7.07 (m, Ph), 7.73 (d, JH,H⫽ 2.1 Hz, 1 H, Tp), 7.80 (d, JH,H⫽ 2.2 Hz, 1 H, Tp), 7.82 (d, JH,H⫽ 2.0 Hz, 1 H, Tp) ppm; minor isomer: δ ⫽ 1.08 (s, 1 H, CH), 1.48 (s, 9 H, Me), 6.13 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.20 (t, JH,H⫽ 2.0 Hz, 1 H, Tp), 6.32 (t, JH,H⫽ 2.2 Hz, 1 H, Tp), 6.72 (br, 1 H, Tp), 6.99 (br, 2 H, Tp), 7.42⫺7.17 (m, Ph), 7.67 (d, JH,H⫽ 2.0 Hz, 1 H, Tp), 7.92 (d, JH,H⫽ 2.1 Hz, 1 H, Tp),8.11 (d, JH,H⫽ 2.0 Hz, 1 H, Tp) ppm.13C NMR [CD3C(O)CD3]: major
isomer: δ⫽ 3.8 (CH), 32.1 (CMe3), 58.7 (CMe3), 119.2 (CN), 126.5
(d, JC,P⫽ 12.1 Hz, C-α), 141.9 ⫺129.3 (Ph, Tp), 163.4 (d, JC,P⫽
21.4 Hz, CN) ppm; minor isomer: δ⫽ 3.5 (CH), 31.2 (Me3), 55.3
(CMe3), 118.9 (CN), 127.1 (d, JC,P⫽ 11.2 Hz, C-α), 129.3⫺146.9
(Ph, Tp), 164.1 (d, JC,P ⫽ 20.1 Hz, CN) ppm. 31P NMR
[CD3C(O)CD3]: δ⫽ 52.9, 53.9 (4:1) ppm. MS (FAB): m/z ⫽ 801.1
[M⫹⫹ 1], 660.3 [M⫹⫺ C2PhCHCN], 577.2 [M⫹⫺ C2PhCHCN
⫺ tBuNC]. C42H40BN8PRu (799.65): calcd. C 63.08, H 5.04, N
14.01; found C 63.12, H 5.10, N 13.97.
Synthesis of 5c: An excess of PhCN (0.21 mL, 2.02 mmol) was
ad-ded to a solution of 4a (1.00 g, 1.02 mmol) in 20 mL of CH2Cl2.
The solution was stirred at room temperature (the color changed from yellow to brown) and then the solvent was removed under vacuum. The solid residue was extracted with diethyl ether, and the extract was filtered. The volume of the resulting solution was re-duced to 5 mL and 40 mL of hexane was added to form an orange precipitate, which was filtered and washed twice with 10 mL of hex-ane. The product was dried under vacuum (0.60 g, 72% yield).1H
NMR [CD3C(O)CD3]: major isomer: δ ⫽ 1.13 (s, 1 H, CHCN),
5.90 (t, JH,H⫽ 2.0 Hz, 1 H, Tp), 6.02 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.06 (br, 1 H, Tp), 6.78 (br, 1 H, Tp), 6.97 (br, 1 H, Tp), 7.69⫺7.03 (m, Ph),7.91 (d, JH,H⫽ 2.2 Hz, 2 H, Tp) ppm; minor isomer: δ ⫽ 0.79 (s, 1 H, CH), 5.92 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.01 (t, JH,H⫽ 1.9 Hz, 1 H, Tp), 6.05 (d, JH,H⫽ 2.0 Hz, 2 H, Tp), 6.43 (d, JH,H⫽ 1.9 Hz, 2 H, Tp), 6.96 (d, JH,H⫽ 2.1 Hz, 2 H, Tp), 7.64 ⫺7.01 (m, Ph), 7.93 (d, JH,H ⫽ 2.0 Hz, 2 H, Tp) ppm. 13C NMR
[CD3C(O)CD3] major isomer: δ⫽ 4.1 (CH), 116.1 (NCPh), 119.2
(CN), 132.6 (d, JC,P⫽ 12.3 Hz, C-α), 147.9 ⫺123.1 (Ph, Tp) ppm. 31P NMR [CD 3C(O)CD3]: δ⫽ 54.5, 54.8 (4:1) ppm. MS (FAB): m/z⫽ 821.4 [M⫹⫹ 1], 718.4 [M⫹⫹ 1 ⫺ PhCN], 577.1 [M⫹⫹ 1 ⫺ PhCN ⫺ C2PhCHCN]. C44H36BN8PRu (819.64): calcd. C 64.47, H 4.43, N 13.69; found C 64.49, H 4.44, N 13.63.
Synthesis of 5d: An excess of CF3C6H4CN (0.14 mL, 2.04 mmol)
was added to a solution of 4a (1.00 g, 1.02 mmol) in 20 mL of CH2Cl2. The solution was stirred for 50 min (the color changed
from yellow to brown) and then the solvent was removed under vacuum. The solid residue was extracted with diethyl ether, and the extract was filtered. The volume of the resulting solution was re-duced to 5 mL and 40 mL of hexane was added to form a orange precipitate, which was filtered and washed twice with 10 mL of hex-ane. The product was dried under vacuum (0.70 g, 77% yield).1H
NMR [CD3C(O)CD3] major isomer: δ⫽ 1.08 (s, 1 H, CH), 5.85
(t, JH,H⫽ 2.2 Hz, 1 H, Tp), 5.98 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.02 (t, JH,H⫽ 1.8 Hz, 1 H, Tp), 6.76 (br, 1 H, Tp), 6.78 (br, 1 H, Tp), 6.86 (br, 1 H, Tp), 7.07⫺7.40 (m, Ph), 7.69 (d, JH,H⫽ 2.0 Hz, 1 H, Tp), 7.81 (d, JH,H⫽ 2.2 Hz, 1 H, Tp), 7.85 (d, JH,H⫽ 2.2 Hz, 1 H, Tp) ppm; minor isomer: δ⫽ 0.82 (s, 1 H, CH), 5.91 (t, JH,H⫽ 2.0 Hz, 1 H, Tp), 6.05 (t, JH,H⫽ 1.9 Hz, 1 H, Tp), 6.07 (d, JH,H⫽ 2.1 Hz, 2 H, Tp), 6.46 (d, JH,H⫽ 2.0 Hz, 2 H, Tp), 6.89 (d, JH,H⫽ 2.0 Hz, 2 H, Tp), 7.01⫺7.64 (m, Ph), 7.97 (d, JH,H⫽ 2.1 Hz, 2 H, Tp) ppm.13C NMR [CD
3C(O)CD3]: major isomer: δ⫽ 5.1 (CH),
110.6 (q, JC,F⫽ 282.0 Hz, CF3), 111.2 (NCPh), 118.1 (CN), 131.7 (d, JC,P⫽ 11.9 Hz, C-α), 148.2⫺126.6 (Ph, Tp) ppm. 31P NMR [CD3C(O)CD3]: δ⫽ 53.6, 54.3 (4:1) ppm. MS (FAB): m/z ⫽ 888.4 [M⫹ ⫹ 1], 718.4 [M⫹ ⫹ 1 ⫺ CF3C6H4CN], 577.1 [M⫹ ⫹ 1 ⫺ CF3C6H4CN⫺ C2PhCHCN]. C45H35BF3N8PRu (887.63): calcd. C 60.88, H 3.97, N 12.62; found C 60.78, H 4.08, N 12.51.
Synthesis of [Ru{ⴝCⴝC(Ph)CH(I)CN}(Tp)(PPh3)2]I (6): CH2Cl2
(30 mL) was added to a solid mixture of 4a (0.51 g, 0.52 mmol) and I2(0.13 g, 0.52 mmol) at 0 °C. The mixture was stirred for
2 min whereupon the color changed from yellow to green; the sol-vent was then removed under vacuum. The residual solid was ex-tracted twice with 20 mL of diethyl ether and, after filtration, the solvent was removed under vacuum to give complex 6 (0.45 g, 69% yield).1H NMR [CD 3C(O)CD3]: δ⫽ 3.23 (s, 1 H, CH), 5.43 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 5.55 (t, JH,H⫽ 2.0 Hz, 1 H, Tp), 5.76 (d, JH,H⫽ 2.0 Hz, 1 H, Tp), 5.65 (d, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.79 (d, JH,H⫽ 1.9 Hz 1 H, Tp), 7.56⫺7.11 (m, Tp, Ph), 7.73 (m, 2 H, Tp), 7.98 (d, JH,H⫽ 2.1 Hz, 1 H, Tp) ppm.13C NMR (CDCl3): δ⫽ 26.1 (CH), 119.4 (CN), 106.8⫺147.2 (Ph, Tp, C-β), 374.5 (t, JC,P⫽ 15.1 Hz, C-α) ppm. 31P NMR [CD3C(O)CD3]: δ⫽ 33.6, 34.3 (AB, JP,P⫽ 26.9 Hz) ppm. MS (FAB): m/z ⫽ 1107.1 [M⫹⫺ I], 980.3 [M⫹⫺ 2I], 839.2 [M⫹⫺ 2 I ⫺ C2PhCHCN], 577.1 [M⫹ ⫺ 2 I ⫺ C2PhCHCN⫺ PPh3]. C55H47BI2N7P2Ru (1233.6): calcd. C 53.55, H 3.84, N 7.95; found C 53.46, H 4.01, N 8.03.
Synthesis of [Ru{ⴝCⴝC(Ph)CH(I)CN}(Tp)(PPh3)(nBuNC)]I (7):
CH2Cl2 (30 mL) was added to a solid mixture of 5a (0.17 g,
0.21 mmol) and I2(0.054 g, 0.17 mmol). The mixture was stirred
for 5 min and the solvent was then removed under vacuum. The residual solid was extracted twice with 20 mL of diethyl ether and, after filtration, the solvent was removed under vacuum to give 7 (0.14 g, 78% yield).1H NMR [CD 3C(O)CD3]: δ⫽ 0.73 (t, JH,H⫽ 7.4 Hz, 3 H, CH3), 1.03 (m, 2 H, CH2), 1.94 (m, 2 H, CH2), 3.46 (t, JH,H⫽ 6.2 Hz, 2 H, CH2), 3.56 (s, 1 H, CH), 5.96 (t, JH,H⫽ 2.3 Hz, 1 H, Tp), 6.10 (t, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.36 (d, JH,H⫽ 2.1 Hz, 1 H, Tp), 6.43 (d, JH,H⫽ 2.1 Hz, 1 H, Tp), 7.39⫺7.01 (m, Tp, Ph), 7.67 (d, JH,H⫽ 2.1 Hz, 1 H, Tp), 7.69 (d, JH,H⫽ 2.3 Hz, 1 H, Tp), 7.75 (d, JH,H ⫽ 2.1 Hz, 1 H, Tp) ppm. 13C NMR [CD3C(O)CD3]: δ ⫽ 14.2 (CH3), 20.0 (CH2), 25.4 (CH), 35.2 (CH2), 44.1 (CH2), 126.3 (CN), 108.4⫺148.9 (Ph, Tp), 167.2 (d, JC,P ⫽ 22.9 Hz, CN), 367.3 (d, JC,P⫽ 16.4 Hz, C-α) ppm. 31P NMR [CD3C(O)CD3]: δ⫽ 45.9 ppm. MS (FAB): m/z ⫽ 926.3 [M⫹ ⫺ I], 800.1 [M⫹⫺ 2 I], 660.2 [M⫹⫺ 2 I ⫺ C 2PhCHCN], 577.2 [M⫹⫺ 2 I ⫺ C2PhCHCN⫺ nBuNC]. C42H40BI2N8PRu (1053.5): calcd. C 47.88, H 3.83, N 10.64; found C 47.81, H 4.02, N 10.70.
Synthesis of [Ru{C(OMe)ⴝC(Ph)CH2CN}(Tp)(PPh3)(nBuNC)]
(8a): A solution of 4a (1.50 g, 1.53 mmol) was dissolved in
meth-anol and nBuNC (0.30 mL, 3.06 mmol) was added. After stirring for 10 min, the yellow solution became bright yellow. The solution was filtered through Celite and the solvent was removed under vac-uum to give 8a (1.01 g, 85% yield).1H NMR [CD
3C(O)CD3]: δ⫽ 0.70 (t, JH,H⫽ 7.5 Hz, 3 H, CH3), 0.86 (m, 2 H, CH2), 1.88 (m, 2 H, CH2), 2.79 (d, JH,H⫽ 16.4 Hz, 1 H, CH), 3.63 (t, JH,H⫽ 6.4 Hz, 2 H, CH2), 3.86 (d, JH,H⫽ 16.4 Hz, 1 H, CHH), 5.63 (t, JH,H⫽ 1.8 Hz, 1 H, Tp), 6.05 (br, 1 H, Tp), 6.11 (br, 1 H, Tp), 7.00 (br, 1 H, Tp), 7.60⫺7.02 (m, Ph, Tp), 7.81 (d, JH,H⫽ 2.0 Hz, 1 H, Tp) ppm.13C NMR [CD 3C(O)CD3]: δ⫽ 13.5 (CH3), 21.1 (CH2), 22.4
(CH2), 31.1 (CH2), 43.1 (CH2), 55.4 (OMe), 115.5 (CN), 123.4⫺147.6 (Ph), 162.1 (d, JC,P⫽ 23.1 Hz, CN),179.1 (d, JC,P⫽ 15.3 Hz, C-α) ppm.31P NMR [CD 3C(O)CD3]: δ⫽ 46.9 ppm. MS (FAB): m/z⫽ 832.3 [M⫹], 660.1 [M⫹⫺ (OMe)C⫽C(Ph)(CH2CN)], 577.1 [M⫹⫺ (OMe)C⫽C(Ph)(CH2CN)⫺ nBuNC]. C43H44BN8
O-PRu (831.69): calcd. C 62.08, H 5.33, N 13.47; found C 62.16, H 5.40, N 13.35.
Synthesis of [Ru{C(OMe)ⴝC(Ph)CH2CN}Tp(PPh3)(tBuNC)] (8b): Complex 4a (0.5 g, 0.51 mmol) was dissolved in CH3OH (20 mL),
and tBuNC (0.30 mL, 3.02 mmol) was added. The mixture was stirred for 50 min to afford a bright-yellow solution. The solvent was then removed under vacuum and the solid residue was ex-tracted with diethyl ether. The extract was filtered, and the filtrate was dried under vacuum to give a bright-yellow solid, which was washed with hexane (2 ⫻ 10 mL), and dried to give 8b (0.327 g, 80.3% yield).1H NMR [CD
3C(O)CD3]: δ⫽ 1.44 (s, 9 H, CMe3),
2.84, 3.87 (2d, JH,H⫽ 15.3 Hz, 1 H, CH2CN), 5.61 (br, 1 H, Tp),
6.15 (br, 1 H, Tp), 6.21 (br, 1 H, Tp), 6.43 (br, 1 H, Tp), 6.96⫺7.56 (m, Ph, Tp), 7.78 (d, JH,H⫽ 2.2 Hz, 1 H, Tp) ppm. 13C NMR
[CD3C(O)CD3]: δ ⫽ 32.7 (CMe3), 55.4 (OMe), 113.2 (CN),
123.4⫺147.6 (Ph), 182.2 (d, JC,P⫽ 16.1 Hz, C-α) ppm.31P NMR
[CD3C(O)CD3]: δ⫽ 46.8 ppm. MS (FAB): m/z ⫽ 832.2 [M⫹], 660.1
[M⫹ ⫺ (OMe)C⫽C(Ph)(CH2CN)], 577.1 [M⫹ ⫺ (OMe)C⫽
C(Ph)(CH2CN) ⫺ tBuNC]. C43H44BN8OPRu (831.69): calcd. C
62.08, H 5.33, N 13.47; found C 62.11, H 5.34, N 13.51.
X-ray Diffraction Analysis: Dark-yellow crystals of 1 suitable for
an X-ray diffraction study were grown directly from CH2Cl2. A
suitable single crystal of dimensions 0.10⫻ 0.40 ⫻ 0.50 mm was glued to a glass fiber and mounted on a Nonius CD4 dif-fractometer. The data were collected using Mo-Kα radiation (at
298 K). The data were processed and the structure was solved and refined with the SHELXTL program. The structure was solved by direct methods and confirmed by Patterson methods. Hydrogen atoms were placed geometrically using the riding model with ther-mal parameters set to 1.2-times that of the atom to which they are attached (1.5-times for the methyl hydrogen atoms). Yellow crystals of 2 suitable for an X-ray diffraction study were obtained as above. A suitable single crystal of dimensions 0.05⫻ 0.10 ⫻ 0.15 mm was mounted on a SMART CCD diffractometer. The data were col-lected using 3-kW sealed-tube Mo-Kα radiation (at 298 K). The
exposure time was 5 s per frame. SADABS (Siemens area detector absorption) absorption correction was applied, and decay was neg-ligible. Data were processed with the SHELXTL program.[21]The
structure was solved by direct methods refining on intensities of all data. Hydrogen atoms were placed geometrically using the riding model. Crystal data for 1 and 2 are listed in Table 1.
Acknowledgments
We thank the National Science Council of Taiwan for financial support.
[1] [1a]S. Trofimenko, Chem. Rev. 1993, 93, 943.[1b]N. Kitajima,
W. B. Tolman, Prog. Inorg. Chem. 1995, 43, 419.[1c]C. Slugovc,
R. Schmid, K. Kirchner, Coord. Chem. Rev. 1999, 185/186, 109.
[1d] S. Hikichi, M. Akita, Y. Moro-oka, Coord. Chem. Rev.
2000, 198, 61.[1e]N. Marques, A. Sella, J. Takats, Chem. Rev.
2002, 102, 2137.
[2] M. D. Curtis, K. B. Shiu, W. M. Butler, Organometallics 1983,
2, 1475.
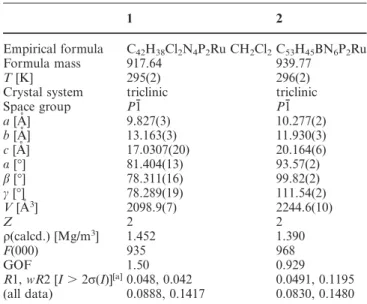
Table 1. Crystal data and structure refinement for 1 and 2
1 2
Empirical formula C42H38Cl2N4P2Ru CH2Cl2C53H45BN6P2Ru
Formula mass 917.64 939.77
T [K] 295(2) 296(2)
Crystal system triclinic triclinic
Space group P1¯ P1¯ a [A˚ ] 9.827(3) 10.277(2) b [A˚ ] 13.163(3) 11.930(3) c [A˚ ] 17.0307(20) 20.164(6) α [°] 81.404(13) 93.57(2) β [°] 78.311(16) 99.82(2) γ [°] 78.289(19) 111.54(2) V [A˚3] 2098.9(7) 2244.6(10) Z 2 2 ρ(calcd.) [Mg/m3] 1.452 1.390 F(000) 935 968 GOF 1.50 0.929 R1, wR2 [I⬎ 2σ(I)][a]0.048, 0.042 0.0491, 0.1195 (all data) 0.0888, 0.1417 0.0830, 0.1480 [a]R1⫽ Σ||F o|⫺ |Fc||/Σ|Fo|; wR2⫽ [Σw(Fo2⫺ Fc2)2/Σw(Fo2)2]1/2.
[3] [3a]Y. Fukumoto, H. Masaoka, T. Dohi, N. Chatani, S. Murai,
Organometallics 2002, 21, 19.[3b]Y. Wang, M. G. Finn, J. Am.
Chem. Soc. 1995, 117, 8045. [3c] C. Bruneau, P. H. Dixneuf,
Chem. Commun. 1997, 507.[3d]M. I. Bruce, Chem. Rev. 1991,
91, 197.[3e]B. M. Trost, Angew. Chem. 1995, 107, 285; Angew.
Chem. Int. Ed. Engl. 1995, 34, 259.[3f]C. Bianchini, M.
Peruz-zini, A. Romerosa, F. Zanobini, Organometallics 1995, 14, 3152. [3g] C. Bianchini, P. Innocenti, M. Peruzzini, A.
Ro-merosa, F. Zanobini, Organometallics 1996, 15, 272. [3h] H.
Werner, R. Wiedmann, P. Steinert, J. Wolf, Chem. Eur. J. 1997,
3, 127.[3i]B. M. Trost, J. A. Flygare, J. Am. Chem. Soc. 1992,
114, 5476.[3j]B. M. Trost, G. Dyker, R. Kulawiec, J. Am. Chem.
Soc. 1990, 112, 7809.
[4] [4a]C. A. Meric, M. E. Pauly, J. Am. Chem. Soc. 1996, 118,
11319.[4b]Y. Wakatsuki, H. Yamazaki, N. Kumegawa, T.
Sa-toh, J. Y. SaSa-toh, J. Am. Chem. Soc. 1991, 113, 9604.[4c]B. M.
Trost, G. Dyker, R. Kulawiec, J. Am. Chem. Soc. 1992, 114, 5476.
[5] [5a]M. I. Bruce, A. G. Swincer, R. C. Wallis, J. Organomet.
Chem. 1979, 171, C5.[5b]C. Bianchini, J. A. Casares, M.
Peruz-zini, A. Romerosa, F. Zanobini, J. Am. Chem. Soc. 1996, 118, 4585.
[6] [6a]P. C. Ting, Y. C. Lin, G. H. Lee, M. C. Cheng, Y. Wang, J.
Am. Chem. Soc. 1996, 118, 6433.[6b]Y. H. Lo, Y. C. Lin, G.
H. Lee, Y. Wang, Organometallics 1999, 18, 982.[6c]Y. C. Lin,
J. Organomet. Chem. 2001, 617⫺618, 141.[6d]C. C. Huang, Y.
C. Lin, S. L. Huang, Y. H. Liu, Y. Wang, Organometallics 2003,
22, 1512.
[7] [7a]G. M. Barreett, N. E. Carpenter, Organometallics 1987, 6,
2249.[7b] P. Normbel, N. Lugan, R. Mathieu, J. Organomet.
Chem. 1995, 503, C22.
[8] D. R. Senn, A. Wong, A. T. Patton, M. Marsi, C. E. Strouse,
J. A Gladysz, J. Am. Chem. Soc. 1988, 110, 6096.
[9] [9a]Special issue on strained organic compounds: Chem. Rev.
1989, 89.[9b] J. F. Liebman, A. Greenberg, Strained Organic
Molecules, Wiley, New York, 1978, p. 91.
[10] [10a]J. F. Liebman, A. Greenberg, Chem. Rev. 1976, 76, 311. [10b]B. Halton, M. G. Banwell, in The Chemistry of the
Cyclo-propnyl group, Part 2 (Eds.: S. Patai, Z. Rappoport), Wiley,
Chichester, 1987, chapter 21, p. 1223.
[11] [11a]R. Gompper, E. Bartmann, Angew. Chem. Int. Ed. Engl.
Hughes, J. Am. Chem. Soc. 1982, 104, 4842.[11c]R. P. Hughes,
W. A. Donaldson, J. Am. Chem. Soc. 1982, 104, 4846.
[12]R. Gompper, E. Bartmann, Angew. Chem. Int. Ed. Engl. 1985,
24, 209.
[13] [13a]U. Kirchassner, H. Piana, U. Schubert, J. Am. Chem. Soc.
1991, 113, 228.[13b]S. Miki, T. Ohno, H. Iwasaki, Z-I. Yoshida,
J. Phys. Org. Chem. 1998, 1, 333.
[14] [14a]R. R. Schrock, Acc. Chem. Res. 1986, 19, 342.[14b]R. P.
Hughes, W. J. Reisch, A. L. Rheingold, Organometallics 1985,
4, 1754.[14c]C. Mealli, S. Midollini, S. Moneti, L. Sacconi, J.
Silvestre, T. A. Albright, J. Am. Chem. Soc. 1982, 104, 59.
[15]T. Khan, N. H. Khan, R. I. Kureshy, K. Venkatasubramanian,
Polyhedron 1992, 4, 431.
[16] [16a]S. Trofimenko, Prog. Inorg. Chem. 1986, 34, 115.[16b]J. P.
Steel, Coord. Chem. Rev. 1990, 106, 227.[16c]J. E. Cosgriff, G.
B. Deacon, Angew. Chem. Int. Ed. 1998, 37, 286.[16d] G. B.
Deacon, E. E. Delbridge, B. W. Skelton, A. H. White, Angew.
Chem. Int. Ed. 1998, 37, 2251.
[17] [17a]G. Lo´pez, J. Ruiz, C. Vicent, J. M. Martı´, G. Garcı´a, P. A.
Chaloner, P. B. Hitchcock, R. M. Harrison, Organometallics
1992, 11, 4090.[17b]M. P. Garcı´a, M. A. Esteruelas, M. Martı´n,
L. A. Oro, J. Organomet. Chem. 1994, 467, 151.[17c]M. Cano,
J. A. Campo, J. V. Heras, J. Lafuente, C. Rivas, E. Pinilla,
Poly-hedron 1995, 14, 1139.
[18] [18a] J. A. Cowan, Inorganic Biochemistry, An Introduction,
VCH, New York, 1993.[18b]N. Kitajama, W. Tolman, Prog.
In-org. Chem. 1995, 43, 419.[18c]B. Govind, T. Satyanarayana, K.
Veera-Reddy, Polyhedron 1995, 15, 1009.
[19] [19a]D. L. Allen, V. C. Gison, M. L. Green, T. F. Skinner, J.
Bashikin, P. D. Grebenik, J. Chem. Soc., Chem. Commum.
1985, 895.[19b]G. Consiglio, F. Morandini, Chem. Rev. 1987,
87, 761.
[20]T. A. Stephenson, G. Wilkinson, J. Inorg. Nucl. Chem. 1996,
28, 945.
[21]G. M. Sheldrick, SHELXTL PLUS User’s Manual, revision
4.1, Nicolet XRD Corporation, Madison, WI, 1991.
Received June 3, 2004 Early View Article Published Online October 7, 2004
![Figure 1. ORTEP drawing of complex 1 (30% probability ellip- ellip-soids); selected bond lengths [A ˚ ] and angles [°]: Ru⫺P(1) 2.3423(20), Ru ⫺P(2) 2.3421(20), Ru⫺N(1) 2.133(5), Ru⫺N(3) 2.127(5), Ru ⫺Cl(1) 2.4283(19), Ru ⫺Cl(2) 2.4261(19);](https://thumb-ap.123doks.com/thumbv2/9libinfo/8679730.197024/2.892.497.773.313.553/figure-ortep-drawing-complex-probability-selected-lengths-angles.webp)
![Figure 2. ORTEP drawing of complex 2 (30% probability ellip- ellip-soids); the carbon atoms of the phenyl groups (except the ipso-carbon atoms) on the triphenylphosphane have been eliminated for clarity; selected bond lengths [A ˚ ] and angles [°]: Ru⫺C(1](https://thumb-ap.123doks.com/thumbv2/9libinfo/8679730.197024/3.892.75.421.88.312/figure-drawing-complex-probability-triphenylphosphane-eliminated-clarity-selected.webp)