Kinetic Model of Thermal Degradation of Polymers
for Nonisothermal Process
BAR-LONG DENQ, WEN-YEN CHIU, KING-FU LIN
Institute of Materials Science and Engineering, National Taiwan University, Taiwan, Republic of China
Received 5 December 1996; accepted 30 December 1996
ABSTRACT: A new kinetic model was developed to describe the thermal degradation behavior of polymers. The model was applied to predict the degradation of poly ( methyl methacrylate ) ( PMMA ) blended with propyl ester phosphazene ( FR ) . The results showed that the thermal degradation mechanism of pure PMMA was dominated by zero- and first-order reactions. For PMMA blended with FR, the thermal degradation mechanism was dominated by first- and second-order reactions due to the formation of anhydride from the ester groups of PMMA. In addition, the major thermal degradation temperature of blends was greater than pure PMMA. By using our model, the activation energy of the thermal degradation PMMA was calculated to be 180 kJ /mol; this activa-tion energy increased as FR was added to PMMA.q 1997 John Wiley & Sons, Inc. J Appl Polym Sci 66: 1855 – 1868, 1997
Key words: poly ( methyl methacrylate ) ; propyl ester phosphazene; thermal degrada-tion kinetics
INTRODUCTION
lent tool for studying the kinetics of thermaldeg-radation. It provides information on frequency
The thermal degradation kinetics in polymers are factor, activation energy, and overall reaction
or-more complicated than in inorganic materials due der.1 – 5
Unfortunately, it does not provide clear
to the nature of polydispersity of polymer chains. information on thermal degradation mechanisms.
Upon thermal excitation, the covalent bonds in One compelling model of thermal degradation
polymer chains undergo complex vibration and ro- is the model, proposed by Ozawa6in 1993, of
non-tation motions within their local space. With fur- isothermal competitive reactions based on the
ther excitation these bonds can break to form a concept of reduced times. The model treats the
variety of fragment radicals or small molecules, conversion of reactant as being dependent only
which may further mutually recombine or break. on the sum of the reduced times of the reactions
Ultimately, the resulting fragments may be va- involved and as being independent of the path of
porized, diffused out, or carbonized. The thermal the process or the temperature change. This
behavior of materials can be improved if the infor- model has not been fully supported by the
litera-mation about the thermal degradation kinetics ture on the thermal degradation mechanism,
how-and degradation mechanisms can be employed to ever, which indicated that the degradation
behav-decrease the thermal degradation rate or increase ior also depends on the experimental conditions,
the heat resistance. such as heating rate, atmosphere, sample size,
Thermogravimetric analysis ( TGA ) is an excel- and sample geometry. In this work we have
devel-oped a model which accounts not only for the
ef-Correspondence to: W.-Y. Chiu. fects of parallel competitive reactions but also for
Contract grant sponsor: Chung Shan Institute of Science the effects of multi-order combined reactions. This and Technology ( CSIST ) , Taiwan, R.O.C.
model is applied to the thermal degradation
be-Journal of Applied Polymer Science, Vol. 66, 1855 – 1868 ( 1997 )
q 1997 John Wiley & Sons, Inc. CCC 0021-8995/97 / 101855-14 havior of poly ( methyl methacrylate ) ( PMMA ) 1855
©CH¤©C©( )n )n CH‹ OCH‹ OC‹ H‡ OC‹ H‡ CO ©N®P©( PMMA
Table I Physical Properties of PMMA and Propyl Ester Phosphazene
Propyl ester phosphazene
aDetermined by differential scanning calorimetry. bBy ASTM Code D-1238. cViscosity . 10§ poise (25ƒC). CM-211 Cyclic / linear Structure 5 65 : 35 Cl , 0.1 wt % 110 2100 Tga (ƒC) 14b c Melt Flow Index
(g /10 min) Materials Formula Remarks
and PMMA blended with propyl ester phospha- side chain, or cyclization. A first-order reaction
would reflect intramolecular transfer and ran-zene ( FR ) . The model’s insights into the thermal
degradation kinetics and mechanisms of PMMA dom scission of the main chain; and a
second-order reaction, weight loss related to two random and PMMA / FR blends are also discussed.
polymer segments colliding simultaneously, such as intermolecular transfer and scission. The thermal degradation rate equation which
KINETIC MODEL
accounted for each of these three reaction orders was then written as follows:
This new kinetic model was developed to ac-count for the type of bond scission and the state
of scission of the polymeric chain at any time. da
dt Åbr da
dT It was assumed that the probability of three
polymer segments simultaneously colliding
Å K0i / K1i( 1 0a) / K2i( 1 0a) 2
( 1 ) could be ignored. Therefore the overall thermal
degradation rate of the polymer at any time
could be represented as a combination of zero- where bis the heating rate;a is the weight loss
order, first-order, and second-order reactions. fraction; da/ dt is the thermal degradation rate at
Physically, a zero-order reaction would reflect any time; da/ dT is the thermal degradation rate
monomer scission from the polymer chain end at any temperature; K0i is the summation of rate
( depropagation ) , small molecule scission from a constants that belong to the zero-order reactions;
K1iis the summation of rate constants that belong
to the first-order reactions; and K2iis the summa-Table II Symbolic Meaning and Phosphorus tion of rate constants that belong to the second-Content of Blended Samples
order reactions.
Each of the above three rate constants change,
Weight
depending on the reaction temperature ( or time ) .
Ratio Pa Pb
They also change depending on the chain scission,
Code (PMMA/FR) (wt %) (wt %)
coupling, and cyclization which take place over the time period of the thermal degradation
pro-PMMA0F 10 : 0 0.00 õ 0.01
PMMA1F 10 : 1 2.01 2.01 cess. In order to solve eq. ( 1 ) simply, the following
PMMA2F 10 : 2 3.68 3.70 assumptions were made:
FR 0 : 10 22.1 22.1
1. Equation ( 1 ) is a continuous function
dur-aPhosphorus content from composition calculation.
bPhosphorus content by colorimetry. ing the thermal degradation process.
THERMAL DEGRADATION OF POLYMERS 1857
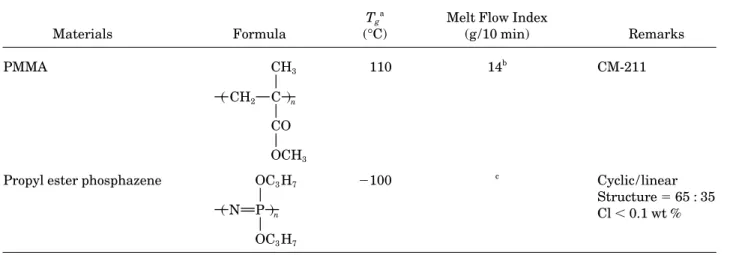
Figure 1 TGA thermograms of FR, PMMA, and their blends at different heating rates under nitrogen atmosphere.
2. The rate constant at any weight loss frac- zero during the thermal degradation
pro-cess. tion,a, is approximately equal to the rate
constant of its neighboring weight loss
fraction. K0i ¢ 0, K1i ¢ 0, K2i ¢ 0 ( 3 )
K0i ,a0 DaÉ K0i ,a É K0i ,a/Da
4. The rate constants for different weight loss K1i ,a0 DaÉ K1i ,a É K1i ,a/Da fractions are mutually independent.
K2i ,a0 DaÉ K2i ,a É K2i ,a/Da ( 2 )
Implementing the above assumptions, the rate equation was then written as
3. All of the rate constants are greater than
b d (a0Da) dT da dT d (a /Da) dT a Å [1 0 (a0Da) ]0 [1 0 (a 0Da) ]1 [1 0 (a 0Da) ]2 [1 0a]0 [1 0a]1 [1 0a]2 [1 0 (a /Da) ]0 [1 0 (a /Da) ]1 [1 0 (a /Da) ]2 K0i( Ta) K1i( Ta) K2i( Ta) ru ( T 0 Ta) ( 4 )
or where [ K ] is the matrix of rate constants; [ f (a) ] is the matrix of concentration; and u ( T 0 Ta) is
the unit function at T Å Ta.
b
F
dadT
G
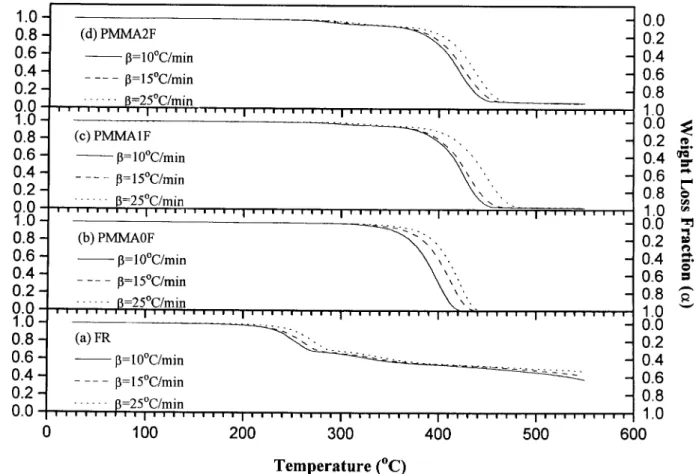
aÅ [ f (a) ] [ K ] u ( T 0 Ta) ( 5 )Solving for the matrix of rate constants, eq. ( 6 ) was obtained: [ [K] Å K0i( Ta) K1i( Ta) K2i( Ta) Å [ f (a) ]01b
F
da dTG
a Åb [1 0 (a 0Da) ]0 [1 0 (a0Da) ]1 [1 0 (a0Da) ]2 [1 0a]0 [1 0a]1 [1 0a]2 [1 0 (a/Da) ]0 [1 0 (a/Da) ]1 [1 0 (a/Da) ]2 01 d (a0Da) dT da dT d (a /Da) dT ( 6 )In order to compare this model with the
experi-mental work of other authors, we defined P1Å
K1i( 1 0a) K0i / K1i( 1 0a) / K2i( 1 0a) 2 ( 8 ) P2Å K2i( 1 0a)2 K0i / K1i( 1 0a) / K2i( 1 0a)2 ( 9 ) P0Å K0i K0i / K1i( 1 0a) / K2i( 1 0a) 2 ( 7 )
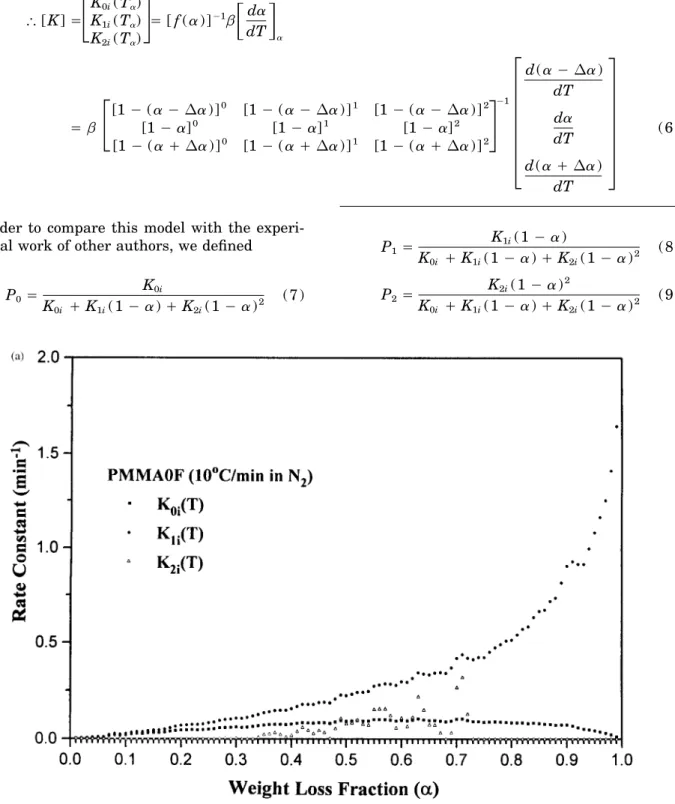
Figure 2 Relationship between the rate constant of different reaction order and weight loss fraction at a heating rate of 107C/min under nitrogen atmosphere for (a) PMMA0F, ( b ) PMMA1F, and ( c ) PMMA2F.
THERMAL DEGRADATION OF POLYMERS 1859
Figure 3 Thermal degradation rate of different reaction order as a function of temper-ature at a heating rate of 107C/min under nitrogen atmosphere for (a) PMMA0F, (b) PMMA1F, and ( c ) PMMA2F.
THERMAL DEGRADATION OF POLYMERS 1861
Figure 3 ( Continued from the previous page )
Figure 4 Relationship of activation energy versus weight loss fraction for PMMA blends, calculated by using Ozawa’s method.
Figure 5 Arrhenius plot of logarithmic value of average reaction rate constant versus inverse of temperature at different heating rates for ( a ) PMMA0F, ( b ) PMMA1F, and ( c ) PMMA2F.
THERMAL DEGRADATION OF POLYMERS 1863
Figure 5 ( Continued from the previous page )
oven at 1067C until the weight of the specimens
nV Å
∑
2
nÅ0
nPn ( 10 ) remained unchanged. The physical properties of
PMMA and FR are listed in Table I, and the sym-bolic meanings of the formulations used in this study appear in Table II. The phosphorus content of the PMMA blends was determined using a col-KU Å
b
S
dadT
D
( 1 0a)nV ( 11 ) orimeter,7and the results are listed in Table II.
Here P0, P1, and P2are the relative contributions Thermal Degradation Analysis of PMMA and
to the entire thermal degradation rate for the Its Blends
zero-, first-, and second-order reactions,
respec-The thermal degradation of the PMMA blends tively; and nV and KV are the averaged reaction
or-was investigated by TGA with a Perkin – Elmer der and rate constant, respectively. Finally, the
TGA-7 thermogravimetric analyzer at various activation energy was calculated from KV and
tem-heating rates under a nitrogen atmosphere. Ki-perature ( T ) using the Arrhenius equation.
netic parameters were calculated from eqs. ( 1 ) – ( 11 ) on a VAX workstation using FORTRAN.
EXPERIMENTAL
RESULTS AND DISCUSSION
Blending MaterialThe samples were prepared from mixtures of
Kinetics of Thermal Degradation of PMMA and PMMA supplied by Chi Mei Industrial Co. Ltd.
Its Blends (Taiwan, R.O.C.), and FR by the Chang Shan
In-stitute of Science and Technology ( Taiwan, In our previous study,8
the relationship between residual weight fraction ( 1 0 a) [ or weight loss R.O.C.) . Samples were mixed in acetone and
pre-pared at room temperature by a solvent-casting fraction (a) ] and temperature of PMMA blends
at different heating rates under nitrogen atmo-technique. The cast films were dried slowly in an
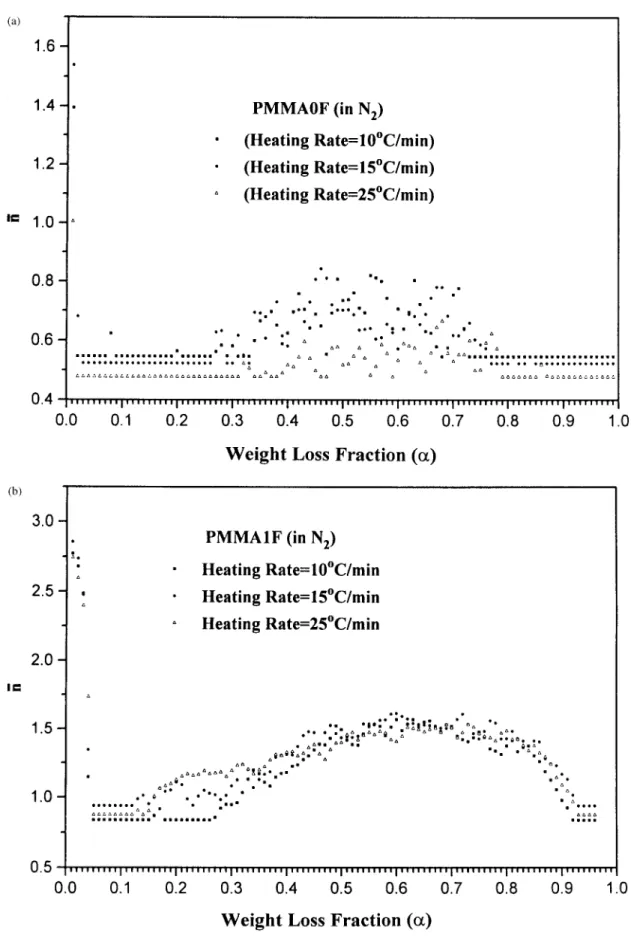
Figure 6 Relationship of average reaction order and weight loss fraction ( a ) at differ-ent heating rates for ( a ) PMMA0F, ( b ) PMMA1F, and ( c ) PMMA2F.
THERMAL DEGRADATION OF POLYMERS 1865
Figure 6 ( Continued from the previous page )
sphere was determined as illustrated in Figure 1. 17 ú 07 ú 27; note that the zero-order contribution
is close to the first-order contribution. The contri-Important phenomena during the thermal
degra-dation of PMMA blends were found, such as a bution to the thermal degradation rate from the
second-order reactions was obvious only at high reduction in the initial thermal degradation
tem-perature of PMMA blends relative to pure PMMA temperature. According to our proposed model,
the thermal degradation rate contributed from and an increase in the major degradation
temper-ature and residual char yield at 5507C with in- the zero-order reaction indicates the scission of
small molecule at side chains or cyclization or creasing levels of FR. These phenomena are
ex-plained as being due to the formation of anhydride monomer breaking away from polymer chain ends.
The thermal degradation rate contribution from the from the ester group of MMA during the thermal
degradation of PMMA blends. The PMMA blends first-order reaction indicates the weight loss due to
the random scission at various locations on the also showed an increase in their major thermal
degradation temperatures with increasing of the main chain; and the thermal degradation rate
con-tribution from the second-order reaction indicates heating rate due to a thermal lag effect, and,
lastly, they showed similar TGA thermograms, the weight loss related to the intermolecular
trans-fer and scission. Considering that our sample mate-which indicate that their thermal degradation
mechanism was similar at different heating rates. rials were polymerized by free radical
polymeriza-tion, and that current literature points out that dur-Using our proposed model, the relationship
be-tween the rate constants, separated by reaction ing the thermal degradation process the important
degradation mechanisms include depropagation, order, and weight loss fraction is shown in Figure
2. The heating rate here was 107C/min and ther- random main chain scission, intra- or
intermolecu-lar transfer, and the scission of small molecules mal degradation took place in a nitrogen
atmo-sphere. The contribution to the thermal degrada- from side chains, the thermal degradation rate data
obtained from our model are reasonable and in tion rates for different reaction orders in our
PMMA blends is shown in Figure 3 as a function agreement with the literature.9,10
The relationship between temperature and the of temperature. In Figure 3 ( a ) the contribution
differ-ent reaction orders for PMMA1F is shown in Fig- the first-order reactions. This phenomenon was ob-served from the analysis of the evolved gases and ure 3 ( b ) . The results indicate that the thermal
degradation rate of the first-order reaction was the residual yield in our previous study8,11
using TGA coupled with Fourier transform infrared spec-always greater than both the zero-order and the
second-order reactions; also that the thermal deg- troscopy and mass spectroscopy, respectively. In
that study it was determined that the evolved gas radation rate of the second-order reaction was
greater than the zero-order reaction only over the was mainly propylene and methanol. The propylene
resulted from the thermal degradation of FR,
temperature range from 415 to 4457C. At other
times the thermal degradation rate contributed whereas methanol resulted from the formation of
anhydride between FR and ester groups of PMMA. from the zero-order reaction was greater than the
In the range above 3507C the number of anhydride second-order reaction. In comparison with the
re-structures formed in PMMA2F was greater than in sults for PMMA0F, the thermal degradation rate
PMMA1F, which restricted direct chain scission of contributed from the zero-order reaction, due to
monomer from chain ends. For this reason the con-monomer breaking away from the polymer chain
tribution to the thermal degradation rate from the end, was drastically decreased as a result of
add-zero-order reaction was smaller than PMMA1F and ing FR. The reason for this decrease was the
for-PMMA0F. The ratio of anhydride and remaining mation of an anhydride structure from the
reac-MMA for Preac-MMA2F could be calculated as fol-tion of FR with the ester groups of PMMA which, in
lows12,13
: turn, restrained monomer scission from the polymer
chain. As for the second-order thermal degradation rate, its contribution was due to the generation of shorter polymer fragment radicals at high tempera-ture from random scission, which then caused weight loss through intermolecular transfer and scission. For PMMA2F, the relationship between thermal degradation rates and temperature for the different reaction orders is shown in Figure 3(c).
Here, the thermal degradation behavior of
PMMA2F depends on the temperature range. In the temperature range below 3507C the contribution to the thermal degradation rate from the zero-order reaction was greater than both the second-order and
©N®P©( )n OC‹ H‡ D OC‹ H‡ (I) (II) ©N©P©( )n 1 nC‹Hfl (12) O H OC‹ H‡ (III) ©N®P©( )n OH OC‹ H‡ 1 nC‹Hfl ©N®P© CH¤©C©CH¤©C© ( )n O2H1 H1 OC‹ H‡ OCH‹ CH‹ O®C OCH‹ CH‹ O®C 1 (13) ©N©P©( )n O H OC‹ H‡ 1CH‹ ©N®P© CH¤©C©CH¤©C© ( )n O2 OC‹ H‡ OCH‹ CH‹ O®C OCH‹ CH‹ O®C 1 ©N©P©( )n O H OC‹ H‡ CH¤©C©CH¤©C© 1 CH‹OH 1 CH‹ O®C O CH‹ C®O (14) ©N®P©( )n O2 OC‹ H‡ ©N©P©( )n O H OC‹ H‡
THERMAL DEGRADATION OF POLYMERS 1867
ing our model were slightly higher than those from Ozawa’s method but still within the values [
repeat number of MMA which formed anhydride structures
repeat number of
remaining MMA structures reported in the literature,
15 – 17
which hints that our proposed model is reasonable and correctly describes the thermal degradation mechanisms. In addition, the relationship between average re-action order of PMMA blends and weight loss frac-Å 20 1631 4 100( a ) 100( b ) 0 20 1631 4 5 1
1 ( 15 ) tion (a) is shown in Figure 6. These graphs show
that the reaction order of PMMA blends increases with increasing the content of FR.
where 100( a )
is the amount of PMMA in polymer blend; 100( b )
is the molecular weight of one unit
CONCLUSIONS
of MMA; 20 is the amount of FR in the polymerblend; 163 is the molecular weight of a propyl
1. A new kinetic model was developed to de-ester phosphazene unit,
scribe the thermal degradation behavior of polymers, which accounted for the effects of parallel competitive reactions and the effects of multi-order combined reactions. 2. The average apparent activation energy of
PMMA0F was about 180 KJ /mol during
©N®P©( )
OC‹ H‡
OC‹ H‡
the thermal degradation process, and the mechanism was dominated by the first-and zero-order reactions.
and 4 is the maximum repeat number of MMA
reacted with one unit of FR to form anhydrides. 3. Addition of FR caused the contribution to
the degradation rate from the first- and From the above calculation, the maximum
probability of the formation of anhydride struc- second-order reactions to become much
more important than the contribution from ture during the thermal degradation of PMMA2F
was about 50%. This indicated that above 3507C the zero-order reactions. Also, the average
apparent activation energy of PMMA the amount of the anhydride was obviously much
increased by the addition of 20 phr of FR and blends was increased by the addition of FR.
4. The major thermal degradation tempera-explained the reduction in the zero-order
contri-bution as a decrease in the amount of MMA mono- ture of PMMA blends increased with
in-creasing the content of FR. This phenome-mer broken from the polyphenome-meric chain end. The
large contribution from the second-order reac- non was due to the formation of an
anhy-dride structure from the ester group of tions above 3507C resulted from random main
chain scission in PMMA2F generating shorter MMA.
and more stable molecules than in PMMA0F and
in PMMA1F. This encouraged the intermolecular The authors thank Mr. Marc DeRosa for his English
correction and the Chung Shan Institute of Science and
transfer and scission.
Technology ( CSIST ) , Taiwan, R.O.C., for their
finan-In our previous study,8
the relationship
be-cial support.
tween apparent activation energy and weight loss fraction (a) was studied using Ozawa’s method14
as shown in Figure 4, and the results showed that
REFERENCES
the activation energies of PMMA0F, PMMA1F,and PMMA2F were about 154, 170, and 220 kJ /
1. A. Jimenez, V. Berenguer, J. Lopez, and A.
San-mol, respectively. In this article, the relationship
chez, J. Appl. Polym. Sci., 50, 1565 ( 1993 ) .
between the average thermal degradation rate
2. J. M. Salin and J. C. Seferis, J. Appl. Polym. Sci.,
constant of PMMA blends versus temperature,
us-47, 847 ( 1993 ) .
ing our proposed method, is shown in Figure 5. 3. J.-D. Nam and J. C. Seferis, J. Polym. Sci., Polym.
From this information the average apparent acti- Phys. Ed., 30, 455 ( 1992 ) .
vation energy of PMMA blends was calculated us- 4. J.-D. Nam and J. C. Seferis, J. Polym. Sci., Polym.
ing the Arrhenius equation; the results were 180, Phys. Ed., 29, 601 ( 1991 ) .
229, and 237 kJ /mol for PMMA0F, PMMA1F, and 5. Z. S. Petrovic and Z. Z. Zavargo, J. Appl. Polym.
Sci., 32, 4353 ( 1986 ) . PMMA2F, respectively. The results calculated
us-6. T. Ozawa, J. Thermal Anal., 39, 1117 ( 1993 ) . of Engineering, National Taiwan University, 1996,
submitted. 7. M. B. Tomson, J. P. Barone, and G. H. Nancollars,
Atomic Absorption Newsletter, 16, 117 ( 1977 ) . 12. S. J. Maynard, T. R. Sharp, and J. F. Haw,
Macro-molecules, 24, 2794 ( 1991 ) .
8. B. L. Denq, Y. S. Hu, W. Y. Chiu, L. W. Chen, and
Y. S. Chiu, Polym. Deg. Stab., to appear. 13. V. S. Papkov, M. N. IL’ima, D. R. Tur, and G. L. Slonimskii, Polym. Sci. USSR, 31, 2509 ( 1989 ) . 9. N. Grassie and G. Scott, Polymer Degradation and
Stabilization, Cambridge University Press, Cam- 14. T. Ozawa, Bull. Chem. Soc. Jpn, 38, 1881 ( 1965 ) . 15. V. A. Brockhaus and E. Jenckel, Makromol. Chem., bridge, 1985, p. 31.
10. I. C. McNeill, in Developments in Polymer Degrada- 18 / 19, 262 ( 1956 ) .
16. S. L. Madorsky, J. Polym. Sci., 11, 491 ( 1953 ) .
tion, Vol. 1, N. Grassie, Ed., Applied Science,
Lon-don, 1977, p. 47. 17. T. Hirata, T. Kashiwagi, and J. E. Brown,
Macro-molecules, 18, 1410 ( 1985 ) .
11. B. L. Denq and W. Y. Chiu, Bulletin of the College