Insights into dynamics of the F + CD 4 reaction via product pair correlation
Jingang Zhou, Jim J. Lin, Weicheng Shiu, and Kopin Liu
Citation: The Journal of Chemical Physics 119, 4997 (2003); doi: 10.1063/1.1592153
View online: http://dx.doi.org/10.1063/1.1592153
View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/119/10?ver=pdfcov
Published by the AIP Publishing
Articles you may be interested in
Lifetime of reactive scattering resonances: Q-matrix analysis and angular momentum dependence for the F + H 2 reaction by the hyperquantization algorithm
J. Chem. Phys. 121, 11675 (2004); 10.1063/1.1814096
Rotationally selected product pair correlation in F + CD 4 DF ( )+ CD 3 (=0, N )
J. Chem. Phys. 120, 5863 (2004); 10.1063/1.1689634
Simulation of the reactive scattering of F + D 2 on a model family of potential energy surfaces with various topographies: The correlation approach
J. Chem. Phys. 120, 800 (2004); 10.1063/1.1631419
Time delay as a tool to identify the signatures of reactive resonance: F + HD and F + H 2 reactions
J. Chem. Phys. 119, 1462 (2003); 10.1063/1.1582172
F + D 2 reaction at ultracold temperatures
J. Chem. Phys. 116, 9222 (2002); 10.1063/1.1472515
COMMUNICATIONS
Insights into dynamics of the F
¿
CD
4reaction via product
pair correlation
Jingang Zhou, Jim J. Lin,a)Weicheng Shiu, and Kopin Liub)
The Institute of Atomic and Molecular Sciences (IAMS), Academia Sinica, P.O. Box 23-166, Taipei, Taiwan 106
共Received 15 May 2003; accepted 27 May 2003兲
To unravel the ‘‘extra-atom’’ complexity of the title reaction, we exploit an experimental approach which, by taking advantage of the correlated information of coincident product pairs, allows us to peel off judiciously the intrinsic complications of a six-atom reaction, extracting the underlying backbone of three-atom dynamics. Examining the collisional energy dependencies of the pair-correlated attributes for a given state共s兲 of CD3products from the title reaction, several of major observations can qualitatively be understood, whereas others await further theoretical investigations. An intriguing possibility for the existence of reactive resonances in this six-atom reaction is
surmised. © 2003 American Institute of Physics. 关DOI: 10.1063/1.1592153兴
The field of gas-phase reaction dynamics has reached a level of maturity where the detailed dynamics of a few-body
system on a single potential energy surface 共PES兲 is
essen-tially a solved problem.1As exemplified from several
bench-mark three-atom exchange reactions,2– 8and soon for a few
favorable four-atom reactions such as OH⫹H2→H2O⫹H,9,10
the exact quantum dynamics can be computed on a highly accurate ab initio PES with the results compared very favor-ably with the most detailed experimental observable, such as the state-to-state differential cross section共DCS兲. These re-ally represent a remarkable accomplishment of the field for both experimentalists and theoreticians in recent years.
As we move into larger reaction systems, the number of degrees of freedom increases rapidly and the situation be-comes far more complex and challenging. For a chemical reaction that forms two molecular products, for example, even if the state-resolved DCS for each product is obtained individually, the coincident attributes of the co-products are still lacking. In a recent report,11we exploited a new method, which combines the state-specific ‘‘tagging’’ of one product by laser spectroscopic detection with a high-resolution ion imaging technique,12 to reveal the coincident information of the product pair in a state-correlated manner. The pair-correlation information is more than another layer of details or specificity. It represents, whatever attribute or distribution one is measuring, the joint probability matrix P(n1,n2) for
finding a product 1 coincidently formed in state n1when the
other product 2 is in state n2 in the same reactive event.
From the viewpoint of the transition-state theory, how a chemical transformation takes place is mostly governed by the transition state property. However, the transition state is
not static, namely, what is relevant are not just the energetics and the structure, etc. It is dynamic in nature, namely, what really counts is the concerted motion of all atoms in the vicinity of the transition state region. It is conjectured that the correlation of coincident product pairs could potentially provide the most revealing imprint of this concerted motion in the transition state region.
Because the correlation information P(n1,n2) is n1⫻n2
in dimension, it can be significantly larger than the sum of
the uncorrelated information, n1⫹n2. Moreover, the
P(n1,n2) represents only the encoded information which
must be decoded to be insightful. The most pressing issue, once the correlated information is available, becomes how to sort the order out of chaos or how to digest these enormous amounts of correlated information. In view of many useful concepts prevailed over the past thirty years for a typical A⫹BC reaction,13 we seek a hierarchical approach that en-ables us to differentiate those attributes, which can be
com-prehended from the extension of the A⫹BC lesson, from
those remained, which could arise from the intrinsic ‘‘extra-atom’’ complexity of a polyatomic reaction.
In this Communication we report our first step toward
understanding the dynamics of the reaction F⫹CD4
→DF⫹CD3. Our recent crossed-beam experiments14,15
in-dicated that the reaction exhibits a threshold of
⬃0.5 kcal/mol, and up to Ec⫽8.5 kcal/mol only the
v2-mode 共the umbrella motion兲 excitation of the CD3
prod-uct has significant populations. Using a newly-developed,
time-sliced ion velocity imaging technique,12 we were able
to reveal the state-correlations of the coincident product pairs of CD3(0v200) and DF(v
⬘
) at one collision energy Ec⫽5.37 kcal/mol, which show striking differences.11
Obvi-ously, this information involves the quantum structures of both products, and the CD3product is not merely a spectator. To unfold the complexity, we now turn our focus into the correlated DF attributes for a given CD3state共s兲 and examine
a兲Also at Department of Applied Chemistry, National Chiao Tung Univer-sity, Hsinchu, Taiwan 300.
b兲Also at Department of Chemistry, National Normal University, Taipei, Taiwan 106.
4997
0021-9606/2003/119(10)/4997/4/$20.00 © 2003 American Institute of Physics
their dependencies on collision energies. By keeping the structural factor of the CD3 product unchanged, CD3 is then
regarded as a pseudospectator so that the situation mimics more closely the familiar three-atom reaction. In other words, an experimental approach to reduce the dimension was adapted, which allows the dynamical attributes thus ob-tained for the present six-atom reaction to be approximated
as a light-atom 共D-atom兲 transfer between two heavy
par-ticles共F and CD3), namely, a heavy–light–heavy system. By
setting the probe laser frequency at the peak of the Q branch of the 000 band, we chose the CD3共0000,
具
N典
⫽4⫾3) statesin this report because they represent a significant fraction of the overall reactivity of F⫹CD4from our previous studies.
15
Similar experiments on collisional energy dependencies for
the vibrationally excited CD3 products were also measured
and will be reported in the future.
Figure 1共a兲 shows a representative raw image for Ec
⫽8.36 kcal/mol. The image was acquired in the same
man-ner as that reported recently.11,12Four ringlike structures are clearly visible. Overlaid on the image is the velocity-vector diagram of the collision system, which is based on the well-established energetics of this reaction. The successive rings can be unambiguously assigned to the correlated vibration states (v
⬘
) of the DF co-products, starting from v⬘
⫽5 forthe innermost feature tov
⬘
⫽2. The clear separation of theserings indicates unequivocally the low rotational excitation of
the DF product. Using the same data analysis procedure,12
the resulted CD3product flux-velocity contour map is shown
in Fig. 1共b兲, which represents the doubly differential cross section (d2/du d(cos)) of the product pairs in the center-of-mass共c.m.兲 polar coordinate (u,). Similarly, the resulted
contours for the two lower collision energies are shown in Figs. 1共c兲 and 1共d兲, respectively. Seemingly counterintuitive is the observation that the harder共i.e., the higher energy兲 the collision is, the colder the vibrational distribution of the cor-related DF product becomes.
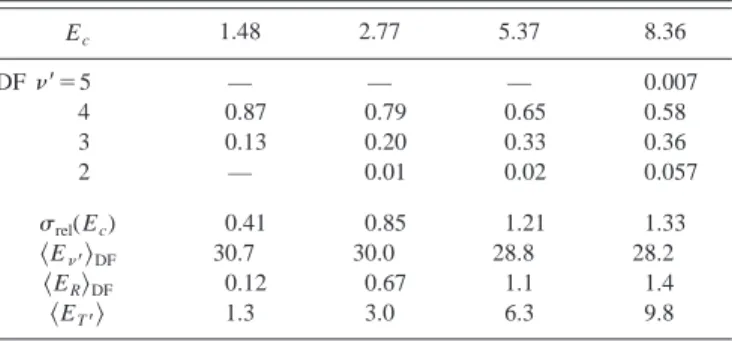
More quantitative information can be obtained through the analysis of the contours. The clear separation of the vibrational features makes it easy, and the resultant
correlated-DCSs 共CDCS兲 are depicted in Fig. 2 along with
that previously reported at Ec⫽5.37 kcal/mol 共Ref. 11兲 for
comparisons. Integrating all angles for each individual CDCS yields the pair-correlated vibrational branching ratio, as summarized in Table I. Apparently, the relative branching
ratio of the two dominant vibrational states, (v
⬘
⫽4)/(v
⬘
⫽3), decreases with the increase in collision energy—atrend was mentioned above. From the vibrational branching ratios and the flux-velocity contours shown in Fig. 1, the complete energy disposal in coincidence with the formation of CD3(0000,
具
N典
⯝4) is obtained 共Table I兲. Despite the sig-FIG. 1. 共Color兲 共a兲 Raw image of the state-selected CD3products from theF⫹CD4→DF⫹CD3reaction at Ec⫽8.36 kcal/mol. The successive ring fea-tures correspond to the labeled vibrational states of the coincident DF prod-uct.共b兲 CD3product state-resolved flux-velocity contour maps derived from
共a兲. The density-to-flux correction has been made. The intensity has been weighted by u2in accordance with conventional representation of the
dou-bly differential cross section关d2/du d(cos)兴. 共c兲 and 共d兲, as 共b兲 but for
Ec⫽2.77 and 1.48 kcal/mol, respectively.
FIG. 2. 共Color兲 Summary of the state-resolved, pair-correlated angular dis-tribution 共CDCS兲. The result for Ec⫽5.37 kcal/mol 共from Ref. 11兲 is in-cluded for comparisons. The scales of the four panels are normalized ac-cording to the excitation function for CD3(0000) shown in Ref. 15. The
black lines represent the vibration-resolved angular distribution of CD3
products.
TABLE I. Correlated vibrational branching ratios and energy disposal into DF(⬘)⫹CD3(0000,具N典⬇4) products. Note: All energies are in kcal/mol,
andrel(Ec) is from Ref. 15.
Ec 1.48 2.77 5.37 8.36 DF⬘⫽5 — — — 0.007 4 0.87 0.79 0.65 0.58 3 0.13 0.20 0.33 0.36 2 — 0.01 0.02 0.057 rel(Ec) 0.41 0.85 1.21 1.33 具E⬘典DF 30.7 30.0 28.8 28.2 具ER典DF 0.12 0.67 1.1 1.4 具ET⬘典 1.3 3.0 6.3 9.8
4998 J. Chem. Phys., Vol. 119, No. 10, 8 September 2003 Zhouet al.
nificant variation in the vibrational branching ratios with col-lision energies, the averaged energy disposal into the DF co-products,
具
Ev⬘典
DF, shows rather minor changes. Moreno-table is the observation of the propensity of T (or Ec)
→T
⬘
. For a heavy–light–heavy collinear reaction, the共mass-weighted兲 skew angle13is small共for the present
reac-tion ⫽25.5°), a strong inertial coupling between the two scaled coordinates is anticipated, which in turn leads to the Polanyi’s rule16 that the reagent translation in excess of the barrier to reaction is channeled principally into product
translation. Hence, the observed propensity of T→T
⬘
has asimple kinematic origin. It is remarkable from Table I how well the extend of T→T
⬘
holds.By conservation of energy, Etotal⫽⌬Hrx⫹T⫽VCD3
⫹RCD3⫹VDF⫹RDF⫹T
⬘
. Since T→T⬘
and both VCD3 andRCD
3 are preselected by laser detection, VDF⫹RDFis
neces-sarily invariant to the change in collision energies. As will be
argued later, RDF is expected to be small. Hence, most of
reaction exoergicity will be deposited into the vibration of the DF co-product. Again, this is anticipated for an exoergic reaction with an early barrier, in particular for a system with small skew angle. On the other hand, the quantitative vibra-tional distribution of the DF co-product must originate from detailed dynamics, thus depending on the actual PES. We
noted in passing that the observed propensity of T→T
⬘
isintimately connected to the previously proposed conserva-tion of vibraconserva-tion acconserva-tion of the two vibrators, DF and CD3.11
The conservation of angular momentum dictates that l
⯝J⫽NCD3⫹jDF⫹l
⬘
. 共The initial rotational angularmomen-tum of CD4 is negligibly small for a supersonic expansion
beam.兲 For a light-atom transfer reaction, both the initial and the final orbital angular momenta 共l and l
⬘
) are mainly car-ried by the orbiting motion of the two heavy particles. Thereaction of F⫹CD4 is fast,
17
and is believed to proceed through a predominantly linear transition-state in the F–D–C
geometry.18As demonstrated by Schulten and Gordon19 for
many direct, three-atom reactions with heavy–light–heavy mass combination, l contributes mostly to l
⬘
, and for a nearly collinear reaction often l⬇l⬘
, i.e., a coplanar dynamics. The estimated l for the title reaction ranges from about 100ប toⲏ200ប over the energies of this study. The probed NCD3 has
been locked to low values, the angular momentum constraint leads naturally to small values of jDF, thus, small RDF, as evidenced from the clear separation of the rings.
The above kinematic constraint on angular momenta, l
⬇l
⬘
, could also have important implication on the observedtotal angular distributions共Fig. 2, black lines兲. In general, for
a long-lived complex-forming reaction, the exact shape of angular distribution can reveal the degree of correlation be-tween reactant and product angular momenta. For example, the observation of sharp forward–backward symmetry in the c.m. angular distribution implies l⬇l
⬘
and an isotropic angu-lar distribution will suggest l⬇j⬘
.20,21In the case of a direct elementary reaction which does not proceed through a long-lived complex, the form of the c.m. angular distribution re-veals the favored reactant geometry,13providing insights into the angular dependence of the PES. For example, a bend approach or a softer bending potential could lead tosubstan-tially more sideways and forward scattering of products. Nevertheless, the above correlation between angular distribu-tion and angular momentum disposal remains largely valid even for a direct reaction. And the sharpness of forward or backward peak is also likely to indicate how closely the ori-entation of l
⬘
lies parallel or antiparallel to that of l. In this sense, the observation of a sharp forward feature for the present reaction over a wide range of collision energies has its kinematic root, at least partially, from l⬇l⬘
.A closer inspection of total angular distributions also re-veals a small backward peak for the two lower energies.
Moreover, the ratio of 共forward peak兲/共backward peak兲
de-creases monotonically with the decrease in collision energy. Both aspects are qualitatively in line with the expectation of the osculating complex model for reaction.22The possibility of the involvement of a short-lived complex in this reaction is not too surprising in view of the mass combination and intrinsic time scales: The reaction time is governed by the slower trajectory of two heavy particles, whereas the faster chattering-motion of the light atom manifests itself as a tran-sient complex. An interesting question then arises: Does there exist any quasibound state or reactive resonance in this reaction? or is the state-specific sharp forward peak the
manifestation of a simple 共transition-state兲 threshold
effect?23 In light of the similar mass combination and
ener-getics of the present reaction to those of F⫹HD→HF⫹D in
which a reactive resonance has recently been
demonstrated,6,7 the above question is perhaps not too far-fetched.
In that regards, one should also keep in mind an alterna-tive viewpoint about the total angular distribution. Since T
→T
⬘
, this reaction is on average like an elastic scattering.Aside from the rainbowlike feature near 30° – 45° 共Fig. 2兲,
the total angular distribution indeed appears reminiscent of that of a hard-sphere collision.24共In this viewpoint, the rain-bowlike features could arise from the van der Waals well, analogous to the elastic rainbow.兲 For a hard-sphere elastic scattering, it is well known that the quantum calculation yields a sharp forward peak superimposed on an isotropic angular distribution. The classical version will be just the isotropic component. This sharp forward peak is essentially geometric in nature resulting from the difference in impact times with different portions of the hard sphere. In other words, it is a consequence of interference phenomena and doesn’t invoke any trapping of quasibound states nor the threshold effect. Nevertheless, time delay is a common char-acteristic of all three mechanisms: a quasibound or trapped state, a reactive threshold effect, and a hard sphere scattering.23–26The time delay allows the collision partners to rotate before forming the products, thus could yield a for-ward peak in angular distribution. Again, like the correlated vibrational branching ratio, the more detailed CDCS will de-pend sensitively on the PES.
In summary, by ‘‘tagging’’ a specific group of CD3
prod-uct states and examining the collisional energy dependencies of thus correlated attributes of the DF coproduct, we eluci-dated in this study how to experimentally decompose a com-plex reaction problem into simpler pieces that preserve the essential physics. Several well established concepts from the
simple A⫹BC reaction were drawn to gain insights into the present six-atom system. The three-atom dynamics revealed here could serve as the underlying backbone to decipher the ‘‘extra-atom’’ complexity when more detailed topography of PES becomes available. Three possible mechanisms were speculated for the origin of the sharp forward peak. The pos-sibility of the existence of reactive resonance in this poly-atomic reaction is intriguing, and deserves further investiga-tions.
This work was supported by the National Science
Coun-cil of Taiwan 关NSC 2119-M-001-011 共KL兲 and NSC
91-2113-M-001-024共JJL兲兴.
1K. Liu, Annu. Rev. Phys. Chem. 52, 139共2001兲.
2L. Schnieder, K. Seckamp-Rahn, E. Wrede, and K. H. Welge, J. Chem.
Phys. 107, 6175共1997兲.
3S. C. Althorpe, F. Fernandez-Alonso, B. D. Bean, J. D. Ayers, A. E.
Po-merantz, R. N. Zare, and E. Wrede, Nature共London兲 416, 67 共2002兲.
4S. A. Harich, D. Dai, C. C. Wang, X. Yang, S. D. Chao, and R. T. Skodje,
Nature共London兲 419, 281 共2002兲.
5
D. Skouteris, D. E. Manolopoulos, W. S. Bian, H. J. Werner, L. H. Lai, and K. Liu, Science 286, 1713共1999兲.
6R. T. Skodje, D. Skouteris, D. E. Manolopoulos, S.-H. Lee, F. Dong, and
K. Liu, Phys. Rev. Lett. 85, 1206 共2000兲; J. Chem. Phys. 112, 4536 共2000兲.
7K. Liu, R. T. Skodje, and D. E. Manolopoulos, Phys. Chem. Comm. 5, 27
共2002兲.
8X. Liu, J. J. Lin, S. Harich, G. C. Schatz, and X. Yang, Science 289, 1536
共2000兲.
9B. R. Strazisar, C. Lin, and H. F. Davis, Science 290, 958共2000兲. 10D. H. Zhang, M. A. Collins, and S.-Y. Lee, Science 290, 961共2000兲. 11
J. J. Lin, J. Zhou, W. Shiu, and K. Liu, Science 300, 966共2003兲.
12J. J. Lin, J. Zhou, W. Shiu, and K. Liu, Rev. Sci. Instrum. 74, 2495共2003兲. 13R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics and
Chemical Reactivity共Oxford University Press, Oxford, 1987兲.
14
W. Teh, W. Shiu, J. Zhou, and K. Liu, Chinese J. Chem. Phys. 15, 218 共2002兲.
15J. Zhou, J. J. Lin, W. Shiu, S.-C. Pu, and K. Liu, J. Chem. Phys.共to be
published兲.
16J. C. Polanyi, Science 236, 680共1987兲, and references therein. 17
A. Persky, J. Phys. Chem. 100, 689共1996兲.
18J. C. Corchado and J. Espinosa-Garcia, J. Chem. Phys. 105, 3152共1996兲. 19K. Schulter and R. G. Gordon, J. Chem. Phys. 64, 2918共1976兲. 20W. B. Miller, S. A. Safron, and D. R. Herschbach, Discuss. Faraday Soc.
44, 108共1967兲.
21
S. K. Kim and D. R. Herschbach, Faraday Discuss. Chem. Soc. 84, 159 共1987兲.
22G. A. Fisk, J. D. McDonald, and D. R. Herschbach, Discuss. Faraday Soc.
44, 228共1967兲.
23
D. E. Manolopoulos, Nature共London兲 419, 216 共2002兲.
24M. S. Child, Molecular Collision Theory共Academic, New York, 1974兲. 25D. Sokolovski, Chem. Phys. Lett. 370, 805共2003兲.
26S. D. Chao and R. T. Skodje, J. Chem. Phys.共to be published兲.
5000 J. Chem. Phys., Vol. 119, No. 10, 8 September 2003 Zhouet al.