行政院國家科學委員會補助專題研究計畫成果報告
※※※※※※※※※※※※※※※※※※※※※※※※※
※ GTP CYCLOHYDROLASE I 基因轉殖小鼠
※
※
啟動子表現及表現型分析
※
※※※※※※※※※※※※※※※※※※※※※※※※※
計畫類別:▓個別型計畫
□整合型計畫
計畫編號:NSC 89-2314-B-002-387
執行期間:89 年 8 月 1 日至 90 年 7 月 31 日
計畫主持人:胡務亮
共同主持人:
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
執行單位:
國立台灣大學醫學院(附設醫院)優生保健部
中
華
民
國 90 年 10 月 31 日
行政院國家科學委員會專題研究計畫成果報告
GTP CYCLOHYDROLASE I 基因轉殖小鼠啟動子表現及表現型分析
計畫編號:NSC 89-2314-B-002-387
執行期限:89 年 8 月 1 日至 90 年 7 月 31 日
主持人:胡務亮 國立台灣大學醫學院(附設醫院)優生保健部
一、中文摘要 GTP cyclohydrolase I (GCH)負責 tetrahydrobiopterine(BH4)合成的第一個步驟。BH4 是一個很重要的分子,它是phenylalaninehydroxylase, tyrosine hydroxylase, tryptophan hydroxylase以及nitric oxide synthase的輔因子。GCH 基因突變會引起惡性苯酮尿症或Dopa-敏感性肌肉 張力不全症,兩者和巴金斯症一樣,都需要接受 L-dopa治療。大部分Dopa-敏感性肌肉張力不全症 是顯性遺傳,其分子機轉為dominant-negative。基 於BH4生理作用的廣泛性,其引發疾病之複雜性, 以及與巴金斯症的相關性,我們希望能製造出缺乏 GCH的動物模型。 我們在過去的時間中,我們自人類基因庫中 篩選到GCH基因前方一斷13kb長的序列。並且證實 2.8kb及5kb(包含部分exon 1)的片段可以表現出啟 動子活性。我們將2.8kb片段後方接上LacZ基因後 注入小鼠受精卵中,結果LacZ F1小鼠腦部無法見 到LacZ基因的表現。因此我們將含有Dopa-敏感性 肌肉張力不全症G201E突變的GCH基因cDNA,接 在5kb片段後方。小鼠F0及F1均已篩選完畢。 在F0及F1的轉殖小鼠中,我們發現有一些小 鼠在Rota Rod中的表現不佳。這些小鼠有可能是腦 中GCH基因的表現受到影響,所以表現出運動上的 障礙。然而這些小鼠的子代並沒有辦法穩定的表現 出這樣的異常,所以無法證實運動障礙和轉殖基因 間的關係。 當轉殖小鼠年齡增大後,我們發現其中有一 些有異常的動作。這是一種持續性的沒有目的的旋 轉。來自同一祖先的好幾株都可以看到這樣的異 常。這可能是一種晚發性的運動障礙,很可能和轉 殖基因,也就是突變型的GCH基因有關。這些小鼠 的生殖力差,新生小鼠死亡率高,相關組織學分析 正在進行中。 關鍵詞:GTP cyclohydrolase I,Dopa-敏感性肌肉張 力不全症,基因轉殖小鼠 Abstr act
GTP cyclohydrolase I (GCH) is responsible for the first step of tetrahydrobiopterin (BH4) biosynthesis.
BH4 is an important molecule, because it is the
synthase. GCH gene mutations are associated with wither malignant phenylketonuria and dopa-responsive dystonia (DRD). Both of them are responsive to L-dopa treatment. DRD is mostly dominantly inherited, and through the dominant-negative mechanism.
In the past years, we have isolated a 13kb DNA from human genomic library containing the 5’ region (including part of exon 1) of GCH gene. We have shown that both a 2.8 and 5kb fragments possessed promoter activity. We connected the 2.8kb fragment to LacZ gene. The construct was injected into mouse fertilized egg. We have screened F0 and F1 for the LacZ mice, but the gene was not expressed. We inserted a GCH cDNA containing the G201E DRD mutation after the 5kb promoter. Both F0 and F1 G201E mice were produced.
The phenotype of the transgenic mice was checked by Rota-Rod. Several mice were found to have poor performance on the Rota. These mice may have decreased GCH activity because of the
expression of the dominant negative GCH gene. However, the offspring of these mice could not express stably the same phenotype. Therefore, we can not make conclusion on the association between motor defect and the transgene.
When the mice were getting old, some of them demonstrated behavior problem. They tended to make purposeless circling movement. This could be a late onset motor abnormality, and may be associated with the GCH gene. Some mice were from the same ancestor. These mice have low fertility power, and many of their babies died shortly after birth. Further studies on them are going now.
Keywor ds: GTP cyclohydrolase I, Dopa-responsive
dystonia, transgenic mice
二、緣由與目的
GTP cyclohydrolase I (GTP-CH) catalyzes the rate limiting step of tetrahydrobiopterin (BH4) biosynthesis
(Nichol, 1985). BH4 is the cofactor of several
important human enzymes including phenylalanine hydroxylase, tyrosine hydroxylase, tryptophan hydroxylase (Kaufman 1959, Nagatsu 1964,
serotonin and nitric oxide. GTP-CH mutations have been found in patients with hyperphenylalaninemia (malignant phenylketonuria), and dopa-responsive dystonia (DRD) (Segawa et al., 1971; Nygaard, 1988).
In hyperphenylalaninemia, patients are not able to metabolize phenylalanine and have multiple neurotransmitter deficiency. In DRD, it looks like only nigro-striatal dopaminergic neurons are affected, and the main symptom is dystonia (Segawa 1976, 1986). The GTP-CH activities in hyperphenylalaninemia patients are very low, while the activities are usually 10 to 20% in HPD/DRD patients (Segawa 1996, Ichinose and Nagatsu, 1997). After the cloning of human GTP-CH gene (Ichinose, 1994), recessive mutations were found in hyperphenylalaninemia and dominant mutations were found in DRD (Ichinose, 1994; Ichinose et al., 1995; Furukawa et al., 1996; Bandmann et al., 1996; Blau et al., 1995). The enzyme is a homodecamer (Nar et al, 1995). In our previous work, we have found a case of DRD who has a homologous GTPCH gene mutation (R249S).
It will be very useful if there is an animal model of GTPCH deficiency to solve the unusual inheritance in GTPCH deficiency, explore the disease mechanism and improve the treatment, and understand more about Dopa metabolism. Our hypothesis is that residual enzyme activity (gene dosage) determines the phenotype, and inheritance depends on the mechanisms of mutations (loss-of-function or dominant-negative).
The hph-1 mouse has BH4 deficiency and reduced levels of dopamine, norepinephrine and serotonin (Hyland, 1996), however, the underlying defect of the hph-1 mouse is still not known (Gutlich et al., 1994). Transgenic mice expressing COLIA1
gene with Gly859Cys mutation died intrauterine or shortly after birth (Stacey et al., 1988). Both the endogenous and mutant pro-α1(I) collagen was expressed, but a striking reduction of the total collagen I content was proportional to the level of mutant gene expression. Mutations in the p53 tumor suppressor gene are found at high frequency in a wide range of cancers (Hollstein et al., 1991). Wild-type p53 acts as a negative regulator of cell growth (Kuerbitz et al., 1992). Mutant forms of p53 no longer possess the ability to arrest cell growth and are unable to bind to specific DNA response sequences (Michalovitz et al., 1990). Mutant p53 actually inhibit wild-type p53 in a “dominant negative” manner through a complex formation between the two forms (Milner et al., 1991).
In this study, we tried to make a mouse model for GCH deficiency. Our strategy is to make
transgenic mice with a dominant negative GCH mutant cDNA.
三、成果及討論
材料及方法
Tissue section pr epar ation
The animal is killed by CO2. It is first perfused with warm normal saline, followed by 2%
paraformaldehyde. Organs are dissected and fixed for another two hours. The organs are then soaked in 30% sucrose in PBS with 2 mM MgCl2. Brain and organs are then embed in OCT compound and ready for frozen section.
Beta-galactosidase staining
To determine the patterns of β-galactosidase
expression, tissue sections will be fixed and stained for β-galactosidase activity.
Fixation and section as described in previous section
The slides are rinsed in PBS with 2 mM MgCl2 Staining buffer PBS 1 mM MgCl2 1.5 mM K4[Fe3(CN)6] 1.5 mM K3[Fe2(CN)6] Substrate: 1 mg/ml 5-bromo-4-chloro-indoyl- -D-galactopyranosi de (X-Gal) Incubation at 37oC for 3 hr
Counter stain with hematoxylin and mount
PCR detection
PCR will be done by primer pair P1F/P4R. This pair of primer specifically amplify human GCH promoter sequence with no cross amplification of the mouse DNA.
Souther n blot analysis
Southern blot will be performed with a 700 bp DNA fragment located at the proximal human GCH promoter. This probe detects no cross-hybridization signal with mouse DNA. A non-radioisotope method will be used in the study. DNA will be cut by different enzyme for the purposes of both verification of integration and estimation of copy number.
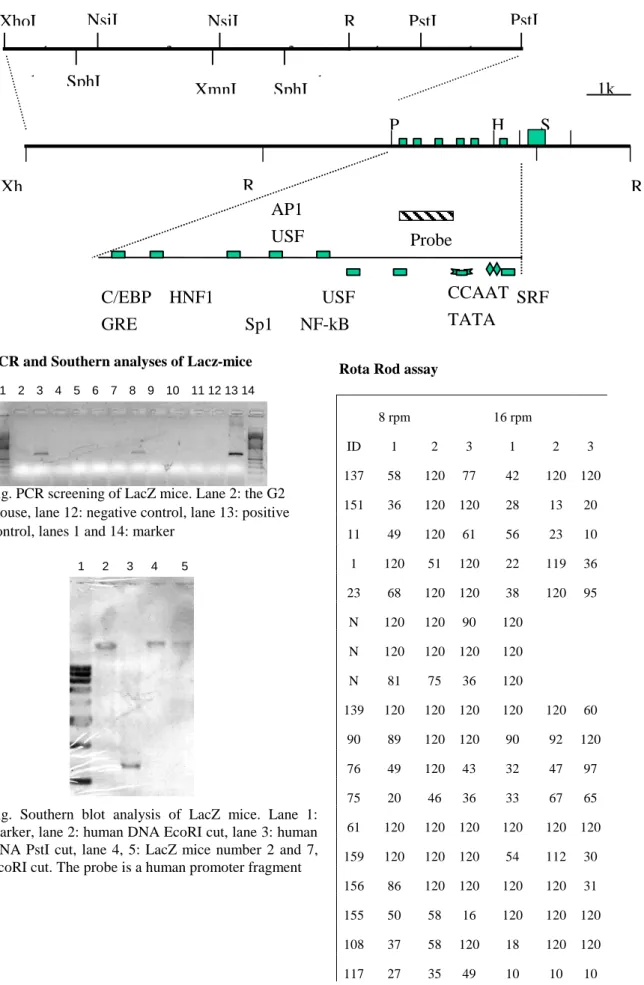
Map of human GCH pr omoter .
One positive phage clone was obtained from Human genomic library (ClonTech, USA). A XhoI/EcoRI 13kb fragment was subcloned into pBluescriptII vector.
Pr omoter activity
Promoter fragments of different lengths were ligated before the CAT reporter gene. The vectors were transfected into BHK cells, and CAT activity was assayed by standard method.
Map of the pr omoter r egion
PCR and Souther n analyses of Lacz-mice
Fig. PCR screening of LacZ mice. Lane 2: the G2 mouse, lane 12: negative control, lane 13: positive control, lanes 1 and 14: marker
Fig. Southern blot analysis of LacZ mice. Lane 1: marker, lane 2: human DNA EcoRI cut, lane 3: human DNA PstI cut, lane 4, 5: LacZ mice number 2 and 7, EcoRI cut. The probe is a human promoter fragment
Rota Rod assay
8 rpm 16 rpm ID 1 2 3 1 2 3 137 58 120 77 42 120 120 151 36 120 120 28 13 20 11 49 120 61 56 23 10 1 120 51 120 22 119 36 23 68 120 120 38 120 95 N 120 120 90 120 N 120 120 120 120 N 81 75 36 120 139 120 120 120 120 120 60 90 89 120 120 90 92 120 76 49 120 43 32 47 97 75 20 46 36 33 67 65 61 120 120 120 120 120 120 159 120 120 120 54 112 30 156 86 120 120 120 120 31 155 50 58 16 120 120 120 108 37 58 120 18 120 120 117 27 35 49 10 10 10 1 2 3 4 5 6 7 8 9 10 11 12 13 14 1 2 3 4 5
Xh
o
R
V
P
H S
P
B
R
1k
b
SphI
NsiI
NsiI
XmnI
SphI
R
V
XhoI
PstI
PstI
1. 1 1. 1 1. 6 2 2 1. 2 1. 8
C/EBP HNF1 USF SRF
GRE Sp1 NF-kB
AP1
USF
CCAAT
TATA
Probe
These results are still under calculation and analysis. The phenotype of the transgenic mice was checked by Rota-Rod. Several mice were found to have poor performance on the Rota. These mice may have decreased GCH activity because of the
expression of the dominant negative GCH gene. However, the offspring of these mice could not express stably the same phenotype. Therefore, we can not make conclusion on the association between motor defect and the transgene.
When the mice were getting old, some of them demonstrated behavior problem. They tended to make purposeless circling movement. This could be a late onset motor abnormality, and may be associated with the GCH gene. Some mice were from the same ancestor. These mice have low fertility power, and many of their babies died shortly after birth. Further studies on them are going now.
四、參考資料
Blau N, Barnes I, Dhondt JL (1996) International database of tetrahydrobiopterin deficiencies. J Inher Metab Dis 19, 8-14.
Furukawa Y, Shimadzu M, Rajput AH, Shimizu Y, Tagawa T, Mori H, Yokochi M, Narabayashi h, Hornykiewicz O, Mizuno Y, Kish SJ (1996) GTP-cyclohydrolase I gene mutations in hereditary progressive and dopa-responsive dystonia. Ann Neurol 39, 609-617.
Griffith OW, Dennis JS (1995) Nitric oxide synthase: properties and catalytic mechanism. Annu Rev Physiol 57, 707-736.
Gutlich M, Ziegler I, Witter K, Hemmens B, Hultner L, McDonald JD, Werner T, Rodl W, Bacher A (1994) Molecular characterization of hph-1: a mouse mutant deficiency in GTP cyclohydrolase I activity. Biochem Biophys Res Comm 203, 1675-1681.
Hasty P, Ramires-Solis R, Krumlauf R, Bradley A (1991) Introduction of a subtle mutation into the Hox-2.6 locus in embryonic stem cells. Nature 350, 243-246.
Hollstein M, Sidransky D, Volgelstein B, Harris CC (1991) p53 mutations in human cancers. Science 252, 49-53.
Hland K, Gunasekera RS, Engle T, Arnold LA (1996) Tetrahydrobiopterin and biogenic amine
mtabolism in the hph-1 mouse. J Neurochem 67, 752-759.
Ichinose H, Ohye T, Takshashi E, Seki N, Hori T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsujis, Fujitak K, Nagatsu T (1994) Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 8, 236-242. Ichinose H, Nagatsu T (1997) Molecular genetics of
hereditary dystonia – mutations in the GTP
cyclohydrolase I gene. Brain Res Bulletin 43, 35-38.
Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB (1992) Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA 89, 7491-7495.
Nichol, C.A., Smith, G.K. &Duch,D.S. Biosythesis and metabolism of tetrahydrobiopterin and molybdopterin.
Ann.rev.Biochem.54,729-764(1985) Nygaard TG, Marsden CD, Duvoisin RC (1988)
Dopa-responsive dystonia, In Fahn S, Marsden CD, Calne DB (Ed) Advances in Neurology, Vol 50, Raven Press, New York, 1988, pp.377-384. Pinkert CA (1994) Transgenic animal technology, a
laboratory handbook. Academic Press.
Segawa M, Ohmi K, Itoh S, Aoyama M, Hayakawa H (1971) Childhood basal ganglia disease with remarkable response to L-Dopa, hereditary basal ganglia disease with marked diurnal fluctuation. Shinryo (Tokyo) 24, 667-672.
Segawa M, Hosaka A, Migagawa F, Nomura Y, ImaiH. Hereditary progressive dystonia with marked diurnal fluctuation. In: Eldridge R, Fahn S, eds. Dystonia, advance in neurology. Vol 14, New York: Raven 1976:215-33.
Segawa M (1996) Segawa disease (hereditary progressive dystonia with marked diurnal fluctuation-HPD) and abnormalities in pteridin metabolism. Rhinsho Shinkeigaku 36,
1322-1323.
Stacey A, Bateman J, Choi T, Mascara T, Cole W, Jaenisch R (1988) Perinatal lethal osteogenesis imperfecta in transgenic mice bearing an engineered mutant pro-a1(I) collagen gene. Nature 332, 131-136.