Kinetics and Mechanisms for the Adsorption, Dissociation, and
Diffusion of Hydrogen in Ni and Ni/YSZ Slabs: A DFT Study
Meng Hsiung Weng,
†Hsin-Tsung Chen,*
,‡Yao-Chun Wang,
†Shin-Pon Ju,*
,†Jee-Gong Chang,*
,§and M. C. Lin*
,∥†Department of Mechanical and Electro-Mechanical Engineering, Center for Nanoscience and Nanotechnology, National Sun Yat-sen
University, Kaohsiung 80424, Taiwan
‡Department of Chemistry, Chung Yuan Christian University, Chungli 32023, Taiwan
§National Center for High-Performance Computing, No. 28, Nan-Ke Third Road, Hsin-Shi, Tainan 74147, Taiwan
∥Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsinchu 30010, Taiwan
*
S Supporting InformationABSTRACT: The adsorption, dissociation, and diffusion of hydrogen in Ni(100) and Ni(100)/YSZ(100) slabs with two different interfaces (Ni/cation and Ni/O interface) have been studied by the density functional theory (DFT) with the Perdew−Wang functional. The H2molecule is found to preferentially absorb on a Top (T) site with side-on configuration on the Ni(100) surface, while the H-atom is strongly bound at a fcc Hollow (H) site. The barrier for the H2dissociation on both surfaces is calculated to be only∼0.1 eV. The potential energy pathways of H diffusion on pure Ni and Ni/YSZ with the two different interfaces are studied. Our calculated results show that the H-atom diffusion occurs via surface path rather than the bulk path. For the bulk path in Ni/YSZ, H-atom migration can occur more readily at the Ni/cation interface compared to the Ni/O interface. The existence of vacancy in the interface region is found to improve the mobility of H-atoms at the interface of Ni/YSZ slab. The rate constants for hydrogen dissociation and diffusion in pure Ni and Ni/YSZ are predicted.
■
INTRODUCTIONNickel (Ni) has attracted a great deal of interest because of its high catalytic activity for hydrogen oxidation in an anode of a solid oxide fuel cell (SOFC).1−3 In a practical SOFC, anode material is usually composed of a mixture of nickel and other oxide ceramics, such as SiO2,
4
doped-CeO2,
5−8 and
Sc-stabilized ZrO2,9 due to their high activity and structural stability. Owing to the high operating temperatures of SOFC (between 800 and 1100 K), the thermal expansion behavior between Ni and oxide ceramics is usually chosen to match each other. The nickel/yttria-stabilized zirconia (Ni/YSZ) cermet, which has been synthesized successfully by Spacil,10is widely used today as an anode material of SOFC10−20due to its good thermal expansion and ion conductivity.
In generally, the mechanism of the H2oxidation reaction was assumed to occur at the three-phase boundary (TPB), which is the composition of gas phase, catalyst, and electrolyte, in the anode of SOFC. The oxidation reaction at the TPB can be described as
+ −→ + −
H2 O2 H O2 2e (1)
It is now widely accepted that the oxidation process of H2at the TPB of the Ni/YSZ cermet electrode can be divided into at least two rate-determining processes.15The first process is the adsorption/dissociation of hydrogen on the nickel surface.15
The second process is the hydrogen migration from nickel to the YSZ surface accompanied by charge transfer. They found that the anode performance of SOFC is determined by that reaction processes. Experimental studies have demonstrated that about 0.1% interstitial H-atoms are present in bulk Ni and YSZ at a high SOFC operating temperature.18−20 The interstitial H-atoms come from the adsorption and dissociation of H2 on Ni.16,17 It is well-known that the performance of SOFC is strongly dependent on the oxygen reduction at the cathode.1,3,21However, the catalytic effect of the anode on the overall efficiency of SOFC is still a critical issue. Many experiments22−25have been carried out to study the tunneling motion of H-atoms in the Ni crystal. Johnson et al. showed that some of H-atoms may diffuse into the bulk Ni at low pressure conditions when H-atoms are adsorbed on the Ni surface.23 The Ni crystalline lattice does not restructure as H embedded in Ni crystals. Kammler’s result shows that the effect of thermal H-atoms with an initial probability of ca. 4.5× 10−2leads to the absorption of H-atoms in subsurface sites of Ni(100).25 However, investigating the H2reaction in Ni/YSZ is difficult experimentally due to the complex reaction involved.
First-Received: August 24, 2011 Revised: March 5, 2012 Published: March 8, 2012
principles calculations provide one technique to elucidate the mechanism of the reaction. By this technique, the adsorption, dissociation, and diffusion of hydrogen on the Ni surface have been studied by Kresse.26,27 Furthermore, Bhatina and Sholl have studied the energy barriers of H-atom migration from a Ni surface (Ni(100), Ni(110), and Ni(111)) into subsurface by density functional theory (DFT) calculations. They found that the activation barriers of 1.12, 0.76, and 0.84 eV need to be overcome for Ni(100), Ni(110), and Ni(111), respectively.28 Recently, Shishkin and Ziegler studied the adsorption and dissociation of H2 on YSZ(111) and YSZ(111)+O surfaces based on DFT calculations.29 Their results indicate that dissociation adsorption of H2 on the YSZ(111)+O surface is an exothermic adsorption, contrary to the endothermic adsorption on YSZ(111). They further employed the DFT computational method to investigate two mechanisms for H2 oxidation proposed for oxygen and hydroxyl spillover on the Ni/YSZ interface.30
To the best of our knowledge, most of previous reports focused on reaction mechanism of H2 molecule on a metal
surface. There are very few studies devoted to the under-standing of interface effect of Ni/YSZ anode on the reaction mechanism of H2 molecule and H-atom diffusion in Ni/YSZ anode bulk. On the other hand, the interface of metal/oxide materials plays a crucial role on catalyst properties which depend on the bonding at the interface. Ab initio calculations have successfully offered microscopic insight into interface structures of metal-ceramics.31−33 In the present paper, two different interfaces (Ni/cation and Ni/O) of Ni/YSZ slab are applied to understand the interface effect on the dissociation of H2and the diffusion of H-atoms by DFT calculations.
■
COMPUTATIONAL METHOD AND MODELAll present calculations are performed with the spin-polarized DFT34 plane wave method utilizing the Vienna ab initio simulation package (VASP).35−37 The ultrasoft pseudopotential38,39in conjunction with the Perdew−Wang (PW91)40,41 functional is applied. The Brillouin
zone is sampled with the Monkhorst-Pack grid.42The calculations are carried out using the 3× 3 × 3 and 3 × 3 × 1 Monkhorst-Pack mesh k-points for bulk and surface calculations, respectively. A 400 eV cutoff Figure 1.Simulation models of Ni(100)/YSZ(100) for Ni-Cation (Zr or Y) and Ni−O interfaces. (a−c) and (d−f) are side views for different Ni/ cation and Ni−O interfaces, respectively; (g) is a top view of Ni-Cation models; (h−j) are the top views of panel (d−f), respectively. Yellow square “V” indicates a vacancy.
energy is used allowing convergence to 1× 10−4eV in total energy for all calculations.
The configuration of YSZ (Y2O3stabilized ZrO2) with cubic CaF2
-type structure is very similar to that of the pure ZrO2. Because Ni and
YSZ crystalline possess different crystal structures and lattice parameters, the high degree of strain in interface can be expected as Ni and YSZ stack each other together in the same cubic space group. In DFT studies by Christensen and Carter for the ideal ZrO2/Ni
interface,31 their results indicate that both Ni(111)/ZrO2(100) and
Ni(110)/ZrO2(100) have larger misfit than Ni(100)/ZrO2(100) in
interface. In addition, they have shown that the bonding between Ni(111) on ZrO2(111) is weak. Moreover, for the Ni(100)/
ZrO2(100) slab, it has been predicted that there are strong chemical
bonds in interface.32,33Therefore, although the Ni(111) surface has the lowest surface energy compared to the Ni(100) surface, we have considered the Ni(100)/YSZ(100) interface as a model of the Ni/ YSZ, first because this interface better matches the surface cell of Ni and YSZ and second because Ni(100)/YSZ(100) interface has a better lattice match between both materials.
On the other hand, Pornprasertsuk et al.43have demonstrated the ionic conductivity in YSZ is sensitive to the surrounding cations. Thus, we consider different kinds of Y-atom doping positions to investigate the diffusion of H-atom in the Ni(100)/YSZ(100) in this study. The side and top views of the p(2 × 2) lateral cell of the Ni(100)/ YSZ(100) with different interfaces, including the Ni/cation interface and the Ni/O interface are shown in Figure 1. Three various kinds of Y-atom doping positions are considered. In the first case, both Y-atoms are in the first layer (denoted as first layer) of the YSZ slab. In the second case, one Y-atom lies in the first layer and the other in the second layer (denoted as first-second layer). In the third case, two Y-atoms lie in the second layer (denoted as second layer). In all six models, three layers of the Ni slab of the (100) faces are stacked on the top of the YSZ(100) surface. Then the Ni slab is rotated by 45° to align with the YSZ slab’s [110] orientation. Two slabs are made of 8 layers (Ni, 12; Y, 2; Zr, 7; O, 4 per supercell) and 9 layers (Ni, 12; Y, 2; Zr, 11; O, 4 per supercell) with a total number of 25 and 29 atoms for Ni/cation and Ni/O interfaces, respectively. To maintain the neutrality (Y is 3+, Zr is 4+), an oxygen vacancy was created as two yttrium atoms substituted the two Zr atoms near the interface, corresponding to the dopant concentration of 21% and 16% mol concentrations of yttria in YSZ for Ni/cation and Ni/O interfaces, respectively. The bottom two atomic layers are kept constrained and set to the estimated bulk parameters, whereas the remaining layers are fully relaxed during the calculations. A vacuum region of thickness about 11 Å in the direction normal to the surface for the surface reaction is used to prevent the interaction between the translational images. However, in this study, we considered some nonoxygen and oxygen terminated YSZ substrate to study the interface effect (for the stack of Ni slab on different lattice mismatched and polar surfaces) on the adsorption, dissociation, and diffusion properties of hydrogen. In addition, the interaction energy of the three-layer Ni metal on the Ni/ cation and Ni/O configurations also has been calculated according to the eq 2. The calculated results show that the adsorption energies of Ni of the Ni/cation models as shown in Figure 1a−c are −6.53, −7.05, and−6.91 eV, and for the Ni/O models as shown in Figure 1d−f are −7.25, −5.21, and −6.95 eV.
In this study, we calculate adsorption energies according to the following equation:
ΔEads=E[slab+adsorbate]−( [slab]E +E[adsorbate]) (2)
where E[slab + adsorbate], E[slab], and E[adsorbate] are the calculated total energy of adsorbed species on the Ni(100)/ YSZ(100) surface, a clean Ni(100)/YSZ(100) surface, and a gas-phase molecule, respectively. The nudged elastic band (NEB) method is employed to obtain a minimum energy pathway (MEP) of a reaction.44,45At least eight images are used to locate each transition state (TS).
■
RESULT AND DISCUSSIONPotential Energy Surfaces for the Adsorption and Dissociation. The structural properties of Ni and ZrO2bulk materials and the H2molecule are taken into account first to verify the reliability of the computational method. The predicted lattice constants of bulk Ni and ZrO2are 3.54 and 5.10 Å, which are in good agreement with the experimental values of 3.5246and 5.09 Å,47respectively. The bond length of H2is calculated in a 10× 10 × 10 Å3cubic box to be 0.74 Å which is also consistent with the available experimental data.48 We initially studied molecular and dissociative adsorption of H2 on selected surface models. In order to locate possible intermediates of H/Ni/YSZ, H2/Ni/YSZ, H/Ni and H2/Ni surfaces, three different adsorption sites are considered: Top (T), Bridge (B), and fcc Hollow (H) sites. For the T site, the molecule adsorbs on top of the first-layer Ni atom of Ni(100) and Ni(100)/YSZ(100) with the Ni/cation and Ni/O interfaces. For the B site, the molecule adsorbs above the center of the Ni−Ni bond of the two nearest Ni sites in between first-layer Ni atoms and second-layer Ni atoms. For the H site, the molecule adsorbs above a second-layer Ni atom. Accordingly, the adsorptions of H and H2are corresponding to 1/4 mole layer (ML) coverage. Table 1 shows the adsorption
energies and geometries of H on the Ni(100) and Ni(100)/ YSZ(100) surfaces. The results indicate that the adsorption energies and geometries on the Ni(100) and Ni(100)/ YSZ(100) surfaces both are similar. Furthermore, the site is the most favorable binding site with the adsorption energies of −2.79, −2.72, and −2.68 eV for the Ni/cation, Ni/O, and pure Ni cases, respectively. These results are also consistent with previous experimental and theoretical values.49,50 In addition, the Ni−H bonding lengths for three cases are between 1.829 and 1.877 Å, which are also in good agreement with the theoretical result of H on the Ni(100), 1.84 Å, for the adsorption of H at a 4-fold hollow site.51
For the adsorption of H2, we have investigated both end-on (with the H−H bond normal to the Ni(100) surface) and side-on (with the H−H bside-ond are parallel to the Ni(100) surface) coordinations of the molecule to various adsorption sites on the Ni(100) surface of Ni and Ni/YSZ. For the end-on configuration, the optimized geometries and adsorption energies of the H2of the H2/Ni/YSZ and H2/Ni have been summarized in Table 2. The calculated H−H bond lengths in three different adsorption sites for three different cases are Table 1. Adsorption Energy and Bond Lengths of H on the Ni (100) and Ni(100)/YSZ(100) Surfaces with Ni/Cation and Ni/O Interfaces
Ni/Y ΔEads(eV) d(Ni−H) (Å)
Top (T) −2.22 2.16
Bridge (B) −2.69 1.631/1.628 fcc Hollow (H) −2.79 1.858
Ni/O ΔEads(eV) d(Ni−H) (Å)
Top (T) −2.19 2.16
Bridge (B) −2.66 1.629/1.648 fcc Hollow (H) −2.72 1.877
pure Ni(100) ΔEads(eV) d(Ni−H) (Å)
Top (T) −2.14 2.16
Bridge (B) −2.59 1.620/1.620 fcc Hollow (H) −2.68 1.842
about 2−6% longer than that in the gas phase. Among all of the calculated adsorption configurations of H2on the Ni(100) and Ni(100)/YSZ(100) surfaces, the adsorption energies of H2in most adsorbed sites vary within 0.0 to −0.02 eV (−0.03 to −0.46 kcal/mol), or some with slightly positive (unstable) values indicating that the adsorption of H2 on the Ni(100) surface with the end-on configuration belongs to physisorption. The side-on coordination results are shown in Table 3. We
found that the H2can absorb stably on the Top site (T) with the calculated adsorption energy of∼ −0.34 eV (−8 kcal/mol). In summary, it is found that the adsorption of H2molecule on the Ni(100) and Ni(100)/YSZ(100) surfaces favors the on Top site (T) with the side-on configuration.
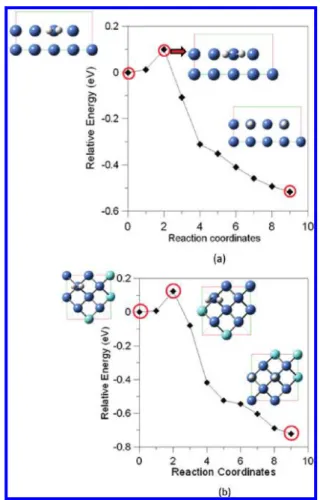
As mentioned above, the side-on adsorption of H2 is energetically more stable than the on adsorption. The end-on cend-onfiguratiend-ons of H2on the Top site of the Ni(100) and Ni(100)/YSZ(100) surfaces are chosen as the starting structures for the study of H2dissociation as shown in Figure 2a and b, respectively. The two separated H-atoms except at the Hsite (its most favorable binding site) are chosen as the final state. The dissociative barriers of H2 on the Ni(100) and Ni(100)/YSZ(100) surfaces are found to be about 0.1 eV, which is also consistent with the result reported by Madix and Hamza52with a molecular beam study. Our result supports the finding that the H2can easily dissociate on the Ni(100) surface due to the low energy barrier. In addition, we found the structure of the pure Ni(100) is unreconstructed after H2 dissociative adsorption on the surface, which is consistent with
other theoretical observations.49,53 Our results show that the interaction between the Ni and the YSZ support does not play a critical role in the reactivity for H2 adsorption and dissociation in view of the similar adsorption and dissociation behavior on both surfaces. We have also performed calculations for H2adsorption on (2× 1), (2 × 2), and (3 × 3) adlayer Ni(100) surfaces, corresponding to coverage of 1/2, 1/4, and 1/9 ML, respectively, using the most stable configuration of H2 on Ni(100). These studies show that the coverage effect on the calculated stability of this specie is negligible (smaller than 0.1 eV).
Energies and Mechanisms for H Migration. The dissociative adsorption of hydrogen results in two H-atoms adsorbed on the surface; the H-atoms may migrate on the surface. The potential energy surface (PES) for H-atom migration on the Ni(100) surface and diffusion into the bulk Ni is shown in Figure 3. For the diffusion of H-atoms on the Ni(100) surface, we found that the diffusion from the fcc Hollow (H) site to the Bridge (B) site (surface→TS1→ surface2) entails a small energy barrier of 0.1 eV. This value is in good agreement with the barrier height of 0.17 eV reported by studies of Truhlar et al.56Therefore, the H-atom can easily move on the clean Ni(100) surface at low temperatures because of the small diffusion barrier. For the tunneling of the H-atom in Ni, an activation barrier of 0.89 eV is required to surmount from a 3-fold site of Ni(100) into an octahedral site of the subsurface (surface→TS2→IM1). We also observed a barrier height of 0.29 eV from a subsurface site to the 3-fold site of Ni(100) which is slightly lower than that of H diffusion in Table 2. End-on Adsorption Energies and Bond Lengths of
H2on the Ni (100) and Ni(100)/YSZ(100) Surfaces with the Ni/Cation and Ni/O Interfaces
Ni/Y ΔEads(eV) d(H−H) (Å) d(Ni−H) (Å)
Top (T) 0.0 (0.01 kcal/mol) 0.77 2.07 Bridge (B) 0.0 (−0.03 kcal/mol) 0.75 2.69 fcc Hollow (H) 0.0 (0.07 kcal/mol) 0.75 2.97
Ni/O ΔEads(eV) d(H−H) (Å) d(Ni−H) (Å)
Top (T) 0.0 (0.02 kcal/mol) 0.76 2.08
Bridge (B) 0.16 0.78 1.95
fcc Hollow (H) 0.01 0.76 3.02
pure Ni(100) ΔEads(eV) d(H−H) (Å) d(Ni−H) (Å)
Top (T) 0.03 0.76 2.21
Bridge (B) 0.05 0.76 2.40
fcc Hollow (H) 0.0 (−0.02 kcal/mol) 0.75 3).05
Table 3. Side-on Adsorption Energies and Bond Lengths of H2on the Ni (100) and Ni(100)/YSZ(100) Surfaces with the Ni/Cation and Ni/O Interfaces
Ni/Y ΔEads(eV) d(H−H) (Å) d(Ni−H) (Å)
Top (T) −0.36 0.87 1.61
Bridge (B) − − −
fcc Hollow (H) − − −
Ni/O ΔEads(eV) d(H−H) (Å) d(Ni−H) (Å)
Top (T) −0.32 0.87 1.60
Bridge (B) − − −
fcc Hollow (H) − − −
pure Ni(100) ΔEads(eV) d(H−H) (Å) d(Ni−H) (Å)
Top (T) −0.34 0.86 1.62
Bridge (B) − − −
fcc Hollow (H) − − −
Figure 2. Potential energy diagrams of H2 dissociation on (a) Ni
(100) surface; (b) Ni(100)/YSZ(100) surface for the Ni-Cation model. Insets of (a) and (b) are the top views of simulation models.
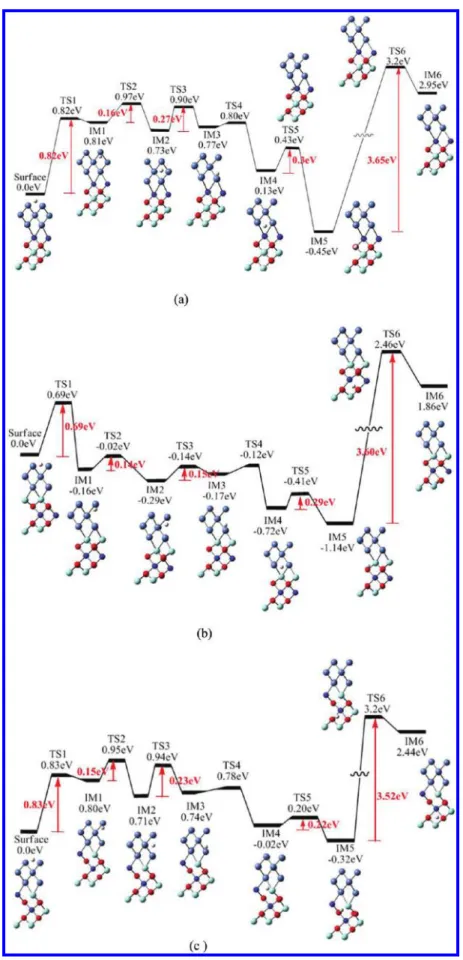
Ni(111).54In addition, H can further migrate in the bulk Ni from the octahedral structure of subsurface to the octahedral structure of the second layer (IM1→TS3→IM2→TS4→IM3), which only has to overcome two small barriers of 0.30 and 0.13 eV. However, the low barriers indicate that H can migrate in the bulk Ni. Furthermore, we have calculated the transverse diffusion of H along the [010] direction in between both tetrahedral structures on the same atomic layer in the bulk Ni. Similar barriers of 0.30 and 0.13 eV were also found. As discussed above, we can conclude that the most favorable diffusion path of H-atoms is the surface path. In addition, H diffusion in the bulk Ni is also possible but the surface barrier of 0.89 eV must first be overcome.
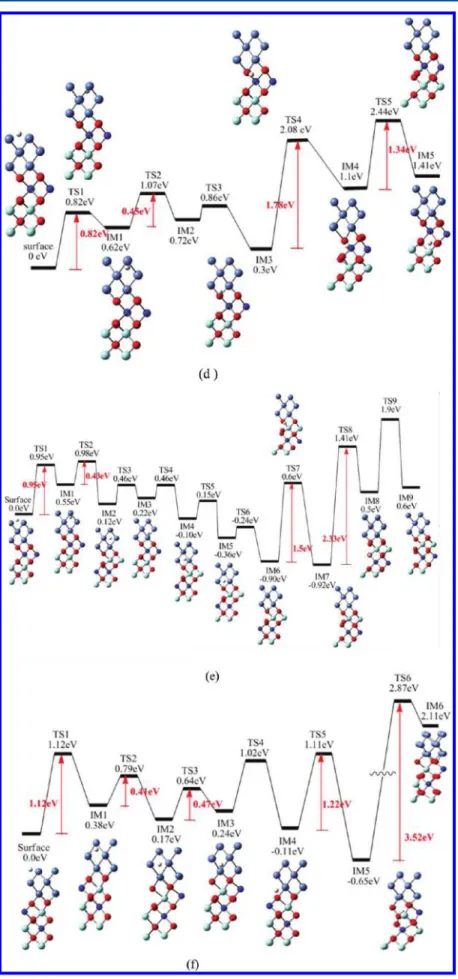
Since interstitial H-atoms may be present in the bulk Ni and YSZ ceramics of the SOFC anode at a high operating temperature,18−20 it is important to understand the migration of H-atoms into the bulk from the Ni surface and the tunneling motion of H-atoms in the Ni/YSZ crystal. In order to examine the role of the Ni/YSZ interface in the diffusion of H-atoms, the diffusion in the Ni/YSZ slab with Ni/cation and Ni/O interfaces was analyzed. Figure 4 shows the PES for the diffusion of H in the Ni/YSZ slab for the six different models given in Figure 2. In Figure 4, the diffusion barrier of H from the surface of Ni to the subsurface for six different models with errors estimated to be about 0.1−0.2 eV are compared to the diffusion result of H in the pure Ni case as shown in Figure 3. In addition, the barriers for H migration inside the bulk Ni from the tetrahedral configuration of subsurface to the octahedral configuration of the second layer are in the ranges of 0.15−0.27 and 0.43−0.47 eV for Ni/cation and Ni/O interfaces which are, respectively, slightly lower and larger than that for migration of H in the pure Ni, stemming from the interaction between the Ni and YSZ. The Ni/cation interface of Ni/YSZ seems to be more active for the H diffusion in the bulk compared to the diffusion in the Ni/O interface. For the path of H-atoms passing through the interface with a vacancy at the Ni/cation interface (IM4→TS5→IM5), the barriers of 0.3 eV, 0.29, and 0.22 eV for the Y doping in first, second, and first-second layers of the YSZ slab were found as shown in panels (a), (b), and (c) in Figure 4, respectively. These results indicate that the migration barrier depends strongly on the local atomic environment. Since the ionic radius of Y3+ ion is bigger than that of Zr4+ ion, the size effect leads to the highest diffusion barrier for Y doping in the first layer rather than in the second layer. In addition, the interface barrier of H diffusion for Y doping in the first-second layer is found to be lower than of that of Y doping entirely in the second layer. This is because the
adjacent edge of the interface of Y doping in the first-second layer may accommodate better the contraction of the lattice in the neighbor of the vacancy when an oxygen vacancy is formed. Therefore, the H-atom passes through most easily the interface of Ni/YSZ with Y doping in the first-second layer comparing to the other two doping cases. In order to study the effect of vacancy on the diffusion behavior, the change of potential energy for H diffusion at the interface of Ni/cation for Y doping in the first-second layer of the YSZ slab without oxygen vacancy is also calculated as shown in Figure 5. A barrier as high as 3.4 eV is found for H passing through the interface. Therefore, our barrier predicitions demonstrated the vacancy influence on H diffusion which improves the mobility of H. In the following process from IM5→TS6→IM6, that is, H diffusion in the YSZ slab, we found the transition structure TS6 for the three different cation edges of the Ni/cation interface, with a large barrier about 3.5 to 3.65 eV. These results suggest that the H-atom cannot move easily in the inner part of YSZ slab at operating temperature. At the Ni/O interface, the process of migration through the interface with 1.78, 1.5, and 1.22 eV barriers are also predicted for Y doping in the first, second, and first-second layers of the YSZ slab, respectively. The same trend can be observed in this process comparing with that of the Ni/cation interface, which is that the H-atom can most easily as through the interface of Y doping in the first-second layer of the Ni/YSZ interface. Comparing the Ni/cation and Ni/O interfaces, we found that the diffusion barriers of H-atoms crossing the Ni/O interface is much larger than that of the Ni/cation interface. These results can be attributed to the stronger interaction between the H-atom and the oxygen atom than that between the H-atom and the cation.
To understand the nature of interaction between the H-atom and the Ni, Y, Zr, and O atoms of Ni/YSZ in the diffusion process, we have calculated the Bader charge58on two Ni/YSZ cases, which are shown in Figures 1(c) and 1(f). We find that the H-atoms are all partially negatively charged wherever in the surface of Ni (100) surface or inside of Ni of Ni/YSZ slab, with the Bader charge of approximately −0.25e and −0.3e for the two cases, respectively. The H-atom in the Ni(100) surface of the Ni/YSZ slab is more negative charged than that in the pure Ni(100) surface. The results demonstrate that the interstitial H-atoms acquire electronic charge from the neighboring Ni(100) surface. We also find that, for H diffusion to the vacancy of the Ni/O interface, the Bader charge of H becomes substantially positive (about 1.0e). On the contrary, for H diffusion to the vacancy in the Ni/cation interface, the Bader charge of H becomes negatively charged (above −0.9e and −0.3e for the Ni/Zr and Ni/Y interfaces, respectively).
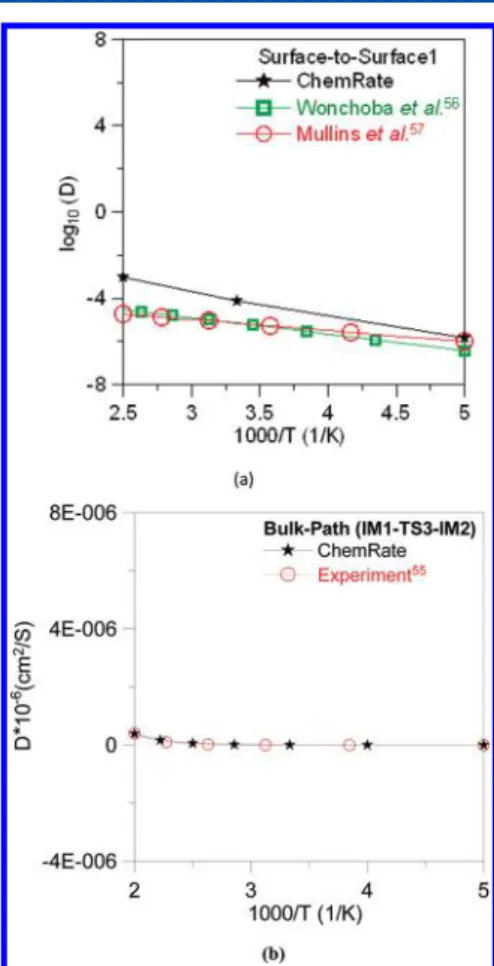
Kinetics for H2Dissociation and Diffusion. Based on the aforementioned PESs, we have calculated the rate constants for the dissociation of H2and diffusion of H-atoms in Ni and Ni/ YSZ by using the ChemRate program59on the basis of Rice− Ramsperger−Kassel−Marcus (RRKM) theory60,61as shown in Table 4. The predicted diffusion rates of H-atoms in Ni for the surface path (surface-IM1-surface2) and the bulk path (IM1-TS3-IM2) are D1 = 1.13 × 108(T/1000)3.32 exp(−3552/T) (cm2/s) and D
3 = 2.61 × 105(T/1000)0.333 exp(−3621/T) (cm2/s), respectively, which are in good agreement with of experimental55,57and theoretical results56as shown in Figure 6. For H-atom diffusion in Ni/YSZ, the predicted diffusivities of D4= 4.83× 104(T/1000)0.323exp(−2915/T) (cm2/s) and D5= 5.29× 104(T/1000)0.151exp(−3017/T) (cm2/s) are obtained for the IM4→TS5→IM5 (panel (c) of Figure 4) and IM4→ Figure 3.PES of H diffusion on the Ni(100) surface and inside the
Figure 4.Potential energy diagrams of H diffusion inside Ni-YSZ slab. Y ions doping in (a) first, (b) second, and (c) first-second layer of the Ni/ cation interface and (d) first, (e) second, and (f) first-second layer of the Ni/O interface, respectively.
TS5→IM5 (panel (f) of Figure 4) models, respectivly. From the rate constant predicitions of Table 4, our results show that H-atoms are more likely to migrate on the Ni surface (surface path) to the three-phase boundary.
■
CONCLUSIONSThe adsorption, dissociation, and diffusion of hydrogen in pure Ni and Ni/YSZ slabs with two different interfaces (Ni/cation and Ni/O interfaces) have been studied by the density functional theory (DFT) with the Perdew−Wang (PW91) functional. The H-atom was found to bind preferentially at the fcc Hollow (H) site on the Ni(100) surface with the adsorption energy of−2.68 eV. The H2molecule absorbed on the Top (T) site of Ni(100) surface with the side-on configuration has the adsorption energy of−0.34 eV (−8 kcal/mol). The calculated barrier energy for H2dissociation on both surfaces is about 0.1 eV. Calculations show that the diffusion energy for the adsorbed H on the Ni(100) surface or on the Ni(100)/ YSZ(100) is only ∼0.1 eV while the diffusion energy for H going through the bulk is as high as 0.89 eV at the first step (from surface to subsurface), suggesting that the surface path is more favorable. Comparing the Ni/cation and Ni/O interfaces, we find that the barrier for H migration through Ni/O is much larger than that through Ni/cation. Our results also demonstrate that the H-atom can pass through the interface most readily with Y doping in the first-second layer in the Ni/ cation case. In addition, the migration of H-atom in the YSZ
slab is found to be very high. On the other hand, the existence of vacancy can enhance the diffusion of H-atoms through the interface of the Ni/YSZ slab. The results of kinetic simulation indicate that H-atoms can possibly diffuse through the Ni bulk to the Ni/YSZ interface.
Since DFT calculation can gain insight into diffusion mechanism of H-atom to explain the effect of structural composition by calculating a set of energy barriers for H-atom migration in Ni/YSZ. The ionic conductivity of H-atom in Ni/ YSZ is interesting and may be investigated by kinetic Monte Carlo calculations based on first-principles quantum simu-lations in the future. On the other hand, Molecular dynamics (MD) simulations at different temperatures are also carried out to verify the interaction of H2 with the Ni surface (see the Supporting Information). The MD results indicate that the adsorption numbers of H2 molecules on the Ni substrate decrease with increasing temperature.
■
ASSOCIATED CONTENT*
S Supporting InformationMD simulations detail. This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: [email protected] (S.-P.J.); changjg@ nchc.org.tw (J.-G.C.); [email protected] (M.C.L.); [email protected] (H.-T.C.).
Figure 5.Potential energy diagram of H-atom passing through the Y− Zr interface in the Ni-YSZ slab without an oxygen vacancy. Table 4. Rate Constants for H2Dissociation (unit: 1/s) and H Diffusion (unit: cm2/s) in Pure Ni and Ni/YSZ Slab
paths rate constants
H2dissociation on pure
Ni(100) surface (Figure 2)
k1= 4.20× 1013(T/1000)0.92exp (−1260/T)
H diffusion in pure Ni (Figure 3)
surface→TS1→surface2 D1= 1.13× 108(T/1000)3.32exp (−3552/T)
surface→TS2→IM1 D2= 2.26× 105(T/1000)0.0475exp (−10545/T) IM1→TS3→IM2 D3= 2.61× 105(T/1000)0.333exp (−3621/T) H diffusion in Ni/YSZ IM4→TS5→IM5 (Figure 4(c)) D4= 4.83× 104(T/1000)0.323exp (−2915/T) IM4→TS5→IM5 (Figure 4(f)) D5= 5.29× 104(T/1000)0.151exp (−3017/T)
Figure 6.Diffusion of H-atoms on and in the pure Ni: (a) the surface path and (b) the bulk path comparing with experimental (refs 55 and 57) and theoretical results (ref 56).
Notes
The authors declare no competing financial interest.
■
ACKNOWLEDGMENTSThe authors would like to thank the ATU Plan of MOE, Taiwan, for partial support of this work. J.-G.C. and H.-T.C. want to acknowledge the support of this study, under Grant Nos. NSC99-2113-M-033-009-MY2 and NSC100-2628-E-110-002, and the National Center for High-performance Comput-ing, Taiwan, for the computer time and facilities. M.C.L. also wants to acknowledge Taiwan Semiconductor Manufacturing Co. for the TSMC Distinguished Professorship and Taiwan National Science Council for the Distinguished Visiting Professorship at the Center for Interdisciplinary Molecular Science, National Chiao Tung University, Hsinchu, Taiwan.
■
REFERENCES(1) Minh, N. Q.; Takahashi, T. Science and Technology of Ceramic Fuel Cells; Elsevier Science: Amsterdam, The Netherlands, 1995.
(2) Singhal, S. C.; Kendall, K. High Temperature Solid Oxide Fuel Cells; Elsevier: Oxford, U.K., 2003.
(3) Ivers-Tiffée, E.; Virkar, A. V. In High Temperature Solid Oxide Fuel Cells: Fundamentals, Design and Applications; Singhal, S. C., Kendall, K., Eds.; Elsevier: New York, 2003.
(4) Takenaka, S.; Kobayashi, S.; Ogihara, H.; Otsuka, K. Ni/SiO2
catalyst effective for methane decomposition into hydrogen and carbon nanofiber. J. Catal. 2003, 217, 79−87.
(5) Li, Y; Zhang, B.; Tang, X.; Xu, Y.; Shen, W. Hydrogen production from methane decomposition over Ni/CeO2catalysts. Catal. Commun.
2006, 7, 380−386.
(6) Maric, R.; Ohara, S.; Fukui, T.; Inagaki, T.; Miura, K. High-Performance Ni-SDC Cermet Anode for Solid Oxide Fuel Cells at Medium Operating Temperature. Electrochem. Solid-State Lett. 1998, 1, 201−203.
(7) Nguyen, T. L.; Kobayashi, K.; Honda, T.; Iinuma, Y.; Kato, K.; Negishi, A.; Nozaki, K.; Tappero, F.; Sasaki, K.; Shirahama, H.; Ota, K.; Dokiya, M.; Kato, T. Preparation and evaluation of doped ceria interlayer on supported stabilized zirconia electrolyte SOFCs by wet ceramic processes. Solid State Ionics 2004, 174, 163−174.
(8) Matsuzaki, Y.; Baba, Y.; Sakurai, T. High electric conversion efficiency and electrochemical properties of anode-supported SOFCs. Solid State Ionics 2004, 174, 81−86.
(9) Sumi, H.; Ukai, K.; Mizutani, Y.; Mori, H.; Wen, C. J.; Takahashi, H.; Yamamoto, O. Performance of nickel−scandia-stabilized zirconia cermet anodes for SOFCs in 3% H2O−CH4. Solid State Ionics 2004,
174, 151−156.
(10) Spacil, H. S. Electrical device including nickel-containing stabilized zirconia electrode, US Patent 3. 1970, 3558360.
(11) Ohara, S.; Fukui, T.; Mukai, K.; Kodera, K.; Kubo, Y. Solid Oxide Fuel Cells. The Electrochemical Society Proceedings Series, Pennington, NJ,1997, 97−40, pp 815.
(12) Fukui, T.; Ohara, S.; Mukai, K. Long-Term Stability of Ni-YSZ Anode with a New Microstructure Prepared from Composite Powder. Electrochem. Solid-State Lett. 1998, 1, 120−122.
(13) Horita, T.; Yamaji, K.; Kato, T.; Sakai, N.; Yokokawa, H. Design of metal/oxide interfaces for the direct introduction of hydrocarbons into SOFCs. J. Power Sources 2004, 131, 299−303.
(14) Atkinson, A.; Barnett, S.; Gorte, R. J.; Irvine, J. T. S.; McEvoy, A. J.; Mogensen, M.; Singhal, S. C.; Vohs, J. Advanced anodes for high-temperature fuel cells. Nat. Mater. 2004, 3, 17−27.
(15) Vogler, M.; Bieberle-Hütter, A.; Gauckler, L.; Warnatz, J.; Bessler, W. G. Modelling Study of Surface Reactions, Diffusion, and. Spillover at a Ni/YSZ Patterned Anode. J. Electrochem. Soc. 2009, 156, B663−B672.
(16) Primdahl, S; Mogensen, M. Oxidation of hydrogen on Ni/yttria-stabilized zirconia cermet anodes. J. Electrochem. Soc. 1997, 144, 3409− 3419.
(17) Holtappels, P.; Vinke, I. C.; de Haart, L. G. J.; Stimming, U. Reaction of Hydrogen/Water Mixtures on Nickel-Zirconia Cermet Electrodes: I. DC Polarization Characteristics. J. Electrochem. Soc. 1999, 146, 1620−1625.
(18) Mogensen, M.; Sunde, S.; Primdahl, S. Proceedings of the 17th Risoe International Sympo-sium on Materials Science, Risoe National Laboratory, Roskilde, Denmark, 1996, pp 77.
(19) McLellan, R. B.; Oates, W. A. The solubility of hydrogen in rhodium, ruthenium, iridium and nickel. Acta Metal. 1973, 21, 181− 185.
(20) Völkl, J.; Alefeld, G. In Hydrogen in metals; Alefeld, G., Völkl, J., Eds.; Springer-Verlag: Berlin, 1978, p 321.
(21) Küngas, R.; Vohs, J. M.; Gorte, R. J. Effect of the Ionic Conductivity of the Electrolyte in Composite SOFC Cathodes. J. Electrochem. Soc. 2011, 158, B743−B748.
(22) Johnson, A. D.; Maynard, K. J.; Daley, S. P.; Yang, Q. Y.; Ceyer, S. T. Hydrogen embedded in Ni: Production by incident atomic hydrogen and detection by high-resolution electron energy loss. Phys. Rev. Lett. 1991, 67, 927−930.
(23) Maynard, M. J.; Johnson, A. D.; Daley, S. P.; Ceyer, S. T. A New Mechanism for Absorption: Collision-induced adsorption. Faraday Discuss. Chem. Soc. 1991, 91, 437−449.
(24) Johnson, A. D.; Daley, S. P.; Ceyer, S. T. The Chemistry of Bulk Hydrogen: Reaction of Hydrogen Embedded in Nickel with Adsorbed CH3. Science 1992, 257, 223−225.
(25) Kammler, Th.; Wehner, S.; Kuppers, J. Interaction of thermal H atoms with Ni(100)-H surfaces: through surface penetration and adsorbed hydrogen abstraction. J. Surf. Sci. 1995, 339, 125−134.
(26) Kresse, G; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251−14269.
(27) Kresse, G. Dissociation and sticking of H2 on the Ni(111),
(100), and (110) substrate. Phys. Rev. B 2000, 62, 8295−8305. (28) Bhatia, B.; Sholl, D. S. Chemisorption and diffusion of hydrogen on surface and subsurface sites of flat and stepped nickel surfaces. J. Chem. Phys. 2005, 122, 204707−204714.
(29) Shishkin, M.; Ziegler, T. The Oxidation of H2and CH4on an
Oxygen-Enriched Yttria-Stabilized Zirconia Surface: A Theoretical Study Based on Density Functional Theory. J. Phys. Chem. C 2008, 112, 19662−19669.
(30) Shishkin, M.; Ziegler, T. Hydrogen Oxidation at the Ni/Yttria-Stabilized Zirconia Interface: A Study Based on Density Functional Theory. J. Phys. Chem. C 2010, 114, 11209−11214.
(31) Christensen, A.; Carter, E. A. Adhesion of ultrathin ZrO2(111)
films on Ni(111) from first principles. J. Chem. Phys. 2001, 114, 5816− 5831.
(32) Beltran, J. I.; Gallego, S.; Cerda, J.; Munoz, M. C. Oxygen vacancies at Ni/c-ZrO2interfaces. J. Eur. Ceram. Soc. 2003, 23, 2737−
2740.
(33) Dong, Y. F.; Wang, S. J.; Chai, J. W.; Feng, Y. P.; Huan, A. C. H. Impact of interface structure on Schottky-barrier height for Ni/ ZrO2(001) interfaces. Appl. Phys. Lett. 2005, 86, 132103−132105.
(34) Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989.
(35) Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15−50.
(36) Kresse, G.; Hafner, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169−11186.
(37) http://cms.mpi.univie.ac.at/vasp/.
(38) Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953−17979.
(39) Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758−1775. (40) White, J. A.; Bird, D. M. Implementation of gradient-corrected exchange−correlation potentials in Car-Parrinello total-energy calcu-lations. Phys. Rev. B 1994, 50, 4954−4957.
(41) Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.; Pederson, M. R.; Singh, D. J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671−6687.
(42) Monkhorst, H. J.; Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188−5192.
(43) Pornprasertsuk, R.; Ramanarayanan, P; Musgrave, C. B.; Prinz, F. B. Predicting ionic conductivity of solid oxide fuel cell electrolyte from first principles. J. Appl. Phys. 2005, 98, 103513.
(44) Ulitsky, A.; Elber, R. A new technique to calculate steepest descent paths in flexible polyatomic systems. J. Chem. Phys. 1990, 92, 1510−1511.
(45) Henkelman, G.; Uberuaga, B. P; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901−9904.
(46) Kittel, C. Introduction to Solid State Physics, 6th ed.; Wiley: New York, 1986.
(47) Zhao, X.; Vanderbilt, D. Phonons and lattice dielectric properties of zirconia. Phys. Rev. B 2002, 65, 075105−075105(10).
(48) Huber, K. P.; Herzberg, G. Molecular Structure and Molecular Spectra IV: Constants of Diatomic Molecules; Van Rostrand-Reinhold: New York, 1979.
(49) Lapujoulade, J.; Neil, K. S. Hydrogen adsorption on Ni(100). Surf. Sci. 1973, 35, 288−301.
(50) Bhawna, B.; Sholla, D. S. Chemisorption and diffusion of hydrogen on surface and subsurface sites of flat and stepped nickel surfaces. J. Chem. Phys. 2005, 122, 204707−204707(8).
(51) Stensgaard, I.; Jakobsen, F. Adsorption-Site Location by Transmission Channeling: Deuterium on Ni(100). Phys. Rev. Lett. 1985, 54, 711−713.
(52) Hamza, A. V.; Madix, R. J. Dynamics of the dissociative adsorption of hydrogen on nickel(100). J. Phys. Chem. 1985, 89, 5381−5386.
(53) Kresse, G.; Hafner, J. First-principles study of the adsorption of atomic H on Ni (111), (100) and (110). Surf. Sci. 2000, 459, 287− 302.
(54) Wonchoba, S. E.; Truhlar, D. G. General potential-energy function for H/Ni and dynamics calculations of surface diffusion, bulk diffusion, subsurface-to-surface transport, and absorption. Phys. Rev. B 1996, 53, 11222−11241.
(55) Edwards, A. G. Measurement of the diffusion rate of hydrogen in nickel. Br. J. Appl. Phys. 1957, 10, 406−410.
(56) Wonchoba, S. E.; Hu, W. P.; Truhlar, D. G. Surface diffusion of H on Ni(100): Interpretation of the transition temperature. Phys. Rev. B 1995, 51, 9985−10002.
(57) Mullins, D. R.; Roop, B.; Costello, S. A.; White, J. M. Isotope effects in surface diffusion: Hydrogen and deuterium on Ni(100). Surf. Sci. 1987, 186, 67−74.
(58) Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys.: Condens. Matter 2009, 21, 084204−084204(7).
(59) Mokrushin, V.; Bedanov, V.; Tsang,W.; Zachariah, M.; Knyazev, V. ChemRate, version 1.19; NIST: Gaithersburg, MD, 2002.
(60) Wardlaw, D. M.; Marcus, R. A. RRKM reaction rate theory for transition states of any looseness. Chem. Phys. Lett. 1984, 110, 230− 234.
(61) Klippenstein, S. J. Variational optimizations in the Rice− Ramsperger−Kassel−Marcus theory calculations for unimolecular dissociations with no reverse barrier. J. Chem. Phys. 1992, 96, 367− 370.