行政院國家科學委員會專題研究計畫 成果報告
分子自組技巧應用於奈米科技
計畫類別: 個別型計畫 計畫編號: NSC91-2113-M-110-020- 執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日 執行單位: 國立中山大學化學系(所) 計畫主持人: 董騰元 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 92 年 10 月 29 日
行政院國家科學委員會補助專題研究計畫成果
報告
※※※※※※※※※※※※※※※※※※※※※※※
※
分子自組技巧應用於奈米科技
※
※
※
※※※※※※※※※※※※※※※※※※※※※※
計畫類別:■個別型計畫 □整合型計畫
計畫編號:
NSC91-2113-M-110-020
執行期間:
91 年 08 月 01 日至 92 年 07 月 31 日
計畫主持人:董騰元
共同主持人:
計畫參與人員:
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
執行單位:國立中山大學化學系
中 華 民 國 92 年 10 月 29 日
Progress Report
This section will begin a listing of papers published in the past year. Following the listing, only the most important results of the works supported by the National Science Council will be mentioned.
(I) Paper Published (2002-2003)
(II) Dong, T.-Y.; Chang, L. S.; Lee, G. H.; Peng, S. H. "57Fe Mössbauer
Spectroscopic and X-ray Structural Analyses on the Mixed-Valence State of Biferrocenium Derivatives with Bromoalkyl Substituents", Inorg. Chem. Comm. 2002, 5, 107-111.
(70) Dong, T.-Y.; Chang, L. S.; Lee, G. H.; Peng, S. M. "Pronounced Effects of Zero-Point Energy Difference on Intramolecular Electron Transfer in Asymmetric Mixed-Valence Biferrocenium Cations : Structural, EPR, and 57Fe Mössbauer Characteristics", Organometallics 2002, 21, 4192-4200.
(71) Dong, T.-Y.; Huang, B. R.; Peng, S. M.; Lee, G. H.; Chiang, M. Y. "Effects of counterions on intramolecular electron transfer in 1',1'''-dinaphthylmethyl-biferrocenium cation: structural, Mössbauer and EPR characteristics", J. Organomet. Chem. 2002, 659, 125-132.
(72) Dong, T.-Y.; Huang, S. J.; Tseng, I. M.; Chang, L. S. " Electroactive Self-Assembled Biferrocenyl Alkanethiol Monolayers on Au(111) Surface and on Gold Nanoclusters", Langmuir. 2003, submitted.
(73) Dong, T.-Y.; Huang, B. R.; Lin, M. C.; Chiang, M. Y. “A functionalized Pyridinyl Ligand Containing Binuclear Biferrocene”, Polyhedron 2003, 22, 1199-1204.
(74) Dong, T.-Y.; Lin, M. C.; Lee, L.; Cheng, C. H.; Peng, S. M.; Lee, G. H. “Pronounced Effects of Grinding on Rates of Intramolecular Electron Transfer in Mixed-Valence 1’,2’,1’’’,2’’’-Tetranaphthylmethyl- and 1’,3’,1’’’,3’’’-Tetranaphthylmethyl- Biferrocenium Triiodides”, J. Organomet. Chem. 2003, 679, 181-193.
(II) Results of Published Works (2002-2003) (a) Preparation of nanoparticle :
想作出均勻的Au nanoparticles 似乎不是想像中的那麼簡單,且其再現
性也是一大挑戰,這也是每一個研究奈米科技group 最頭痛的問題,我們也
作出一些較均勻的Au nanoparticle 其 TEM 如下圖,而且某些 condition 下似
乎也可以長出棒狀之Au 棒(化學方法)。
(b) 最近結果
(i) Monolayers at Au(111) and Au nanoparticle
The self-organization of various functionalized alkanethiols chemisorbed on two-dimensional Au surfaces has been extensively investigated. The chemisorbed of ferrocene-terminated alkanethiols with nonelectroactive n-alkanethiols on evaporated gold films improves the overall ordering of the adsorbed layer by constraining the ferrocene groups to a fixed position. An important recent report describing the stabilization of Au cluster with chemisorbed alkanethiols opens the way to study of three-dimensional monolayers by conventional methodologies such as NMR spectroscopy. In particular, alkanethiolate Au clusters are stable in air, soluble in
nonpolar organic solvents, and capable of facile modification with other thiols through exchange reactions. Following the initial report by Nishihara on a synthesis of biferrocene derivative-modified Au cluster 1, this paper describes the spectroscopic and electrochemical characterizations of a stable biferrocene-modified Au cluster 2, and chemisorbed biferrocenylalkanethiols on Au(111) surface 3.
The biferrocene-modified Au cluster 1 undergoes two-step one-electron oxidation processes in CH2Cl2 solution. Furthermore, the shape of the voltammogram changes
gradually during the potential scans, and it converges finally into one broaden redox wave. We suspect the stability of the Au cluster 1 in CH2Cl2 solution during the
potential scans. Our results indicate that the chemically oxidized dication of acylbiferrocenium is not quite electrochemically stable in organic solvents. Our strategy is the use of redox stable biferrocenylalkanethiol attached to the Au cluster and Au(111) surface.
S R S R S S S R S S R S R S R S S S R SR S R S R S R S R S S S S S S R C S (CH2)7 O S Fe Fe (1) : -C8H17S S R : S (CH2)6 S Fe Fe (2) : -C6H13S S R : S (CH2)6 S Fe Fe (3) : -C6H13S S R :
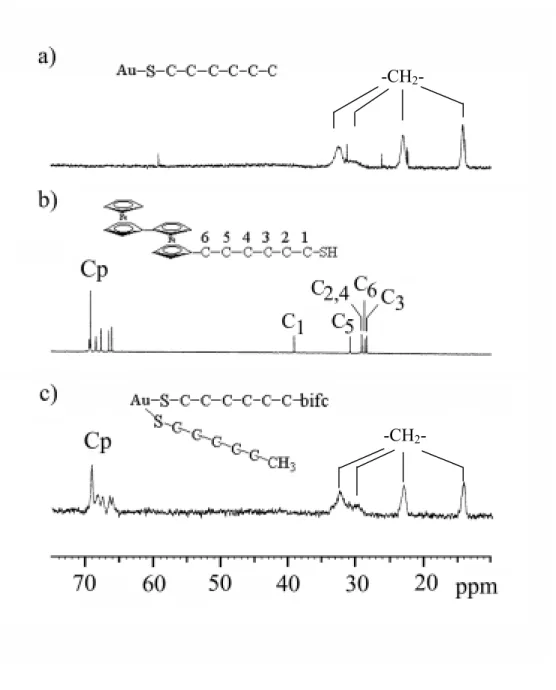
Gold cluster 2 was prepared by substitution method of hexanethiol-stabilized Au cluster developed by Murray. A TEM image of 2 indicates that the core size of the cluster is ~2.2 nm corresponding to 309 Au atoms. It is proposed that the number of hexanethiolate units on one cluster is in the range 63~65 estimated from the elemental analysis data, the number of biferrocenylhexanetholate units on the cluster is 9 estimated from the ICPMS. The IR spectra of the Au cluster 2 and of biferrocenylhaexanethiol are similar, which indicates that the biferrocenylthiol is indeed part of the composite. Figure 1 shows the 13C NMR spectra of hexanethiol (a), biferrocenylhexanethiol (b), and the Au cluster2 (c). The chemical shift assignments
for the carbons (and proton, not shown) on the hexanethiol chain were based on 2D H-H, C-C, and C-H correlation experiments.
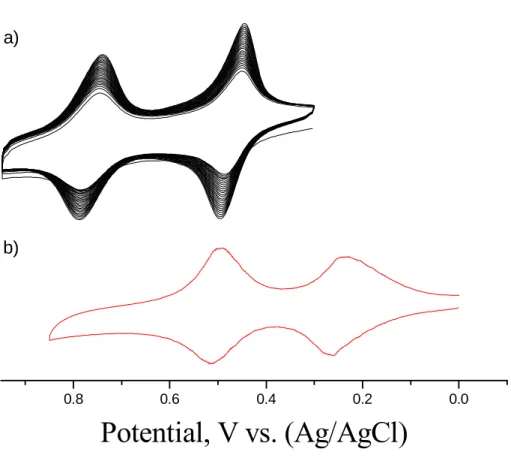
When the cyclic potential scans are carried out in DME solution of 2, two successive reversible one-electron redox waves were observed (E1/2 = 0.47; 0.78 V vs
Ag+/Ag). As shown in Figure 2(a), the two redox waves increase slightly in size with repeated scanning (100 mvs-1), donating electrodeposition of a redox-active film. When the voltammetry is carried out at different scan rates, the positive and negative current peaks for each redox wave occur at almost the same potential (peak-to-peak separation ~30 mv) and the peak current increases almost linearly with the sweep rate, implying that the Au cluster is adsorbed on the electrode surface. The magnitude of the peak separation between two waves (△E1/2) gives an indication of the interaction
between the two Fe sites. In the case of free biferrocenylhexanethiol, the two reversible redox waves (E1/2) were observed at 0.45 and 0.78 V. A comparison of the
magnitude of △E1/2 of biferrocenylhexanethiol (0.33 V ) and the Au cluster 2 (0.31
V ) reveals that the magnitude of the interaction between the two Fe sites in the Au cluster is smaller. We suggest that this difference in Fe-Fe interaction between solution state and structurally defined self-assembled Au cluster is a result of
difference in the degree of reorganization energy.
Electrochemical measurements were also made for the chemisorbed biferrocenyl- hexanethiol on Au(111) surface (3) in a cell in which a 0.61cm2 glass tube was pressed down against the Au(111) surface. The electrolyte (1 M HClO4), the counter
electrode (Pt), and the reference electrode (Ag+/Ag) were placed in the tube. In Figure 2b is shown cyclic voltammogram of the monolayer for the biferrocenylhexanethiol on Au(111). Two reversible redox waves were observed (E1/2 = 0.25 and 0.51V).
Repeated rescanning (100 mVs-1) does not change the voltammograms, demonstrating that the monolayer is stable to electrochemical cycling in 1M HClO4 solution. The
peak-to-peak separation for each wave is ~20 mV and the peak current increases linearly with the sweep rate. From the similarities of the electrochemical measurements between 3D-SAM Au cluster and biferrocenylhexanethiol monolayer on 2D-SAM Au(111) surface, it arises the question whether the character on alkane chain ordering on the planer Au(111) surface shows intrinsic differences from the Au cluster (owing to the surface curvature) or not. A comparison of the △E1/2 value of
the biferronylhexanethiol on Au(111) ( 0.26V ) and the Au cluster 2 ( 0.31V ) indicates that the interaction between the two Fe centers in the monolayer on Au(111) surface is smaller. We suggest that this is a result of difference in the degree of reorganization energy between “solid-like” biferrocenylhexanethiol on Au(111) surface and the “solution-like” Au cluster.
The coverage of biferrocenyl sites on the Au(111) surface evaluated from the peak area of cyclic voltammogram is 3.5×10-10 molcm-2. A study of coadsorption of ferrocene-terminated alkanethiols with unsubstituted alkanethiols on evaporated Au films was supported by Chidsey. Lower concentrations of alkanethiols linked to ferrocene (Fc) by a polar ester group (FcCO2(CH2)nSH) show ideal surface
linked directly to the nonpolar ferrocene group (Fc(CH2)nSH) lead to broadened and
asymmetry electrochemical features. In Figure 2b, as the mole fraction of biferrocenylhexanethiol (χbifc) is equal to 1, the first and second redox waves are
nearly symmetric with a full width at half maximum of ~120 and 150mV, respectively, and with ~20 mV peak-to-peak separation. Using biferrocenylhexanethiol and hexanethiol as coadsorbates results on very similar cyclic voltammograms for χbifc =
0.25, 0.1, and 0.067. Ideal redox peaks are symmetric with ~90 mV full width at half maximum and with no splitting between the oxidation and reduction peaks.
In conclusion, we have prepared stable biferrocenylhexanethiol of Au nanoparticles and on Au(111) surface. This might be useful in molecular electronic devices.
Figure 1. The 13C NMR spectra : a) hexanethiol-Au; b) biferrocenylhexanethiol; c) the Au cluster 2. The NMR spectra were obtained in CDCl3 solution.
-CH2
-Figure 2. Cyclic voltammograms : a) the Au cluster 2 in DME solution; b) biferrocenylhexanethiol on Au(111) surface in 1M HClO4 solution.
0.8 0.6 0.4 0.2 0.0
b) a)
(ii) Molecular Wire Containing Redox Active Subunit
The manufacture of supramolecular wire is an active area of research in chemistry and material science. However, how to arrange the various components into an ordered wire that permits controlled transfer of stored information along the molecular wire is an important issue. The research groups of Harrimann and Ziessal reported a photoactive molecular wire on stiff ditopic ligands and oligomers derived from terpyridines bridged by an alkyne spacer. The polyalkynylene chain allows very fast exchange of electron between photoactive terminals.
The ruthenium(II) bis(2,2’:6’,2”-terpyridine) complexes are normally only weakly luminescent at room temperature but attaching an alkynylene group at the 4’-position switches on the desired emission.
Our interest in the design of materials for applications in molecular electronics has focused on the molecules having spatially remote, redox-active subunits in good electronic communication. These redox-active subunits, as a consequence of specific interactions after undergoing electrochemical processes, can be the sites for the storage of energy. Thus, the binuclear biferrocene appears to be promising bridge which can ensure fast and quantitative exchange of information within the array. Here, we describe the preparations of multinuclear complexes assembled from 1’,1’’’-bis- (terpyridyl)biferrocene redox-active subunit attached to a ruthenium(II) metal center.
The new functionalized oligopyridine ligand was prepared by the reaction of biferrocene-1’,1’’’-dicarbaldehyde with 2-acetylpyridine to give the bis-chalcone as indicated in Scheme 1. The reaction of bis-chalcone with N-[1-oxo-2-(2-pyridyl)-ethyl]pyridinium iodide in ethanol and an excess of ammonium acetate yielded 1’-1’’’-bis(terpyridyl)biferrocene. We have also investigated the coordination behavior of this novel ligand, which is expected to show behavior similar to that of 2,2’:6’,2’’-terpyridine. The reaction of 1’,1’’’-bis(terpyridyl)biferrocene with 4’-substituted (terpyridyl)ruthenium(II) chloride gave a dark coloured solution, from which the desired complexes was precipitated as a solid by the addition of NH4PF6. Analytical and spectroscopic data
are in agreement with the proposed formulation.
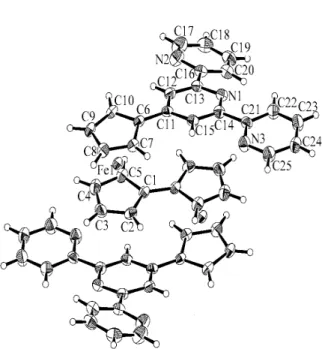
The structure of 6 was confirmed by single crystal X-ray structure determination. An ORTEP view of 6 is shown in Figure 3, and confirms the molecular structure with
N N N N N N Ru N N N N N N Ru alkyne spacer
the ferrocenyl group directly linked to the 4’-position of the 2,2’:6’, 2’’-terpyridine. The 2,2’:6’, 2’’-terpyridine group adopts the expected trans-trans conformation about the interannular C-C bonds. The terpyridine group is not completely coplanar and exhibits dihedral angles between the N1-C11-C12-C13-C14-C15 and
N2-C16-C17-C18-C19-C20 planes, and between the N1-C11-C12-C13-C14-C15 and
N3-C21-C22-C23-C24-C25 planes of 14.52 and 9.18°, respectively. The directly bonded
Cp ring of the ferrocene group is not coplanar with the central ring of terpyridine, but is slightly twisted with a dihedral angle of 11.87°. This degree of non-planarity is within the range expected for crystal packing effects, and represents no significant loss of conjugation between the two rings.
Compound 6 is redox active and exhibits reversible processes in the CV (CH2Cl2
solvent; AgCl /Ag as reference; [NBu4][PF6] as supporting electrolyte ) at 0.51 and
0.94 V respectively, with ∆Ep value of 72 mV and an anodic-cathodic intensity ratio equal to unity. The oxidation processes correspond to the formation of the appropriate ferrocenium salts. A comparison of the magnitude of ∆E1/2 of 6 (0.43 V) with that of
biferrocene (0.31 V) indicates that the magnitude of interaction between the two Fe sites in 6 is larger to that in biferrocene. The comproportionation constant equilibrium calculated from ∆E1/2 is 1.94 × 107.
The reaction of 6 with RuLCl3 (a: L = 2,2’:6’, 2’’-terpyridine; b: L=
4’-ferrocenyl-2,2’:6’,2’’-terpyridine; c: L= 4’-biferrocenyl-2,2’:6’,2’’-terpyridine), followed the reduction with N(C2H5)3 and the precipitation with [NH4][PF6], gave a
dark violet-blue solid (7). The 1H-NMR and mass spectrum (ES-MS) are in good agreement with the proposed structure. As shown in Figure 4, the Ru(II) complex 7a shows three reversible oxidation processes on sweeping at anodic potentials, corresponding to the oxidation of biferrocenyl moiety and Ru(II)-Ru(III) processes at 0.61, 0.92 and 1.35 V respectively. As expected, the intensity of the wave at 1.35 V is twice that of the first wave (0.61 V) and the second wave (0.92 V). As shown in Figure 4, two consecutive reduction waves attributed to the reduction of the Ru(terpy)22+ core at –1.16 and –1.43 V. The smaller ∆E1/2 value (0.3 V) of the
biferrocenyl moiety than that for free ligand 6 is as expected on the basis of charge build-up after the coordination of Ru(II) ion. In the case of 7b, the presence of additional ferrocenyl group dose not change the redox potentials appreciably. Three reversible oxidation processes at anodic potentials, corresponding to the oxidation of ferrocenyl and biferrocenyl moieties and Ru(II)-Ru(III) processes at 0.65, 0.93, and 1.35 V respectively. Furthermore, two consecutive reduction waves attributed to the reduction of the Ru(terpy)22+ core at –1.18 and –1.42 V were also observed. The
intensity of the process at 0.65 V obtained from the differential pulse voltammetry indicates that this process involves the oxidation of ferrocenyl group and one of Fe
centers of biferrocenyl group at the same potential. The use of the functionalized 4’-biferrocenyl-2,2’:6’,2’’-terpyridine ligand (bifcterpy) in the complex 7c introduces into the system a new biferrocenyl group. The CV of this complex exhibits four reversible oxidation processes at 0.47, 0.59, 0.87, and 1.35 V. The presence of a 4’-substituted group on the terpy ligand in complexes 7a-c does not change the Ru(II)-Ru(III) potential (1.35 V). However, there were appreciable variations detected in the potentials associated with the Fe(II)-Fe(III) processes of the ferrocenyl moieties. The intensity obtained from the differential pulse voltammetry and the comparison of the oxidation potentials indicate that the oxidations of the spacer 6 occur at potentials of 0.59 and 0.87 V. The oxidations of the terminal bifcterpy occur at potentials of 0.47 and 0.87 V which are comparable to the oxidation potentials of the free ligand bifcterpy (0.50 and 0.90 V). The variations of the ∆E1/2 values (0.31 V in 7a; 0.28 V in
7b; 0.28 V in 7c) and the appreciable variations detected in the Fe(II)-Fe(III)
oxidation potentials strongly indicate that there is a interaction between the spacer and terminal ferrocenyl moieties.
Most important result, we find that the redox-active binuclear biferrocene subunit is a promising bridge in which the two Fe centers show quantitative electron-transfer process after undergoing electrochemical oxidation. The mixed-valence compound [7a]+[PF6]- was prepared. The room temperature EPR
spectrum of powder sample of [7a]+[PF6]- is an isotropical signal at gav=2.00. The 77
K EPR spectrum is clearly a typical axial-type spectrum (g⁄⁄=3.32; g⊥=1.99). The
value of ∆g tensor anisotropy (∆g=g⁄⁄-g⊥) is considerable reduced. This is a reflection
of considerably reduced orbital angular momentum in the ground state that results from admixture of the S=0 Fe(II) electronic state into the Fe(III) electronic ground state. Furthermore, the admixture of electronic states could be the origin of the EPR signal which can be observed at room temperature. The mixed-valence cation [7a]+ was also examined by 57Fe Mössbauer technique. At 80 K, the Mössbauer spectrum shows two doublets, one for the Fe(II) metallocene (∆EQ=2.00 mms-1) and the other
for the Fe(III) metallocene (∆EQ=0.49 mms-1). Both doublets have the same area. This
pattern of two doublets is what is expected for a mixed-valence biferrocenium cation which is valence trapped on the Mössbauer technique (electron-transfer rate < ≈107 s-1 at 80 K).
In conclusion, we have prepared a series of polynuclear redox active supramolecule with the functionalized biferrocenyl ligand. All these data show that the use of suitable metal ions such as Ru(II) will prove to be versatile in building “electron- reservoir’’ molecular wire.
n-BuLi DMF Fe Fe Br Br Fe Fe CHO Fe Fe CHO CHO + 1 2 1 (i) (ii) N N N Fe Fe N N N Fe Fe N N N (ii) 6 7a 8a 7b 8b 8c + + + (iii) (iii) (iii) 7c
: ferrocenyl ; : Ru (II) ; : terpyridinyl ligand
(i) 2-acetyl pyridine, NaOH, EtOH (ii) Py+I-, NH4OAc, EtOH
(iii) RuLCl3, N(Et)3, NH4PF6 (L= terpy (7a); fcterpy (7b); 4 (7c))
6 4 Fe Fe 3 Fe Fe 5 O 2 (i) N O N O N Scheme 1.
Figure 3. ORTEP drawing for compound 6.
Figure 4. Cyclic voltammograms of 7a- c (Insert: DPP for 7c).
1000 500 0 -500 -1000 -1500