Excimers in light-emitting conjugated polymers
Hsin-Fei MengInstitute of Physics, National Chiao Tung University, Hsinchu 300, Taiwan ~Received 19 December 1997!
The electronic structures of the low-lying excited states for two parallel chains of poly
( p-phenylene-vinylene! with arbitrary relative positions are studied. The interchain interaction is treated
mi-croscopically. The energy and charge transfer fraction of the ~light-emitting! lowest singlet state are found
to depend sensitively on the horizontal shift of the two chains, which is in turn determined by the
pack-ing geometry. Our predictions of the excimer-formpack-ing geometry and the relative~dipole-forbidden!
photolu-minescence lifetimes are in excellent agreement with experiments.@S0163-1829~98!07529-8#
The physics of electroluminescent conjugated polymers, especially poly( p-phenylene vinylene! ~PPV!, has been of major interests since the first demonstration of a light-emitting diode ~LED! based on such materials.1Despite the ensuing extensive studies, the photophysical process under-lying both the photoluminescence ~PL! and electrolumines-cence ~EL! in solid state is still not completely settled. Re-cently, it has been observed that the light-emitting species in some PPV derivatives ~CN-PPV and Br-PPV! are found to be quite different from the others, both in terms of spectrum and lifetime.2,3The fact that a major difference in the spectra exists between chemically very similar derivatives is surpris-ing. Since no such spectral difference is observed in the so-lutions, the interchain interaction is believed to be the origin. Excimer formation4,5has been proposed as a possible expla-nation.
Assuming the intrachain electronic structure is not af-fected by the side groups, the question is how does the pack-ing geometry determine the emission process. In order to answer the question, we study in this paper a two-chain model including microscopic interchain interactions. Con-figuration interaction among exciton ~EX! and charge-transfer ~CT! states, common in smaller organic molecules,6,7are found to be indispensable in obtaining the correct excited states for polymer films as well. Our key result is that the electronic structure of the emitting state
~lowest singlet excited state! depends sensitively not only on
the interchain distance, but also the relative horizontal shift of the two chains in a dramatic way. Based on the packing geometry obtained from computer simulation by Conwell
et al.,8a significant CT component is found in the emitting state for CN-PPV, but it is negligible in MEH-PPV and PPV. These results completely agree with the conjecture based on experimental data that excimers are formed in the first but not in the latter two.2,3Furthermore, three experimental sig-natures of excimers, large Stokes shift, broad emission spec-trum, and, most importantly, long radiative lifetime,2 all agree with our calculation. Our results are opposite to the intuitive interpretation8in one crucial aspect. We found that the emitting state is always of even parity, implying the tran-sition to the ground state is dipole forbidden. Vibrational coupling to a higher odd parity state is necessary for the transition to happen, with the transition rate inversely pro-portional to the square of the energy splitting of the two
states due to interchain interaction. This implies that the larger the interchain interaction ~and therefore the CT com-ponent!, the smaller the transition rate is, instead larger as Conwell et al. claimed. This explains the extraordinarily long radiative lifetime of CN-PPV,3 which has a quite large CT component by our calculation.
Consider two chains a and b, such that b is displaced from a by a rigid translation vector dez1Dh. ez is the unit vector normal to the symmetry plane ~approximately the benzene plane! of chain a. Dh is the horizontal shift parallel to the plane. The Hamiltonian for the p electrons contains four pieces:
H5H0a1H0b1Hi1Hc. ~1!
The first two are intrachain tight-binding Hamiltonians for chains a and b, respectively. Hiis the interchain hopping and
HC the Coulomb interaction. In second quantized form,
H0a,b52
(
~i j!,s ti jcˆisa,b†
cˆa,bjs1H.c. ~2!
Here cˆia,bs is the annihilation operator of electrons on carbon site i and spin s of chain a and b, respectively. We include only the nearest-neighbor intrachain hopping. ti jis equal to t for benzene bonds, t1 for single bonds, and t2 for double
bonds@see inset of Fig. 1~b!#. For the interchain hopping,
Hi52
(
n51 N(
l51 8(
s561i(
51 m tli@cˆn fb†~l,i!scˆnals1H.c.#. ~3!Here n is the index for the unit cells and N is the total number of cells in each chain.l is the index for the carbon atoms within each unit cell@see inset of Fig. 1~b!#. One-to-many interchain electron hopping is allowed from carbon site l on chain a to site f (l,i) on chain b with hopping integral tli. m is the maximal number of i. Hopping from b to a is in the H.c. part of Hi. Note that the p electrons are assumed to be superpositions of carbon pz orbitals only. Both intrachain and interchain carbon-hydrogen coup-lings are ignored. The eigenstates of Ha and Hb are the Bloch states, with the annihilation operators Aˆkas
5(nleiknazla(k)cˆnls a
, Bˆkas5(nleiknazla(k)cˆnls b
, where a is the lattice constant. k lies in the first Brillouin zone PRB 58
@2p/a,p/a#. a51•••8 is the band index. In terms of Aˆkas and Bˆkas, Ha,b is in diagonized form: Ha
5(ksaEa
0
(k)Aˆk†asAˆkas, Hb5(ksaEa
0
(k)Bˆk†asBˆkas. The band structure Ea0(k) is well known.9The lower four bands are filled in the ground state. In terms of the Bloch states, Hi becomes Hi52
(
kaa8s Taa8~k!Aˆk†asBˆka8s1H.c., Taa8~k!5N(
l51 8(
i tlizla~k!zaf~l,i!8* ~k!. ~4!The hopping integral is assumed to be the form tli
5A exp(2mrli), where rli is the distance between site l
on chain a and site f (l,i) on chain b, which depends on, besides l and i, both the vertical distance d and horizontal shift Dh between the two chains.m is the size of the carbon
pz orbital and A is a fitting parameter.10In order to simplify our problem, we include only the middle two bands, the conduction (a5c) and the valence band (a5v), and rewrite the Hamiltonian in terms of the electron operators
ak,s[Aˆkcs,gk,s[Bˆkcs, and hole operatorsb2k,2s[Aˆk†vs,
d2k,2s[Bˆkvs
† . Within such a two-band approximation, the
total Hamiltonian becomes H5H˜a1H˜b1H˜C1H˜i. Here
H˜a5(ksEe(k)aks
† a
ks1Eh(k)bks
† b
ks. H˜b can be obtained by making the substitutiona→g andb→d. The renormal-ized single-particle spectra for electrons and holes are
Ee(k)5Ec(k)1DEc, Eh(k)52Ev(k)1DEv. The band edge renormalizations DEc andDEv are integrals involving the Bloch wave functions and the Coulomb potential V(r). The Coulomb part H˜C splits further into five pieces: H˜C
5HC1 a 1H C2 a 1H C1 b 1H C2 b 1H C ab , with HC1a 51 2 kk
(
8qÞ0V1~q!~ak1qs † a k82qs8 † ak8s8aks 1bk1qs † b k82qs8 † bk8s8bks 22ak1qs † b k82qs8 † bk8s8aks! ~5! and HC2a 5(
kk8q V2~q!ak†1qsb2k2s† b2k81q2s8ak8s8. ~6! As before, HC1,2b can be obtained by the substitution a→g andb→d. The interchain Coulomb interaction HCa,bisHCab51 2 kk
(
8q Vab~q!~ak1qs † g k82qs8 † gk8s8aks 1bk1qs † d k82qs8 † dk8s8bks2ak1qs † d k82qs8 † dk8s8aks 2gk1qs † b k82qs8 † bk8s8gks!. ~7! In order to get the eigenstates of the Hamiltonian, we first break the total Hamiltonian into ‘‘free’’ and ‘‘interaction’’ parts: H5H01H˜i, with H05H˜a1H˜b1H˜C. There are four low-lying singlet excited states for the ‘‘free’’ HamiltonianH0. They are the exciton in chain a, the exciton in chain b,
and two CT states for which the electron and hole reside on different chains. H˜icauses mixing~configuration interaction! among those four states. The resulting 434 eigenvalue prob-lem can be further reduced to two 232 problems, for even and odd parity subspaces, respectively. Diagonization of the two 232 matrices gives the physical eigenstates.
The basis states for singlet elementary excitations in the two-band approximation are uk
&
A, uk&
B, uk&
C, anduk
&
D, which are defined as11 uk
&
A[(1/A
2)(ak↑ † b 2k↓ † 1ak↓ † b 2k↑ † )u0&
,uk&
B[(1/A
2)(gk↑ † d 2k↓ † 1g k↓ † d 2k↑ † )u0&
, uk&
C[(1/
A
2)(gk†↑b2k↓† 1g†k↓b2k↑† )u0&
, and uk&
D[(1/A
2)[~1/2!~gk↑†b2k↓†1gk↓†b2k↑†!u0&, and uk
&
D[(1/A
2)3(ak↑ † d 2k↓ † 1a k↓ † d 2k↑
† )u0
&
. The ground stateu0&
is the statewith no electrons and holes. The electron and hole are in the
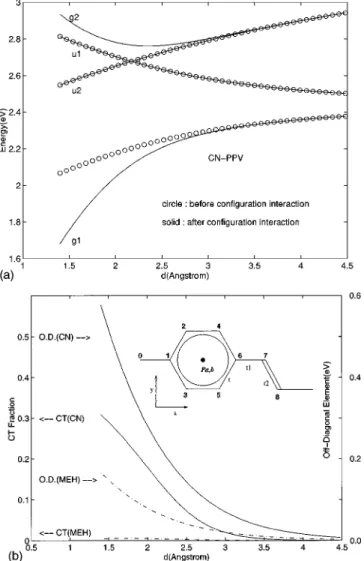
FIG. 1.~a! The energies of the low-lying excited states «u1,2and
«g1,2~solid lines! are shown as functions of vertical interchain
dis-tance d. The horizontal shift Dh for CN-PPV is used. Exciton
en-ergies «EX6 ~lower two circle lines! and CT energies «CT ~upper
circle line! before configuration interaction are also shown. In
prac-tice, A512.8 eV,m51.18 Å are used for tli. The result is almost
independent on m, the maximal number of interchain hoppings
from each site, as long as m.3. ~b! The CT fractions u^CT
2ug1&u2 and the off-diagonal matrix element
^CT2uH˜iuEX2&~real! of ug1,2& for CN-PPV~solid! and
same chain for uk
&
A and uk&
B, while they are in different chains foruk&
Canduk&
D. All the basis states have zero total momentum. They span four subspaces, which are decoupled from one another without H˜i. The matrix elements of H0within each subspace are
A
^
k8
uH0uk&
A5B^
k8
uH0uk&
B5@2Ec~k!1DEg#dk8k2V~k
8
2k!, C^
k8
uH0uk&
C5D^
k8
uH0uk&
D5@2Ec~k!1DEg#dk8k2Vab~k
8
2k!, where V~k8
2k!5V1~k8
2k!1V2~0! 51 NF
n(
Þ0 e2iR~k82k! V R~n!1UG
, Vab~k8
2k!51 NF
(
n e2iR~k82k! V R'~n!G
. ~8!We have used the relation Ev(k)52Ec(k) for PPV band structure. R(n)@R'(n)# is the distance between two unit cells in the same~opposite! chain~s! differing in indices by n. Anisotropy of the dielectric constant is also included.12The bandgap renormalization DEg5DEc1DEv, together with the on-site and off-site Coulomb energies U and V are treated as adjustable parameters. The lowest states, denoted byuA
&
,uB&
uC&
uD&
in these subspaces can be expanded asuA,B
&
5(kFEX(k)uk&
A,B, uC,D&
5(kFCT(k)uk&
C,D. The energies are «EX for uA&
,uB&
and «CT for uC&
,uD&
. Parityeigenstates can be constructed out of them: uEX6
&
[
A
1/2(uA&
6uB&
),uCT6&
[A
1/2(uC&
6uD&
). As mentioned before, there is no matrix element between 1 and 2 states. We can, therefore, consider them separately. For the diago-nal matrix elements, we have H0uEX6&
5«EX6uE6&
,H0uCT6
&
5«CT6uE6&
. Using Eq.~4!, the off-diagonal matrix element becomes^
CT6uH˜iuEX6&
5 12
(
k FCT* ~k!FEX~k!@2Tcc*~k!6Tvv*~k!
6Tvv~k!2Tcc~k!#. ~9!
It turns out that
^
CT1uH˜iuEX1&
50 because the transition amplitudes Taa is real and the same for a5c,v. Conse-quently, there is no configuration interaction between EX and CT states for the1 subspace, and we need to diagonize the 232 matrix for the 2 subspace only:S
«EX2^
CT2uH˜iuEX2&
^
CT2uH˜iuEX2&
* «CT2D
. ~10! Note that«EX65«EXand«CT65«CT so far. Davydov splitting between «EX6 13 is phenomenologically included by the re-placement«EX6→«EX6m2/d3, where m is the transition di-pole and d is the interchain distance.
Davydov splitting is a purely many-body Coulomb effect, which couples the exciton states uA
&
and uB&
. In order tocoupleuA
&
anduB&
, a term proportional tog†d†ab ~simul-taneous interchain transfer of an electron-hole pair due to Coulomb interaction! is required in H. The coefficient of such a term is hard to know without the detailed knowledge of some integrals involving the the atomic wave functions. Since our treatment of the Coulomb interaction is not very microscopic, with parameters U and V so chosen such that the intrachain exciton energy«EXis equal to theexperimen-tal value, the transition moment m for the «EX6 splitting can only be introduced as a fitting parameter. On the other hand, the interchain hopping Hamiltonian Hi is a purely one-body effect. The configuration interaction due to Hi are then treated much more microscopically. Note that, unlike Hi, Davydov splitting does not mixuEX
&
withuCT&
. Therefore, it does not affect the CT fraction as much as Hi does.Let us turn to the inversion symmetry~parity! of the prob-lem now. Consider the symmetry of a single chain, say, a, first. The lattice structure is invariant under inversion with respect to the point Pa, the center of benzene @see inset of Fig. 1~b!#. For Bloch states, we have uk,a
&
↔hu2k,a&
, h561. ↔ means inversion with respect to Pa. In terms of the Wannier functions, the symmetry manifests itself as
zl¯a(k)5hzla(2k)5hzla*(k), with l↔l¯ under inversion. More explicitly, (ll¯)5(1,6),(2,5),(3,4),(7,0), and (8,21).
l less than 1 means carbon site in the previous unit cell.
Explicit calculation ofzla(k) shows thath51 fora5v, and
h521 fora5c, which implies uk
&
A↔2u2k&
A. Combined with the fact that the envelope functionsFEX,CT(k) are both even functions of k, we haveuA&
↔2uA&
, i.e., the exciton is of odd parity~dipole allowed!. In the case of two chains, the point of inversion symmetry becomes the middle point P of the symmetry points Pa of chain a and Pb of chain b. The parity of the states can be easily seen to be uA&
⇔2uB
&
, uC&
⇔2uD&
. ⇔ means inversion with respect to P. Finally, we have uEX,CT1&
⇔2uEX,CT1&
anduEX,CT2
&
⇔uEX,CT2&
. The interesting point here is that the 2 subspace, which contains the lowest state, is of even parity and dipole forbidden to decay radiatively to the ground state. Quantum lattice fluctuations must be consid-ered to yield a finite radiative lifetime. In fact, similar situa-tion has been reported for a pair of thiophene oligomers.14In the following we present our results. First of all, since the energies of intrachain excitations have been studied intensively9 and is not our main concern here, we simply choose V(5U) such that the exciton creation energy «EXis 2.43 eV. The renormalized bandgap Ee(0)1Eh(0) is chosen to be 3.4 eV. We consider two PPV derivatives CN-PPV and MEH-PPV, with packing geometries determined from com-puter simulations.8 The horizontal shift Dh is (2,
A
3)b for CN-PPV and (9/4,2A
3/4)b for MEH-PPV.8 b51.4 Å isthe carbon bond length. Consider CN-PPV first. In Fig. 1~a!, we plot the energies«u1,2and«g1,2of the lowest four excited states uu1,2
&
and ug1,2&
for a range of vertical interchain distance d with fixed Dh. u,g means ungerude ~odd! and gerude ~even!. At large d, we have uu1,2&
.uEX1&
,uCT1&
and ug1,2&
.uEX2&
,uCT2&
.ug1&
is always the lowest ex-cited state. Even though it is dipole forbidden, it must be responsible for the PL photon emission. By definition, an excimer is formed if such a state has a significant CT component.6,7The CT fraction ofug1&
is shown in Fig. 1~b!.Indeed, the excimer character is clear when d<3 Å, which is not far from the the ground state equilibrium interchain distance d053.3 Å.8Since«g1(d) decreases rapidly with d, the two chains must come closer to each other due to the attractive force when excited. The new equilibrium distance
d* depends on both the exciton density and the molecular force field between these two chains. Two characteristic en-ergy differences are of particular interest here: D«1
[«g1(`)2«g1(d*) and D«2[«g1(d0)2«g1(d*). Experi-mentally,D«1 is the difference in solution and film PL pho-ton energies, while D«2 is the net Stokes shift due to inter-chain lattice relaxation, i.e., the apparent Stokes shift minus the optical phonon energy. For CN-PPV,D«1. 0.35 eV and
D«2. 0.25 eV.2,3Since the transition dipole m responsible
for Davydov splitting is unknown in our model, we adjust it such that «g1(`)2«g1(d0)5(0.3520.25) eV. The
corre-sponding transition diople length is 1.6 Å. The actual final lattice configuration in films is, however, very difficult to predict due to two reasons.~a! Polymers are heavy molecules with large atomic weight. It is not likely that a significant shrinkage of interchain distance can be caused by the exci-tation of a single electron-hole pair. A finite exciton densi-ty~exciton number per monomer! is required. d*is therefore determined by the exciton density, instead of a single exci-ton. Because the magnitude of exciton density is presently unknown, a reliable prediction seems unlikely. ~b! In the solution we need only to consider a pair of polymers in order to determine d*. In films, however, the force experienced by a particular chain is affected by many neighboring chains and is much more complicated. Strictly speaking, our two-chain approach is accurate in predicting only the initial ten-dency toward excimer formation, but not the final configura-tion, which may contain distortions involving more than two chains, or relaxations other than vertical distance shrinkage, e.g., rotation and horizontal displacement. We can, however, give a simple estimate of d* based on the Leanard-Jones form of the molecular force field U(d)5C@(1/2)(d0/d)12 2(d0/d)6#. d*is the minimum point of«g1(d)1U(d). C is so chosen such that D«250.25 eV, from which we predict
d*.2.35 Å, where the CT fraction is about 15 %. The
ac-tual equilibrium configuration may be even lower in energy and have an even larger CT component.
Surprisingly, when we apply the same procedure to MEH-PPV, with Dh5(9/4,2
A
3/4)b, the story becomes totally dif-ferent. For the same range of d as before, the off-diagonal element in Eq.~10! is nearly zero, and the ug1&
state remains basically uEX2&
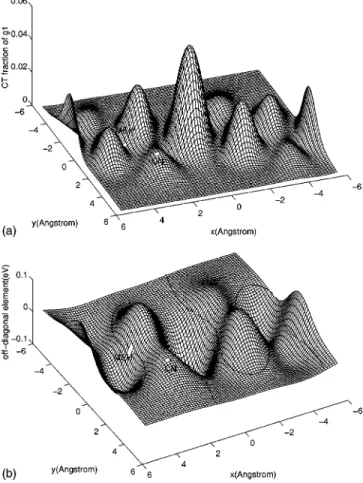
@see Fig. 1~b!#. Consequently, the light-emitting state remains basically the exciton as in the solution (d5`). Since the slope of «g1(d) at d054.05Å is vanish-ingly small, d0 and d*almost coincide with each other. Thisexplains the very small solution-film PL difference and Stokes shift observed in MEH-PPV.15Similar results are ob-tained for PPV, with Dh5(21.79,3.17) Å and d053.26 Å,16 agreeing with the fact that excimers are also absent in the PL spectrum of PPV.2The difference between these three PPV derivatives becomes conspicuously transparent when we scan the off-diagonal term
^
CT2uH˜iuEX2&
, which is real due to certain symmetry, and the CT fraction of ug1&
, over a range of Dh for some fixed d @see Figs. 2~a!,2~b!#. Since^
CT2uH˜iuEX2&
is the superposition of hoppingterms, which involves the complex Wannier wave fuctions
zla(k), between many different pairs of carbon orbitals, large
variations are expected due to the quantum interferences
~cancelations!. The landscapes of all the figures changes little
as long as d is in the physical range of 2.524.5 Å. CN-PPV happens to be close to a local maximum, while MEH-PPV is close to the contour of
^
CT2uH˜iuEX2&
50. PPV is already in the flat region away from all the peaks. D^
CT2uH˜iuEX2
&
/Dd can be viewed as the effective attractive force due to one excitation, which is equal to20.094 eV/Å for CN-PPV and 20.0095 eV/Å for MEH-PPV, if Dd is chosen to be 0.5 Å. In addition to the spectra, our model also explains the lifetimes of PL and photoinduced absorption ~PA!,17 in par-ticular the large difference in PL lifetimes between MEH-PPV ~1.2 ns! ~Ref. 18! and CN-PPV ~17 ns!.3The symmetry-forbidden transition rate rg from ug1
&
tou0
&
, via vibrational coupling touu1&
, is given by19rg5ru
S
1 «u12«g1D
2(
i^
qi2&
S
] ]qi^
u1uDH~q!ug1&
D
0 2 . ~11!FIG. 2. ~a! The CT fraction u^CT2ug1&u2of the light-emitting
stateug1&are shown for an area of Dh. d is fixed at 3.3 Å. The CT
fraction for CN-PPV is about 10 times larger than that of
MEH-PPV. Surface elements at Dh of CN-PPV and MEH-PPV are
re-moved for clarity. The central peak at~0,0! corresponds to the
ge-ometry that two chains are right on top of each other. ~b! The
off-diagonal matrix element ^CT2uH˜iuEX2& for the2 subspace
are plotted for the same area of Dh. The contour of zero is shown to
emphasize that MEH-PPV happens to be in a geometry of vanishing CT-EX mixing due to inteference.
q is the collective lattice coordinates. ru is the transition rate fromuu1
&
tou0&
, which is actually equal to the rate fromuA&
tou0&
for a single chain.«u1 and«g1 are excited state ener-gies at the relaxed lattice configuration.^
qi2&
is the mean square lattice quantum fluctuation. Consider, for example, the lattice displacements in chain a only. Since they effect the electronic states in chain a but not in chain b, the exact inversion symmetry of uu1&
is broken, and the emission is slightly dipole allowed. ruis inversely proportional to the PL radiative lifetime tPLf in films, while rg is inversely propor-tional to the PL radiative lifetimetPLs in solutions. Using the relation 1/tPLf ;(«
u12«g1)22, we can predict the relativetPL
f of the derivatives. For MEH-PPV, we simply take d*5d0
54.05 Å, at which «u12«g150.17 eV from our calculation. For CN-PPV, the lattice is relaxed substantially and the ac-tual value of d* is hard to find. However, we can approxi-mate «u12«g1 by 2D«150.7 eV, assuming that the even and odd states split evenly off the the unperturbed value
«g1(`). The ratio between their tPL
f
is predicted to be (0.7/0.17)2517, consistent with the experimental value 17 ns/1.2 ns514.2.
The author is grateful for the support of the National Science Council of Taiwan, R.O.C., under Contract No. NSC 86-2112-M-009-001. The hospitality and support of the Center for Theoretical Science in Taiwan is also appre-ciated.
1J.H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K.
Mackay, R. H. Friend, P. L. Burn, and A. B. Holmes, , Nature
~London! 347, 539 ~1990!.
2N.T. Harrison, D. R. Baigent, I. D. Samuel, and R. H. Friend,
Phys. Rev. B 53, 15 815~1996!.
3I.D.W. Samuel, G. Rumbles, and C.J. Collison, Phys. Rev. B 52,
R11 573~1995!.
4S.A. Jenekhe and J.A. Osaheri, Science 263, 765~1994!.
5S. Webster and D.N. Batchelder, Polymer 37, 4961~1996!.
6J. Michl and V. Bonacic-Koutecky, Electronic Aspects of
Or-ganic Photochemistry~Wiley, New York, 1990!.
7T. Azumi, A. Armstrong, and S. McGlynn, J. Chem. Phys. 41,
3839~1964!; J. Murrell and J. Tanaka, Mol. Phys. 7, 363 ~1964!.
8E.M. Conwell, J. Epstein, and S. Shail, Phys. Rev. B 54, R2308
~1996!.
9M. Chandross, S. Mazumdar, S. Jeglinski, X. Wei, Z. V.
Vard-eny, E. W. Kwock, and T. M. Miller, Phys. Rev. B 50, 14 702
~1994!; Y. Shimoi and S. Abe, Synth. Met. 78, 219 ~1996!.
10H.A. Mizes and E.M. Conwell, Synth. Met. 68, 145~1995!.
11S. Nakajima, The Physics of Elementary Excitations ~Springer,
Berlin, 1980!.
12H. Mizes and E. Conwell, Phys. Rev. B 50, 11 243~1994!.
13M. Klessinger and J. Michl, Excited States and Photochemistry of
Organic Molecules~VCH, New York, 1990!.
14J. Fagerstro¨m and S. Stafstro¨m, Phys. Rev. B 54, 13 713~1996!.
15I. D. W. Samuel, B. Crystall, G. Rumbles, P. L. Burn, A. B.
Holmes, and R. H. Friend, Chem. Phys. Lett. 213, 472~1993!.
16P. Gomes da Costa, R. Dandrea, and E.M. Conwell, Phys. Rev. B
47, 1800~1993!.
17M. Yan, L. J. Rothberg, F. Papadimitrakopoulos, M. E. Galvin,
and T. M. Miller, Phys. Rev. Lett. 72, 1104 ~1994!; J. W. P.
Hsu, M. Yan, T. M. Jedju, and L. J. Rothberg, Phys. Rev. B 49, 712~1994!.
18G. Hayes, I. Samuel, and R. Phillips, Phys. Rev. B 52, R11 569
~1995!.
19G. Herzberg and E. Teller, Z. Phys. Chem. Abt. B 21, 410~1933!;
J. Murrell and J. Pople, Proc. Phys. Soc. London, Sect. A 69, 245~1956!.