Graduate Institute of Microbiology College of Medicine
National Taiwan University Doctoral Dissertation
To identify novel modulators of miRNA biogenesis and function
Yu-De Chu
Advisor: Shih-Peng Chan, Ph.D.
105 7
July 2016
(tB) tlt±^^C
^AH^: ^^^a^gt^S^^^L^
^-^t^a^
^A^§: To identify novel modulators of miRNA biogenesis and function
-Af-rVB
^t^ ^f^ ^ (^^ F99445120 ) i£^^S^^-^^^
^-^^T^^^I (t^) ±-^^i^, ^a 105 ^ 7 ^ 12 B7?<T
N^g^M^^?SM^n^X^, ^ittH^
n^^^: ^ ^
(^^)A^
? ^
A
^/
yA^
J^f. ^f
^^ ^rcm ^ ^ <^)
(miRNA) 70%
(RNAi) DEAD/H-box
let-7(mg279) DDX-23
DDX-17 let-7 (phenotype)
let-7
let-7 let-7 (primary-let-7)
let-7
(primary miRNA)
let-7 heterochronic lin-4 miR-48 miR-84
miR-241
heterochronic lsy-6 DDX-23 DDX-17
DDX-23 DDX-17
hnRNP hnRNP Q HRP-2 let-7(n2853)
HRP-2 (miRISC)
lin-41 3’UTR let-7
poly-A HRP-2 lin-41
HRP-2
HRP-2 3’UTR
let-7 lin-41
HRP-2 let-7 lin-41
hnRNP Q let-7 lin-41
TRIM71 HRP-2 lin-41
3’UTR TRIM71 3’UTR
hnRNP Q let-7
poly-A 3’UTR let-7
TRIM71 HRP-2
hnRNP Q lin-41
TRIM71 3’UTR let-7
let-7 DDX-23 HRP-2 hnRNP Q lin-41/TRIM71
Abstract
As post-transcriptionally gene regulators demonstrated by numerous studies, microRNAs have been predicted to control more than 70% of human coding genes expression. However, studies regarding modulators for miRNA biogenesis and/or function remain relatively few. Here, we performed a candidate-based RNAi screen in C. elegans to identify DEAD/H-box proteins involved in miRNA biogenesis and/or function. In the let-7(mg279) sensitized genetic background, knockdown of a homolog of yeast splicing factor Prp28p, DDX-23, or a homolog of human helicases p68 and p72, DDX-17, enhanced let-7 loss-of-function phenotypes, suggesting that these helicases play a role in let-7 processing and/or function. In both ddx-23(RNAi) and ddx-17(RNAi), levels of mature let-7 were decreased while pri-let-7 was found accumulated, indicating that the helicases likely act at the level of pri-let-7 processing. DDX-23 and DDX-17 were also required for the biogenesis of other known heterochronic miRNAs, including lin-4 and the let-7 family members miR-48, miR-84 and miR-241. Their function was not confined to the heterochronic pathway, however, since they were both necessary for down-regulation of cog-1 by the spatial patterning miRNA, lsy-6. Therefore, we present a novel function for C. elegans DDX-23 in pri-miRNA processing, and also suggest a conserved role for DDX-17 in this process. On the other hand, we also show that RNAi knockdown of C. elegans HRP-2, the homolog of mammalian hnRNP Q, relieved the heterochronic phenotypes in let-7(n2853) mutant animals, indicating an involvement of HRP-2 in let-7-lin-41 regulation. In addition, we detected an RNA-dependent
miRNA-mediated silencing complex (miRISC). Moreover, we identified an HRP-2 response element in the lin-41 3’UTR at a position, downstream of the two let-7 complementary sites (LCSs), close to the poly(A)-tail. Deletion of this response element caused further down regulation of a GFP reporter carrying the lin-41 3’UTR in a let-7-dependent manner. Thus, we propose that HRP-2 impedes let-7/miRISC activity when binding to the lin-41 3’UTR. Interestingly, we found that depletion of human hnRNP Q also enhanced let-7-mediated down-regulation of TRIM71. Similar to the case in C. elegans, hnRNP Q binds to a response element adjacent to the poly(A)-tail in the TRIM71 3’UTR. Deleting this element from the 3’UTR significantly enhanced let-7 repression. Taken together, our findings uncover a novel evolutionarily conserved function for HRP-2/hnRNP Q to inhibit let-7/miRISC activity when they bind to specific response elements in the lin-41/TRIM71 3’UTRs.
Key words: miRNA, let-7, DDX-23, biogenesis, HRP-2, hnRNP Q, lin-41/TRIM71
………I
………..II
……….III
English abstract……….V
………...1
Chapter 1 Introduction ………11
1.1 microRNAs……….11
1.1.1 Discovery of miRNAs……….11
1.1.2 miRNA family………...12
1.1.3 Functions of miRNAs……….13
1.1.4 Biogenesis of miRNAs………14
1.2 The let-7 miRNA………14
1.2.1 Regulation of let-7 biosynthesis………..14
1.2.3 The evolutionary conserved let-7/lin-41 regulatory pathway……….17
1.3 The DExD/H-box RNA helicases ………..18
1.3.1 Functions of the DExD/H-box RNA helicases………...18
1.3.2 The implication of DExD/H-box RNA helicases in the miRNA
pathway……….……..19
1.4 Heterogeneous nuclear proteins (hnRNPs) ……….….…..20
1.4.1 Functions of hnRNPs………..20
1.4.2 The potential roles of hnRNPs in the miRNA pathway………….….21
1.5 The animal model—Caenorhabditis elegans……….…22
1.5.1 The advantages of using C. elegnas as animal model……….…22
1.5.2 The heterochronic gene pathway and let-7 miRNA in C. elegans…..22
1.5.3 Controlling of vulval development by miR-84-mediated repression of
let-60/RAS……….……..24
1.5.4 The lsy-6 miRNA-mediated repression of cog-1…….….…………..25
1.6 Project aims……….26
Chapter 2 Materials and Methods………...27
2. 1 Caenorhabditis elegans……….….….……….27
2.1.1 Strains………….….….……….27
2.1.2 Culture………...28
2.1.3 RNA-interference (RNAi) ………....28
2.2 Construction of plasmids………...30
2.2.1 C. elegans RNAi clones……….………...30
2.2.2 C. elegans expressing plasmids……….………...30
2.2.3 TRIM71 3’UTR fused luciferase plasmids…….………...32
2.2.4 hnRNP Q1 plasmids……….………...32
2.2.5 Ago2-GFP plasmid……….………...33
2.3 Microscopy analysis……….………...34
2.4 RNA isolation……….………...34
2.4.1 From C. elegans……….………...34
2.4.2 From human cell cultures………….……...35
2.5 Northern blot analysis………...35
2.6 RT-PCR and RT-qPCR…………...37
2.6.1 Poly-A tail-based reverse transcription and qPCR...37
2.7 Lysate preparation…………...45
2.7.1 For C. elegans western blot analysis………...45
2.7.2 For C. elegans immunoprecipitation experiment………...46
2.7.3 For mammalian cell lines western blot analysis………...46
2.7.4 For mammalian cell lines immunoprecipitation experiment………...47
2.8 Western blot analysis………....………...47
2.9 Co-immunoprecipitation……….….….……...48
2.10 RNA-immunoprecipitation (RIP)……….….….……...49
2.11 In vitro transcription………...50
2.12 Biotinylated RNA affinity selection pull-down……….………….…...54
2.13 Generation of transgenic C. elegans……….………….…...55
2.13.1 Conventional microinjection……….………….…...55
2.13.2 Mos1 mediated single copy transgene insertion (MosSCI)...55
2.14 Integration of extra-chromosomal array in C. elegans...56
2.15 Cell culture and transfection……….………….…...57
2.16 Lenti-virus infection……….………….…...58
2.17 Luciferase assay……….………….…...60
2.18 DAPI staining……….………….…...61
Chapter 3 Results……….………….…...63
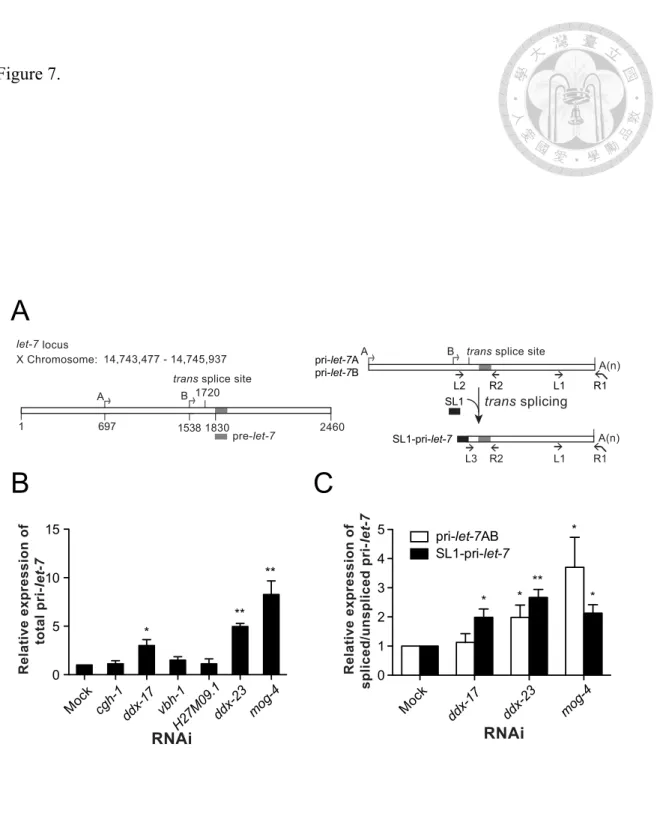
3.1 The role of DDX-23 in primary microRNA processing in Caenorhabditis elegans……….………….…...63
3.1.1 The potential roles for several DEAD-box RNA helicases in let-7-mediated gene regulation……….…...63
3.1.2 DDX-23 and DDX-17 promote let-7 maturation...67
3.1.3 DDX-23 and DDX-17 support the processing of primary let-7...67
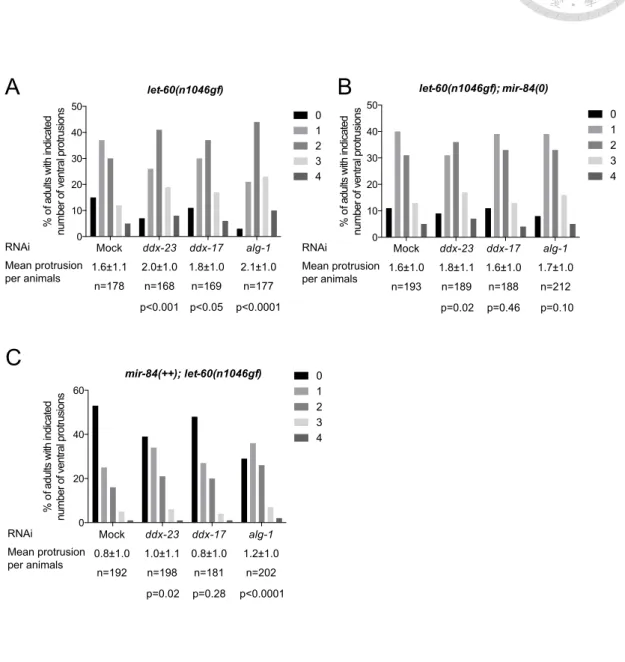
3.1.4 DDX-23 and DDX-17 facilitate regulation of let-60/RAS by miR-84...68
3.1.5 DDX-23 and DDX-17 support pri-miR-84 biogenesis...70
3.1.6 DDX-23 and DDX-17 are also required for other heterochronic miRNAs processing...…...71
3.1.7 Expression of miRNA pathway components is not affected by depletion of DDX-23 or DDX-17...…...71
3.1.8 DDX-23 is localized to the nucleus and functions with miRNAs in multiple tissues...…...73
3.2 HRP-2/hnRNP Q impede let-7-mediated repression of lin-41/TRIM71 in
Caenorhabditis. elegans and humans...75
3.2.1 HRP-2 functions upstream of lin-29...75
3.2.2 Depletion of HRP-2 causes precocious lin-29 expression...77
3.2.3 Depletion of HRP-2 enhances let-7-mediated repression of lin-41...78
3.2.4 Depletion of HRP-2 does not alter let-7 level...79
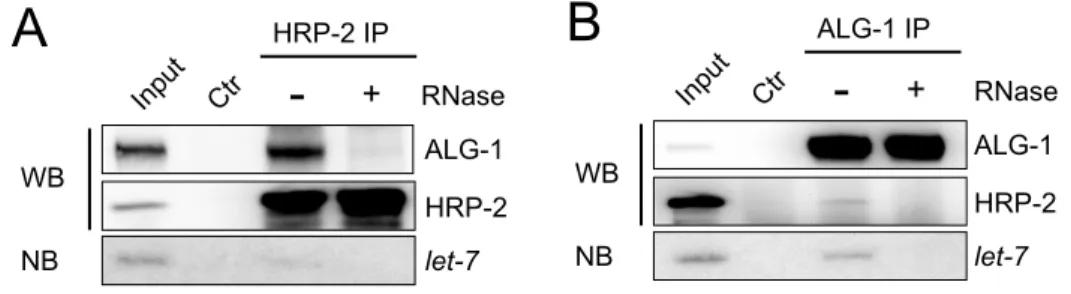
3.2.5 C. elegans HRP-2 interacts with ALG-1 in an RNA-dependent manner...80
3.2.6 HRP-2 directly interacts with lin-41 3’UTR...80
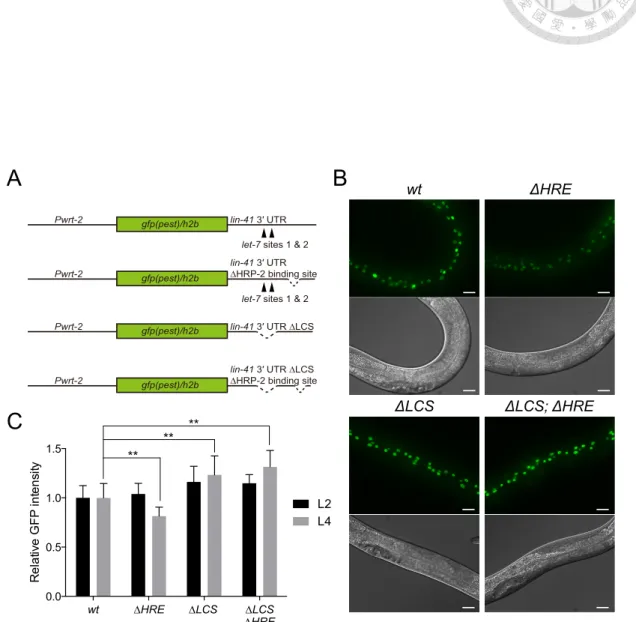
3.2.7 HRP-2 binds to lin-41 3’UTR at a position between LCS and poly-(A)…..81
3.2.8 Deletion of HRE enhances let-7-mediated repression of lin-41...84
3.2.9 HRP-2 locates in both nucleus and cytoplasmic fractions...85
3.2.10 hnRNP R does not involved in let-7-mediated repression of TRIM71...87
3.2.11 hnRNP Q blocks repression of TRIM71 by let-7...88
3.2.12 hnRNP Q is associated with miRISCs...90
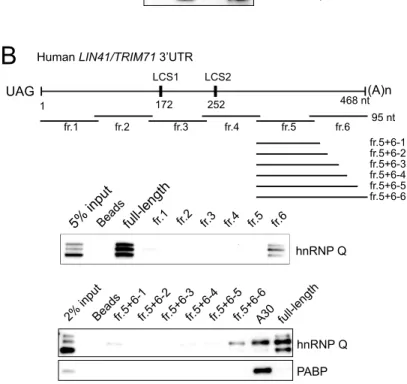
3.2.13 hnRNP Q binds to an element at a position near poly-A tail in TRIM71 3’UTR...92
3.2.14 QRE in TRIM71 3’UTR is required for hnRNP Q binding...93
3.2.15 Deletion of QRE enhances let-7-mediated repression of TRIM71...94
Chapter 4 Discussion...96
4.1 A novel function for the DEAD-box RNA helicase DDX-23 in primary miRNA
processing in C. elegans...96
4.2 HRP-2/hnRNP Q impedes let-7 miRNA-mediated repression of lin-41/TRIM71 in
C. elegans and humans...102
Chapter 5 Table...108
Table 1. Using let-7(n2853) mutant to screen for hnRNP(s) involved in let-7/lin-41
pathway...108
Chapter 6 Figures...109
Figure 1. The vulval bursting phenotype caused by RNAi knockdown of DEAD-box
RNA helicase genes...109
Figure 2. The retarded seam cell development caused by RNAi knockdown of
several DEAD-box helicases...111
Figure 3. Depletion of DDX-17 or MOG-4 causes discontinued clusters of seam
Figure 4. Adult hermaphrodites show disrupted seam cell syncytia or unfused seam
cells upon RNAi against several RNA helicase genes...113
Figure 5. col-19::GFP expression is down-regulated upon RNAi against several
RNA helicase genes...115
Figure 6. RNAi knockdown of ddx-23, ddx-17 and mog-4 reduced mature let-7
levels...117
Figure 7. DDX-23 and DDX-17 are required for pri-let-7 processing……….118
Figure 8. Inactivation of ddx-23 and ddx-17 enhance the MUV phenotype in let-60
gain-of-function mutant...120
Figure 9. Reduced expression of ddx-23 and ddx-17 increases accumulation of
pri-miR-84...122
Figure 10. DDX-23 and DDX-17 are required for biogenesis of several heterochronic
miRNAs...123
Figure 11. Reduction of DDX-23 or DDX-17 does not affect the expression of
machineries for miRNA biogenesis and function...124
Figure 12. DDX-23 and DDX-17 are required for efficient regulation of cog-1::gfp
expression by ectopic lsy-6 miRNA in the uterus...125
Figure 13. The spatial expression pattern of DDX-23...126
Figure 14. The model for DDX-23 and DDX-17 in miRNA biogenesis………...127
Figure 15. Depletion of HRP-2 relieved let-7(n2853) retarded seam cell development...128
Figure 16. HRP-2 genetically acts upstream of LIN-29...130
Figure 17. RNAi knockdown of HRP-2 promotes precocious LIN-29 expression at early stage...131
Figure 18. Reduction of HRP-2 enhances let-7-mediated repression of lin-41...133
Figure 19. hrp-2(RNAi) does not alter let-7 and miRISC levels...135
Figure 20. HRP-2 interacts with miRISC in an RNA-dependent manner...136
Figure 21. The let-7 binding site is not required for HRP-2 to interact with lin-41 mRNA 3’UTR...137
Figure 22. HRP-2 is associated with a 14-nt U-rich motif in lin-41 3’UTR...138
Figure 23. Deletion of HRP-2 response element (HRE) enhances let-7-mediated lin-41 repression...140
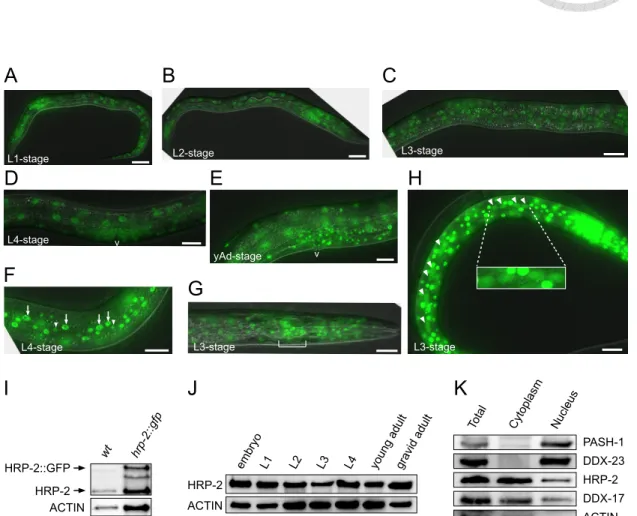
Figure 24. HRP-2 distributes either in nucleus and cytoplasm in C. elegans...142
lin-41/TRIM71 repression...144
Figure 26. hnRNP Q impedes let-7-mediated repression of TRIM71...146
Figure 27. hnRNP Q is associated and co-localized with miRISC in P-bodies...148
Figure 28. hnRNP Q binds to an element in TRIM71 3’UTR...150
Figure 29. The hnRNP Q response element (QRE) is required for hnRNP Q binding...151
Figure 30. Deletion of hnRNP Q response element increases repression of TRIM71 by let-7...153
Chapter 7 References...154
Appendix 1...170
Appendix 2...172
Appendix 3. Brief summary of the previous ‘RACK-1’ project...173
Appendix 4. The published article for the ‘RACK-1’ project...175
Appendix 5. The published article for the ‘DDX23’ part in this thesis...191
Chapter 1 Introduction
Chapter 1.1 miRNAs
1.1.1 Discovery of miRNAs
MiRNAs are around 22 nucleotides in length, small non-coding RNAs.
Initially, the first miRNA—lin-4 was discovered by genetically phenotypic analysis and
identified in C. elegans as a heterochronic gene, with fluctuating expression pattern 1.
Later, Lee et al. demonstrated that the lin-4 gene locus encodes a small non-translated
RNA fragment, which can partially complement to lin-14 mRNA 3’UTR 2. In the same
year, Wightman et al., also proposed that lin-4 post-transcriptionally down-regulates
LIN-14 expression by targeting these sites. Such a lin-4-mediated repression of lin-14
was therefore believed to play an important role in controlling developmental timing of
C. elegans 3. It was initially considered as a specific event in the nematodes until 2000
when the second small RNA, let-7, was characterized. At that time, let-7 was emerging
as a new member in heterochronic gene pathway. In addition, repression of its target
lin-41 by targeting the lin-41 3’UTR promotes the downstream adult stage-specific
transcription factor—LIN-29, which is important for determination of developmental
progresses 4,5. let-7 was also identified in other species, including Drosophila, Zebrafish,
mouse and human 6,7. So far, a growing body of miRNAs have been identified and their
expression are frequently correlated with diseases, development, and tumorigenesis 6,8,
leading to the interest of investigating functions and mechanisms of miRNAs involving
in various biological progresses.
1.1.2 miRNA family
Following identification of miRNAs in different species, the researchers
extracted some rules for the complementarity between miRNAs and their targets.
According to the computational method and experimental validation, they found that the
2-7 nucleotides of miRNAs are often completely complementary to the targets 9.
Afterward, these few nucleotides were defined as an critical motif for target recognition
and named as the ‘seed region’. Furthermore, they also demonstrated that a single target
could be regulated by the miRNAs sharing same sequences in the seed region. Based on
this property, these miRNAs therefore were classified into the same miRNA family, in
which they potentially serve as a modulator to control the same genes expression, such
as the let-7 family, lin-4/miR125 family and so on.
1.1.3 Functions of miRNA
Currently, over 70% of human protein-coding genes are predicted to be
regulated by miRNAs. Dysfunction or misregulation of miRNAs lead to severe
developmental defects, diseases or carcinogenesis. How do miRNAs perform their
functions? Post-transcriptionally targeting of cognate mRNAs by miRNAs restrains
their translation or promotes mRNAs degradation 10. Following the recognition and
targeting of the mRNAs in the cytoplasm, they will aggregate at a foci called the
Processing-body (P-body) for efficiently performing their functions 11. The miRNAs are
not working alone, they are associated with several components, including the core
protein Argonaut, GW182 and other partner proteins to form the microRNA-induced
silence complex (miRISC). Previous studies have demonstrated that the PABP (poly
adenine binding protein) has the ability to recruit miRISCs to the target mRNAs and
then facilitate their function 12. When miRISCs are brought close to mRNAs, the
GW182 protein facilitates the dissociation of PABP from the poly-A tail and also
recruits the CCR4-NOT1 deadenylase complex 13-15. Not only GW182, but also a
DEAD-box RNA helicase DDX6 directly interacts with deadenylase and modulates
16-18
complex to promote mRNA degradation19. Overall, the functions of miRNAs are
primarily to repress cognate mRNAs translation and subsequently promote turnover.
1.1.4 Biogenesis of miRNAs
A majority of miRNA biosynthesis starts with transcription of
primary-miRNAs (pri-miRNA) by RNA polymerase II or III. The products are
equipped with m7G cap and polyadenylated tail and featured with stem-loop secondary
structure 20-22. Recognition and digestion of it by a complex, composed of Drosha and
DGCR8, called “Microprocessor”, leads to generation of precursor-miRNAs, which
generally contains 60-80 nt and still retains the stem-loop secondary structure 23-27.
Subsequently, exportation of the pre-miRNAs into cytoplasm is done by the XPO5, an
exportin associated on the nucleus membrane. Then, the stem-loop structure can be
detected and processed by the Dicer complex to produce an approximately 22-nt long
miRNA duplex 28-31. One strand of the double stranded miRNA will be degraded and
the other one will be incorporated into functional miRISCs 32.
1.2 The let-7 miRNA
1.2.1 Regulation of let-7 biosynthesis
The biosynthesis of some miRNAs is more complicated and stringently
regulated, such as let-7. The phylogenetic conserved miRNA—let-7 had been identified
in a number of species with a high similarity in sequence 6. As other miRNAs, the
pri-let-7 is transcribed by RNA polymerase II and equipped with the cap and poly-A tail
structure both in mammals and nematode 33. Different to most other miRNAs, the
stem-cell specific regulator LIN28, an RNA-binding protein, recognizes pri-let-7 and
pre-let-7 intermediates and promotes degradation. Two LIN28 isoforms, A and B,
inhibit let-7 maturation in different cellular compartments via distinct mechanisms. In
the nucleus, LIN28A reduces the activity of Drosha/DGCR8 complex and thus blocks
the intermediate processing by targeting the terminal loop of pri-let-7 34-36. In the
cytoplasm, as a result of the association between LIN28B and pre-let-7, the terminal
uridyl transferase (TUTase) is recruited to the LIN-28-pre-let-7 complex to uridylate the
3’ of pre-let-7 37,38. Later, the uridylated intermediate will be degraded by a 3’-5’
exonuclease, DIS3L2 39. The way to regulate let-7 biosynthesis by LIN28 is highly
conserved among species, indicating the significance of regulating let-7 biogenesis
during evolution 38,40,41. In addition, the hnRNP A1 and KSRP also reported as
pre-let-7 42. Taken together, these evidences suggest that let-7 biogenesis requires
stringently regulated and the detail mechanism is worth to further investigate.
1.2.2 The role of let-7 in maintaining stem cell stemness
Determination of stem cell differentiates or divides has long been a big
question for scientists. Well-controlled proliferation by the cell cycle is associated with
proper development, organogenesis and numerous aspects of biological progresses 43.
Mis-regulation of cell growth leads to diseases and carcinogenesis 44. The tumor
suppressor let-7 miRNA modulates expression of a bundle of proteins—most
of them are important for cell cycle progression 45,46. For example, let-7 regulates the
MYC transcription factor for several cell cycle genes 46, and LIN28, which both are
required for the induced pluripotent stem cell (iPSCs) transformation 33,47. As
mentioned above, LIN-28 negatively controls let-7 miRNA biogenesis. Therefore, let-7
and LIN-28 form a negative feedback regulation loop to modulate the cell cycle. In
addition, the high mobility group AT-hook 2 (HMGA2), a transcriptional positive
regulator to promote cell cycle, commonly associated with tumorigenesis 48, is also a
let-7 target. Interestingly, the HMGA2 3’UTR contains eight let-7-complementary sites
(LCS), implying the potential for being a competing endogenous RNA (ceRNA) to
reduce the impact of let-7-mediated repression to other targets 49. RAS, another let-7
target, functions as a binary molecular switch that controls intracellular signaling
networks. RAS activates several pathways, of which the mitogen-activated protein
(MAP) kinase cascade has been well-studied. This cascade transmits signals
downstream and results in the transcription of genes involved in cell growth and division 50. Beside to other let-7 targets, the potential E3 ubiquitin ligase
LIN-41/TRIM71 controls the degradation of Argonaut protein, the core enzyme in
miRISC 51. Expression of LIN-41/TRIM71 is required for appropriate development in
animals. However, overexpression of LIN-41/TRIM71 in adults leads to carcinogenesis
52,53. Recently, the let-7/LIN-41 pathway had been reported as an important factor for
the fates decision of stem cells 54. Taken together, these targets of let-7 serve as
important roles in modulating cell cycle progression, reflecting the significant role for
let-7 in regulating stemness maintenance.
1.2.3 The evolutionary conserved let-7/lin-41 regulatory pathway
LIN-41 was first identified in C. elegans as a phenotype suppressor of
let-7(n2853) and thus classified as a member in the heterochronic gene pathway, in
Subsequently, the LIN-41 homologs, called TRIM71 in mammals, were found
evolutionarily conserved in diverse species, such as in D. melanosgater, C. elegans,
Zebrafish, mouse and human 55. Over the past decades, the investigation in controlling
LIN-41/TRIM71 expression shows that let-7-mediated regulation seems to be the major
way to modulate its levels in numerous species 4-6,52. Developmental defects were
observed in animals without proper tuning of the let-7/lin-41 pathway, indicating the
importance of this pathway in development for these species and showing that the detail
mechanism in let-7/lin-41 pathway can be studied by using a simple model organisms,
such as C. elegans.
1.3 The DExD/H-box RNA helicases
1.3.1 Functions of DExD/H-box RNA helicases
The DExD/H-box RNA helicases family, including the DEAH, DExH, DExD
subfamilies, was highly conserved among species from bacteria to humans. According
to the current knowledge, they are involved in nearly all processes of RNA metabolism
steps such as transcription, mRNA splicing, ribosomal biogenesis, mRNA export,
translation, and RNA turnover 56. Initially, they were named based on the amino acid
sequence D-E-A-D (Asp-Glu-Ala-Asp) or D-E-A-H (Asp-Glu-Ala-His) in their
conserved motif II. With extensive homology, there are eight conserved motifs that
form the helicase core within the ATPase and helicase activities. Depending on the
auxiliary domains, which are often located in the amino-terminus and carboxyl-terminus
ends, and bound cofactors, the helicase core of the DExD/H-box proteins can catalyze
different types of conformational changes and nucleic acid rearrangements, such as
single strand translocation, duplex annealing or displacement. Many DExD/H-box
proteins also displace RNA-protein interaction and facilitate the remodeling of large
ribonucleoprotein (RNP) complexes 57,58.
1.3.2 The implication of DExD/H-box RNA helicases in the miRNA pathway
During the past decade scientists have attempted to understand the molecular
mechanisms of miRNA biogenesis and function. It is reasonable to expect great
DExD/H-box RNA helicase activities associated with miRNA since there are numerous
conformational changes in multiple-step miRNA processes. However, it seems that only
few helicases have been implicated for their roles in the miRNA pathway so far.
p68/p72 and DDX1 are components of the larger Microprocessor complex and are
required for processing of a subset of miRNAs 23,59,60. RCK/p54/CGH-1 interacts with
target repression 18. Recently, C. elegans CGH-1 has been shown in association with a
TRIM-NHL protein, NHL-2, and this complex interacts with miRISC components and
modulates miRNA function 61. Overall, these evidences suggest the critical role for
DExD/H-box RNA helicases in miRNA biogenesis or function and raise the possibility
that maybe there are novel helicases in the pathway.
1.4 The heterogeneous nuclear proteins (hnRNPs)
1.4.1 Functions of hnRNPs
The heterogeneous nuclear ribonucleoproteins (hnRNPs) are RNA-binding
proteins with critical roles in almost all aspects of nucleic acid metabolism, including
transcription, protecting of nascent RNAs, mRNA splicing, translational control and so
on. Despite they share some general characteristics, the variations of the domain
composition and the functions of hnRNPs are large between each others. There are ~20
proteins (named hnRNP A-U) consisting of multiple RNA-binding domains, including
RRM (RNA recognition motif), KH (K homology), atypical RRM and glycine-rich
domains (for a review see ref. 62). During the past decades, hnRNPs gain-of-function or
loss-of-function have been shown to be associated with carcinogenesis 63. For example,
several hnRNPs are required for the length maintenance of telomere. Shortening of the
telomere results in tumorigenesis and cancer progression 64. Defects in mRNA splicing
and alternative splicing are common events in cancer development, such as that
mis-splicing of the cell proliferation regulator c-Src by hnRNP A1 and H leads to
carcinogenesis 65-67. These evidences indicate the important roles for hnRNPs in tumor
progression, however, little is known about the involvement of hnRNPs in miRNA
pathway and miRNA-related tumorigenicity.
1.4.2 The potential role of hnRNPs in the miRNA pathway
To date, a few hnRNPs had been implicated in miRNA biogenesis and
function. For example, the nuclear hnRNP A1 stabilizes pri-miR-18a stem-loop
structure to promote Drosha-mediated processing 68. By contrast, the binding of hnRNP
A1 to the terminal loop of pri-let-7a-1 inhibits the Drosha-mediated processing by
directly competing with an RNA-binding protein KSRP that facilitates the processing 69.
In addition, a recent study showed that hnRNP E1 binds to several evolutionarily
conserved CU-rich elements of human endothelial nitric oxide synthase (eNOS) mRNA
to prevent miR-765-mediated repression 70. Moreover, hnRNP Q has been shown to
compete with the poly(A) binding protein (PABP) for binding to the poly(A) tail in
with GW182, a core component of miRISC 72,73, hnRNP Q has been proposed to play a
negative role in miRNA-mediated repression of poly(A)+ mRNAs 74. However, whether
any else hnRNPs are required for miRNA biogenesis and/or function remain elusive.
1.5 The animal model—Caenorhabditis elegans
1.5.1 The advantages of using C. elegnas as an animal model
As an excellent animal model, Caenorhabditis elegans is used to study in a
variety of biological processing, including apoptosis, signaling pathway, cell cycle, cell
polarity, gene regulation, metabolism, ageing, sex determination and miRNA regulation.
With several characteristics, such as the completely sequenced genome composition, the
light-transmittable body for easy observation and the short life cycle, the C. elegans has
become a useful tool for investigators 75. Furthermore, during the process of
development, the heterochronic gene pathway is essential for proper determination of
developmental timing 1. Involvement of miRNAs in this pathway helps scientists, who
are interesting in investigation of miRNA biogenesis and functions, to get an easier way
to reach their goals.
1.5.2 The heterochronic gene pathway and let-7 miRNA in C. elegans
In C. elegans, the heterochronic gene pathway is an excellent model for
studying miRNAs because it acts as binary switches to control temporal development,
which can be easily monitored. In the early larval development, the lin-4 miRNA
represses its target lin-14 and mediates the L1-to-L2 switch. The L2-to-L3 switch is
mediated by lin-4 and the let-7 family (mir-48, -84, and -241) repressing lin-28 and
hbl-1. Finally, let-7 repression of lin-41 and hbl-1 mediates the L4-to-adult switch and
controls terminal differentiation. Since the let-7 mutants cause traceable phenotypes,
they have been used in genetic analysis to determine the interaction with other genes in
the miRNA pathway, including functional partners or downstream targets 76-78. The
temperature-sensitive mutant allele let-7(n2853) carries a G-to-A substitution in the
seed region of the mature let-7 miRNA that reduces target silencing and also the level of
let-7 miRNA 79. let-7 loss-of-function results in heterochronic phenotypes such as
retarded hypodermal cell differentiation and vulval abnormalities. The mutant animals
die by bursting through the vulva at the L/A switch 4,80. Knocking down of let-7
downstream targets usually suppresses the phenotypes 76. Knocking down negative
functional partners of let-7 has also been shown to suppress the lethality of the
modulating target silencing 81,82. On the other hand, a weak allele of let-7, let-7(mg279),
slight reducing the level of mature let-7 due to processing defects, not severe enough to
cause the vulval bursting phenotype, could be a sensitized background in which to
detect other defects in the miRNA pathway 20,83 . Indeed, the let-7(mg279) has been
utilized in synthetic-lethal screens by which several functional factors in the miRNA
pathway have been identified 78.
1.5.3 Controlling of vulval development by miR-84-mediated repression of
let-60/RAS
The oncogenic protein RAS activates proteins necessary for the propagation
of growth factor and other receptors signal such as c-Raf and PI 3-kinase and therefore
promote tumorigenesis (for a review see reference 84) . LET-60 is the homolog of RAS
in C. elegans. Initially, scientists found that the let-60 gain-of-function animals
exhibited extra-vulva or also called multi-vulva phenotype 85,86. Following that, they
found it is a target of miR-84, one of the members in let-7 family. The relieved
protrusion number of vulva by overexpressing miR-84 also supported this idea 87,88.
Finally they found that, during the process of vulva development, only the P6.p cell, one
of the vulva precursor cell, continuously expresses LET-60 and finally it will develop
into the vulva structure. Expression of miR-84 in other precursor cells down-regulates
LET-60 level to ensure only one precursor cell turn into vulva tissue 87. The multi-vulva
phenotype caused by let-60 gain-of-function is easy traceable and observable, making it
a useful tool to monitor the miR-84 activity in C. elegans 61,87,88.
1.5.4 The lsy-6 miRNA-mediated repression of cog-1
The lsy-6 miRNA is one of the most famous miRNAs in C. elegans and not
included in the heterochronic pathway. It functions as a determinant to control the
specification and differential gene expression of two morphologically similar and
bilaterally symmetric neurons, ASEL and ASER. The expression of lsy-6 miRNA is the
ASEL-specific and promotes down-regulation of a key lsy-6 target, the transcription
factor COG-1. In animals without lsy-6 expression, the ASEL neurons employ the
ASER fate due to derepression of COG-1 89. In addition to ASEL/R, the expression of
COG-1 also observed in tissues that do not normally express lsy-6 miRNA, including
the vulva and uterus 90. Utilization of ectopic expressed lsy-6 by the cog-1 promoter can
down-regulate COG-1, for example, in vulval and uterine tissues 89. Therefore, it could
serve as a useful tool to examine the impact of interested genes RNAi on lsy-6-mediated
1.6 Project aims
Since miRNAs regulate more than 60% of human protein-encoding genes and
mis-function of miRNAs directly associates with various types of cancers, to better
understand the mechanisms of miRNA action is certainly important. In the thesis, I use
genetic analysis of the alleles let-7(2853) or let-7(mg279) to discover novel factors,
including DExD/H-box RNA helicases and hnRNPs, which potentially function in the
miRNA pathway.
Chapter 2. Materials and methods
2.1 Caenorhabditis elegans
2.1.1 Strains
The C. elegans strains used in this thesis are listed below:
Strain name Genotype Source
Bristol N2 Wild type Dr. Wu, Y.-C., Taiwan
SPN004 eri-1(mg366) IV; let-7(mg279) X Dr. Ruvkun, G., USA SPN007 eri-1(mg366) IV; wIs51[SCM::gfp,
unc-119(+)] V; let-7(mg279) X Homemade SPN008 wIs51[SCM::gfp, unc-119(+)] V;
let-7(mg279) X Homemade
FT250 xnIs96[pJN455(hmr-1p::hmr-1::GFP::u
nc-54 3'UTR) + unc-119(+)] CGC, USA
maIs105[col-19::gfp] Dr. Grosshans, H., Switzerland
MT2124 let-60(n1046gf) CGC, USA
GR1689 let-60 (n1046gf); mir-84(tm1304) Dr. Ruvkun, G., USA GR1690 mgIs45[mir-84++]; let-60(n1046gf) Dr. Ruvkun, G., USA PS3662 syIs63[cog-1::GFP + unc-119(+)] CGC, USA
OH7310
syIs63[cog-1::GFP + unc-119(+)];
otIs193 [cog-1p::lsy-6hp + rol-6(su1006)]
CGC, USA
SPN133 ddx-23::GFP::3xFLAG Homemade
MT7626 let-7(n2853) X Dr. Slack, F. J., USA
SPN016 wIs51[SCM::gfp, unc-119(+)] V;
let-7(n2853) X Homemade
2.1.2 Culture
Except the worms carrying the let-7(n2853) mutant allele were cultured at its
permissive temperature 15°C, the others were maintained under standard condition at 20
°C as previously described 91. In brief, the animals were placed onto the nematode
growth media plate (NGM plate: 17 g bacto-agar, 2.5 g tryptone and 3 g NaCl for 1 L)
with sufficient E. coli OP50 on the surface of agar.
lin-29(n333) II; wIs51[SCM::gfp,
unc-119(+)] V Homemade
lin-29(n333) II; maIs105[col-19::gfp] Homemade MT19756 nIs408 [lin-29p::lin-29::mCherry +
ttx-3p::GFP] CGC, USA
HW769 pwrt-2::gfp::lin-41 3′ UTR Dr. Grosshans,
H., Switzerland
HW783 pwrt::gfp::lin-41 3′ UTR∆LCS Dr. Grosshans,
H., Switzerland
HW786 pwrt-2::gfp::unc-54 3′ UTR Dr. Grosshans,
H., Switzerland EG3366 ttTi5605 II; unc-119(ed3) III; oxEx1578
[eft-3p::GFP + Cbr-unc-119] CGC, USA SPN062 pwrt-2::gfp::lin-41 3′ UTR Homemade SPN063 pwrt::gfp::lin-41 3′ UTR∆HRE Homemade SPN064 pwrt::gfp::lin-41 3′ UTR∆LCS Homemade SPN065 pwrt::gfp::lin-41 3′ UTR∆LCS∆HRE Homemade
Phrp-2::gfp::hrp-2 Homemade
2.1.3 RNA-interference (RNAi)
All the RNAi experiments for C. elegans used in this thesis were feeding
RNAi. The E. coli strain HT115 carrying IPTG inducible expression vectors, in which
the dsRNA can be expressed to against specific genes by driving the two T7 promoter
located at each side of interested gene fragments. In order to get the same growth rate of
worms, we harvested the embryo from gravid adults as mentioned in previous study 92.
In summary, animals were collected and precipitated by gravity in M9 buffer.
Discarding the supernatant and supplementing 0.75 mL 5N NaOH and 1.5 mL bleach to
the final volume is 7.5 mL in the 15 mL centrifuge tube. It was immediately undergone
the vigorously votex for around 3-5 minutes to dissolve worm bodies and release
embryos. Following 3000 rpm centrifugation for 1 minute, the supernatant was removed
and supplemented with M9 buffer to wash the embryo pellet. After three times repeat of
centrifugation and washing pellet, the embryos were soaked in a few volume (usually
3-5 mL) of M9 buffer and left in 20 °C with constantly rotating overnight. The next day,
synchronized L1 animals were then subjected into plates that were seeded with bacteria
carrying control or RNAi clone against genes of interest two days before. Following 30
subsequent assays.
2.2 Construction of plasmids
2.2.1 C. elegans RNAi clones
The pL4440 vector was digested with restriction enzyme EcoRV to produce a
blunt-end cutting site. Following the alkaline phosphatase (CIP) (NEB, #M0290)
treatment, the linearized vector was incubated with the PCR-amplified interested gene
fragment, which had been blunted by the kit from NEB (#E1201) according to the
manufacture’s instructions, and then ligated by T4 DNA ligase at 16°C overnight. The
detail information of RNAi clones and the primer sets used for newly generated RNAi
clones in this thesis are listed in Appendix 1 and 2.
2.2.2 C. elegans expressing plasmids
The Multisite Gateway system (Invitrogen) was used to generate genomic
DNA fragment-based expression vector for C. elegans and the procedure was according
to the instruction mentioned previously 93. Briefly, as a promoter region, the ~3000 bp
upstream of the start codon (ATG) was PCR-amplified and carrying attB4 and attB1r
site for BP clonase (Invitrogen) recognition. Following the BP reaction, recombining of
the promoter region into pDONRP4P1r vector leads to generation of the 5’ entry clone.
The universal tag, such as HA and GFP, with start codon was amplified and inserted
into the pDONR221 middle entry clone. The fragment for 3’ entry clone was amplified
by the primers start from the coding region without start codon and stop with the end of
3’UTR, which was according to the sequence published in NCBI database. After
insertion of this fragment into pDONRP2rP3 vector, the 3’ entry clone was ready for
subsequent steps. For BP reactions, 150 ng of vector and 10-150 ng of insert were mix
and supplemented with sterile water to the final volume is 8 µL. For each reaction, 2 µL
BP clonase (Invitrogen, #11789020) is sufficient. Following overnight incubation at
room temperature, the reaction was transformed into E. coli DH-5α strain by heat shock.
After sequence validation of these three entry clones, the 5’, middle and 3’, they were
used as the materials for LR reaction. 150 ng of each entry clone were mixed in a
microcentrifuge tube and plus 50 ng of the destiny vector, pCFJ150 for MosSCI and
pGC188 for conventional injection method in this thesis, to the final volume is 8 µL.
Following incubation at room temperature overnight after supplemented 2 µL LR
clonase (Invitrogen, #11791100), the reaction was transformed into E. coli DH-5α strain
ready for subsequent use to inject into worms to express the gene of interest.
2.2.3 TRIM71 3’UTR fused luciferase plasmids
The TRIM71 3’UTR and an LCS-deleted version were artificially synthesized
and inserted into a vector (IDT custom gene synthesis). Following digested with NotI
and XhoI, the purified TRIM71 3’UTR fragments were subjected into the ligation
reaction with the linearized psicheck2 luciferase vector at 16 °C overnight in water bath.
To delete the hnRNP Q response element, we designed a primer pair contains the
restriction enzyme site for XhoI in forward primer:
ATCGCTCGAGTTGCATTTCCTAGGTTTCTGTGT and NotI in reverse primer:
GCCAGCGGCCGCAGTAGTTTTTTTTTGTGTTTCCT to generate the fragment
without this element (24 bp deletion). After amplified and digested the PCR fragments,
the ligation reactions were performed as the same condition as the full-length TRIM71
3’UTR.
2.2.4 hnRNP Q1 plasmids
For constructing HA-hnRNP Q1, the primer set complement to hnRNP Q1
coding region was used for PCR amplification. The DNA oligo sequence for the
forward primer is TTCAATCGATGGGCTACAGAACATGTTAAT and reverse primer
is TCCCGATCGATTCATTGTAACAGGTCAGGA. The ClaI recognition sites (the
sequence underlined) were artificially introduced to each end of primers. Following
amplified the coding region and digested by ClaI, the fragment was placed into the
ligation reaction with linearized pKH3 vector at 16 °C overnight in water bath. For
constructing hnRNP Q1-mCherry, the primer pair, forward:
ATCCGCTAGCATGGCTACAGAACATGTTAA and reverse:
ATGCAGATCTTTGTAACAGGTCAGGACCGG, were designed and used to amplify
the hnRNP Q1 cDNA with the recognition sites for NheI at 5’ and BglII at 3’ end,
respectively. Following the digestion reaction by these restriction enzymes, the PCR
amplified fragment was subjected into ligation reaction with the linearized
pDEST-mCherry-N1 vector, digested by NheI and BamHI, at 16 °C overnight in water
bath. Single colonies of these constructs after E. coli transformation were selected
following sequenced.
2.2.5 GFP-Ago2 plasmid
The pEYFP-C1-Ago2 was a kind gift from Dr. Chia-Ying Chu (National
restriction enzymes, we subjected the purified Ago2 fragment into the ligation reaction
with the digested pEGFP-C1 vector at 16 °C overnight in water bath. The single
colonies were selected and sequenced to get the pEGFP-C1-Ago2 construct.
2.3 Microscopy analysis
For numbering the worms burst from vulva, we directly counted the animals
on plates by using a dissecting microscopy. For imaging, animals were collected and
mounted on the agar pad contains 2% Agarose I (amresco, #9012-36-6). Prior to cover
the coverslips, the worms were soaked in 20 µL 1 mM Tetramisole Hydrochloride
(Sigma, #L9756) for anesthesia. The image were obtained and analyzed by the Zeiss
Axiovision system.
2.4 RNA isolation
2.4.1 From C. elegans
The animals for total RNA purification were collected and washed
sufficiently by M9 buffer. The worm pellet was resuspended by 1 mL of Genezol
(Geneaid, #GZR100) and put into a 2-mL screw cap tube contains 0.5 mL sterile
Zirconia beads. Following the homogenization of the mixture by the homogenizer (M.P.
Biomedicine, FastPrep 24), 200 mL chloroform was added into each tube. Subsequently,
the centrifugation was performed at 12000 g for 15 minutes at 4 °C to separate the water
phase and oil phase. The water phase was then moved to a new 1.5 mL microcentrifuge
tube. Following 10 minutes incubation of the equal amount isopropanol, the
centrifugation was performed at 12000 g for 20 minutes at 4 °C. After removing the
supernatant and washing the pellet by 70% ethanol at 12000 g for 5 minutes at 4 °C, the
RNA was dissolved by proper amount of DEPC treated water.
2.4.2 From human cell cultures
The pelleted cells or cell lysates were used to isolate total RNA by
introducing 1 mL Genezol (Geneaid, #GZR100). After incubation at room temperature
for 10 minutes, each tube was added 200 mL chloroform and mixed thoroughly. The
supernatant was moved into a new tube following the 15 minutes centrifugation at
12000 g at 4 °C. After 10 minutes incubation of the equal amount isopropanol, the
centrifugation was performed at 12000 g for 20 minutes at 4 °C. After removing the
supernatant and washing the pellet by 70% ethanol, the RNA was dissolved by proper
amount of DEPC treated water.
2.5 Northern blot analysis for small RNAs
For detection of miRNAs, 10 µg of total RNA samples, per lane, were
separated by 12% polyacrylamide gels (8 M urea, Acrylamide/Bis 19:1) and transferred
onto Hybond-N+ membranes (GE Healthcare, #RPN119B). RNA was cross-linked to
the membrane by 254 nm UV light irradiation (120000 microjoules/cm2) for each side
of the membrane and then baking at 80 °C for 1 hour. Short RNA probes
complementary to miRNAs or U6 snRNA were prepared by in vitro transcription using
T7 RNA polymerase as previously described 94. Generally, the reaction was mix up with
3 µL master mix, contains 6.67 µM ATP, UTP, GTP and 1X T7 RNA polymerase
buffer, 10.5 µL 5 µM DNA template, 1 µL Superas-In (Ambion, #AM2694), 4 µL
32P-alpha-CTP (3000 uCi/mole) and 1.5 µL T7 RNA polymerase (NEB, #M051) to the
final volume is 20 µL. To generate the DNA templates for in vitro transcription, the
independent DNA oligos were synthesized and heat annealed. For annealing of two
independent but complement oligos, the mixture contains 5 µM of each DNA oligos
was heated at 95 °C for 5 minutes and then put at room temperature to allow the oligos
anneal together. The sequence of DNA oligos used in this thesis were summarized and
listed below.
T7Top
GAA ATT AAT ACG ACT CAC TAT AGG
T7let-7 btm
TGA GGT AGT AGG TTG TAT AGT TCC TAT AGT GAG TCG TAT TAA TTT C
T7lin-4 btm
TCC CTG AGA CCT CAA GTG TGA CCT ATA GTG AGT CGT ATT AAT TTC
T7miR-48 btm
TGA GGT AGG CTC AGT AGA TGC GAC CTA TAG TGA GTC GTA TTA ATT TC
T7miR-241 btm
TGA GGT AGG TGC GAG AAA TGA CCT ATA GTG AGT CGT ATT AAT TTC
T7miR-84 btm
TGA GGT AGT ATG TAA TAT TGT AGA CCT ATA GTG AGT CGT ATT AAT TTC
T7U6 btm
GGA TGA CAC GCA AAT TCG TGC CTA TAG TGA GTC GTA TTA ATT TC
*Note: The sequence underlined is the site for T7 RNA polymerase recognition.
Hybridization was performed at 55 °C in 0.36 M Na2HPO4, 0.14 M NaH2PO4,
1 mM EDTA, 10% SDS, 25% Formamide and 0.1 mg/ml salmon sperm DNA. Washes
were done at 55 °C twice in the low stringency buffer (4× SSPE and 4% SDS) and
twice in the high stringency buffer (0.1× SSC and 0.1% SDS). Radioactive signals were
detected by a storage phosphor image plate and the Typhoon Trio Variable Mode
Imager (GE Healthcare) or exposed onto the radioisotope-sensitive films (Kodak,
#Z363073).
2.6 RT-PCR and RT-qPCR
2.6.1 Poly-A tail-based reverse transcription and qPCR
For detecting the total pri-let-7 (the sum of pri-let-7A, pri-let-7B and
SL1-pri-let-7) by qPCR, up to 1 µg of total RNA was used and subjected into poly-A
tail-based reverse transcription. Briefly, the sample was mixed with reagents listed as
follows and heat at 65 °C then cool-down to 4 °C to anneal the universal RT-primer
containing a VN anchor, oligo(dT) and the universal reverse PCR primer sequence,
called URT primer, as previously described approach 95.
Annealing of poly-A tail-based URT primer
RNA sample (up to 1 µg) 10 µL
50 µM URT primer 1 µL
10 mM dNTP mix 1 µL
Nuclease free water 1 µL
Subsequently, following waiting at least 1 minute for incubation of the
reaction at 4 °C, reagents listed below were added into the tube and then the reaction
was subjected into reverse transcription.
Reagents for poly-A tail-based reverse transcription
Primer annealed RNA sample 13 µL
5X First-strand buffer 4 µL
0.1 M DTT 1 µL
Superase-In (Ambion, #AM2694) 0.5 µL
Superscript III (Invitrogen, #18080044) 0.5 µL
Nuclease free water 1 µL
Final volume 20 µL
The program for reverse transcription was initiated from activating the
enzyme by keeping the temperature at 25 °C for 5 minutes. Next, for best activity of
RTase, the temperature was set at 55 °C for one hour. Finally, to stop the reaction, heat
inactivation was performed by setting the temperature at 70 °C for 15 minutes. The
RNA templates were eliminated by adding 1 µL RNase mixture, which contains 2.5 unit
RNase H (NEB, #M0297) and 5 µg RNase A (QIAGEN, #19101), and incubating at 37
°C for 20 minutes.
To perform the quantitative-PCR, the KAPA SYBR FAST qPCR kit was used
according to the manufacture’s instructions. The reagents used in qPCR were listed in
the table below.
Reagents for poly-A tail-based quantitative-PCR
50X diluted cDNA 5 µL
Forward primer (10 µM) 0.4 µL
Reverse primer (10 µM) 0.4 µL
Nuclease free water 4.2 µL
Final volume 20 µL
For detecting the signals, the ABI Prism 7000 system was used and the
program was set as follows.
Program used for detecting poly-A tail based quantitative-PCR
Step 1 95 °C 3 minutes Activate enzyme
Step 2.1 95 °C 30 seconds Denaturing DNA
Step 2.2 60 °C 30 seconds DNA synthesis
Detect the signals at the end of this step at each cycle and repeat step 2 for 40 cycles
Step 3 60-95 °C Dissociation
The sequences of oligonucleotides used in this assay are listed in the table below.
URT primer
5’-AAC GAG ACG ACG ACA GAC TTT TTT TTT TTT TTT VN-3’
Total pri-let-7 forward (L1)
5’-GCA TCT ACC TCG ATT GGA CCT A-3’
eft-2 forward
5’-GTG CTA ATC CAC CTC TGG AA-3’
Universal reverse primer (R1)
5’-AAC GAG ACG ACG ACA GAC TTT-3’
2.6.2 Random priming-based reverse transcription and qPCR
This assay was used to examine the presence or abundance of mRNAs or
primary miRNAs. 10 µg of total RNA was treated with DNase I (Ambion, #AM1907),
in the final volume is 20 µL for at least 30 minutes at 37 °C. The DNase was inactivated
by adding 4 µL of inactivation reagent with constantly tapping of the tubes for 5
minutes at room temperature. Following brief centrifugation for a few seconds, 10 µL of
the supernatant was used for subsequent steps. The cDNA was synthesized by
SuperScript III reverse transcriptase (Invitrogen, #18080044) using random hexamers.
The detail information of reagents used in cDNA synthesis is listed in the table below.
Annealing of random priming-based primers
DNase treated RNA sample (around 5 µg) 10 µL
100 ng/µL Random hexamers 1 µL
10 mM dNTP mix 1 µL
Nuclease free water 1 µL
Following adding reagents, the tube was put into a PCR cycler and the
program was set as 65 °C for 5 minutes and cool-down to 4 °C at least 1 minutes to
allow the primers to properly attach the RNA molecules. Prior into the cDNA synthesis,
several reagents were added into the tube as listed below.
Reagents for random priming-based reverse transcription
Random hexamers annealed RNA sample 13 µL
5X First-strand buffer 4 µL
0.1 M DTT 1 µL
Superase-In (Ambion, #AM2694) 1 µL
Superscript III reverse transcriptase (Invitrogen, #18080044) 1 µL
The program for cDNA synthesis was initiated from 5 minutes at 25 °C to
activate RTase and then one hour at 50 °C for enzyme functioning and finally stopped
by inactivating the enzyme for 15 minutes at 70 °C. The RNA templates were
eliminated by adding 1 µL RNase mixture, which contains 2.5 unit RNase H (NEB,
#M0297) and 5 µg RNase A (QIAGEN, #19101), and following incubation at 37 °C for
20 minutes.
To perform the quantitative-PCR, the KAPA SYBR FAST qPCR kit was used
according to the manufacture’s instruction. The reagents used in qPCR were listed in
the table below.
Reagents for poly-A tail-based quantitative-PCR
10-50X diluted cDNA 5 µL
2X SYBR FAST master mix (KAPA, #KK4600) 10 µL
Forward primer (10 µM) 0.4 µL
Reverse primer (10 µM) 0.4 µL
Nuclease free water 4.2 µL
Final volume 20 µL
For detecting the signals, the ABI Prism 7000 system was used and the program was set
as follows.
Program used for detecting poly-A tail based quantitative-PCR
Step 1 95 °C 3 minutes Activate enzyme
Step 2.1 95 °C 30 seconds Denaturing DNA
Step 2.2 60 °C 30 seconds DNA synthesis
Detect the signals at the end of this step at each cycle and repeat step 2 for 40 cycles
Step 3 60-95 °C Dissociation
The sequences of oligonucleotides used in this assay are listed in the table below.
Primers used in random priming-based quantitative-PCR For C. elegans
SL1-pri-let-7_forward (L2) 5’-GGTTTAATTACCCAAGTTTGAGGC-3’
pri-let-7 and
SL1-pri-let-7_reverse (R2)
5’-CGCAGCTTCGAAGAGTTCTG-3’
pri-let-7_forward (L3) 5’-TCCTAGAACACATCTCCCTTTGA-3’
pri-miR-84_forward 5’-ATTTGGCGATGCGAGAAAGT-3’
pri-miR-84_reverse 5’-AGGCAGACGTATGATGAATA-3’
pri-lin-4_forward 5’-GACAATTTCTAGAGTTTTGGTTGG-3’
pri-lin-4_reverse 5’-CCTTTTCCCCGAATACCATT-3’
pri-miR-241_forward 5’-GTTCGGAATGGATTTTGGTTG-3’
pri-miR-241_reverse 5’-AGTGATGTTTCGATCTCCAC-3’
drsh-1_forward 5’-CGGACAAGACCGGAGAAGTA-3’
drsh-1_reverse 5’-CGTTTCCCAAACCTTTTTCA-3’
pash-1_forward 5’-GATTGCAGCGAATGATGAGA-3’
pash-1_reverse 5’-TCCTCAACCATTCCATCACA-3’
dcr-1_forward 5’-TGGTGGTGATGTCTCGAAAA-3’
dcr-1_reverse 5’-TCCCAACGTCAGCAAATGTA-3’
alg-1_forward 5’-TGCGCAGAAAGTATCGTGTC-3’
alg-1_reverse 5’-CTCTGGTGGCAGGTAGGTGT-3’
tbb-2_forward 5’-CAAATTCTGGGAGGTCATCTC-3’
tbb-2_reverse 5’-CATACTTTCCGTTGTTGGCT-3’
lin-42_forward 5’-TCTTGTTCACGTGCACCTTC-3’
lin-42_reverse 5’-GGCTCCGTCTGGCATAGTAA-3’
eft-2_forward 5’-TGTGTTTCCGGAGTGTGTGT-3’
eft-2_reverse 5’-CCATCGTCGTCTCCGTAAGT-3’
lin-41_forward 5’-GGATTGTTCGACACCAACG-3’
lin-41_reverse 5’-ACCATGATGTCAAACTGCTGTC-3’
gfp_forward 5’-ACCAGACAACCATTACCTGTCC-3’
gfp_reverse 5’-TCCCAGCAGCTGTTACAAACTC-3’
For mammalian cells
TRIM71_forward 5’-TGTGAGCTGCTGTGGAAGGT-3’
TRIM71_reverse 5’-GTCTTCAGCTCCTGCACCTG-3’
Renilla_forward 5’-GCCTAAGATGTTCATCGAGT-3’
Renilla_reverse 5’-TACTGCTCGTTCTTCAGCAC-3’
2.7 Lysate preparation
2.7.1 For C. elegans western blot analysis
The lysates were made according to previously described with little
modifications 96. Briefly, packed animals were collected and washed twice with 4-fold
volumes of wash buffer (50 mM Tris-HCl at pH 7.5 and 10 mM potassium acetate)
followed by centrifugation at 3000 rpm for 1 min. The animals were resuspended in
4-fold volume of homogenization buffer [50 mM Tris-HCl at pH 7.5, 10 mM potassium
acetate, 5 mM DTT, 10 U/ mL Superase-In (Ambion, #AM2694), and 1X Complete
protease inhibitor (Roche, #11697498001)] and incubated for 20 min on ice with
intermittent agitation. The animals were then rubbed by using a 1.5 Pellet Pestle and a
handy motor for 1 minute, with 10 seconds pulsing for each 10 seconds rubbing. The
homogenate was incubated for 20 min on ice after 2 mM magnesium acetate was added
and the concentration of potassium acetate was adjusted to 100 mM. Then centrifuged
at 13200 rpm for 10 minutes at 4 °C. The supernatant was used as cell extracts in
western blot analysis.
2.7.2 For C. elegans immunoprecipitation experiment
Worm lysates for immunoprecipitation (IP) were made according to
previously described with a few modifications 97. In brief, the pelleted worms were
homogenized in 5 volumes of IP buffer [10 mM Hepes-KOH, 250 mM NaCl, 0.5%
NP-40, 10% glycerol, 5 mM EDTA, 1 mM DTT, 1X Protease Inhibitors (Roche,
#11697498001)] for three times 30 seconds homogenous at 4 °C, with 5 minutes
interval for each action. The homogenate were then centrifuged at 10,000 rpm for 20
min at 4 °C. The supernatant was moved to a new microcentrifuge tube and used as cell
extracts for immunoprecipitations. The total protein concentration was determined using
the Bradford reagent (Amresco, #M172).
2.7.3 For mammalian cell lines western blot analysis
Lysis of cultured mammalian cells was performed as described in previous
study 98. In summary, the collected cells were washed twice by 1 mL cold PBS buffer
(137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, adjusted the final pH
value to 7.4). The packed cell pellet was lysed by adding appropriate volume of RIPA
lysis buffer (150 mM NaCl, 5 mM EDTA pH 8, 1M Tris pH 8, 1% NP-40, 0.5% sodium
deoxycholate and 0.1% SDS). Following incubation on ice for 30 minutes, the lysate
was centrifuged at 13200 rpm for 15 minutes at 4 °C. The supernatant was moved into a
new tube and used as cell extract for western blot analysis.
2.7.4 For mammalian cell lines immunoprecipitation experiment
The lysates for immnoprecipitation were prepared according to the description
in previous study 99. Briefly, cells were collected and washed twice with PBS buffer.
Then, the cell pellet was resuspended by appropriate volume of NET buffer [50 mM
Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100 and 1X protease
inhibitor (Roche, #11697498001)], usually 0.5 mL is sufficient for the cells collected
from a 10 cm dish, and incubated on ice for 30 minutes. Following that, centrifugation
was performed at 13200 rpm for 10 minutes at 4 °C. The supernatant was transferred to
a new tube and served as cell lysate for immunoprecipitation.
2.8 Western blot analysis
20-50 µg total proteins were used and mixed with 5X SDS sampling buffer
(375 mM Tris-HCl pH6.8, 10% SDS, 50% glycerol, 10% 2-mercaptoethanol and 0.03%
Bromo Blue) and then heat at 95 °C for 5 minutes to denature proteins. Following
centrifugation at 13200 for 30 second, the samples were then separated by 10% Bis-Tris
polyacrylamide gels and transferred to PVDF membranes for western blotting analysis.
In this thesis, antibodies and their working condition used for western blot were listed in
the table below.
Antibody against
Secondary antibody
Dilution/blocking reagent
Source
C. elegans
ALG-1 Rabbit 1:5000/ 5% Milk Thermo Fisher, PA1-031X PASH-1 Rabbit 1:1000/ 5% Milk From Dr. Helge Grosshan DDX-17 Rabbit 1:3000/ 5% Milk Home made by LTK Cop.
DDX-23 Rabbit 1:1000/ 3% BSA Home made by LTK Cop.
ACTIN Mouse 1:5000/ 5% Milk Proteintech, 66009-1-Ig
HRP-2 Rabbit 1:5000/ 5% Milk From Dr. Alan M. Zahler Mammalian cell lines
TRIM71 Sheep 1:3000/ 3% BSA R&D systems, AF5104 hnRNP Q/R Mouse 1:3000/ 5% Milk Sigma, R5653
ACTIN Mouse 1:10000/ 5%
Milk
Proteintech, 66009-1-Ig Ago2 Rabbit 1:5000/ 5% Milk Abcam, ab186733 HA-tag Mouse 1:1000/ 5% Milk Covance, MMS101R GFP-tag Mouse 1:1000/ 5% Milk Clontech, 632375
PABP Mouse 1:1000/ 5% Milk Santa Cruz, sc-32318
2.9 Co-immunoprecipitation
Immunoprecipitations were initiated by binding 1-2 µg antibody to Protein A
Sepharose beads (GE healthcare, #17-0780-01) in TBST buffer for 30 minutes at room
temperature. In this thesis, the antibodies used for IP experiments are anti-HRP-2,
anti-ALG-1 and anti-HA (3F10). It was subsequently washed twice by IP buffer and
supplemented with 500-1000 µg lysate into each reaction. For RNase treatment, 20 µg
RNase A (QIAGEN, #19101) was added into lysate and incubated at room temperature
for 20 minutes to digest total RNAs. Following incubation at 4 °C for at least 2 hours,
beads were washed by IP buffer five times and the precipitated proteins were eluted by
boiling in SDS sampling buffer. The samples were then centrifuged 5 min at 13200 rpm
to separate Protein A beads, and the supernatants were loaded into 10% Bis-Tris gel.
proteins.
2.10 RNA-immunoprecipitation (RIP)
The first step in RIP experiments was performed as the condition mentioned
above in IP experiment. After that, the pellet was subjected into RNA purification step
by adding 1 mL Genezol (Geneaid, #GZR100) for each reaction. Following addition of
200 µL chloroform and vigorously shaking the tube, the mixture was centrifuged at
12000 g for 15 minutes at 4 °C. The supernatant was moved into a new tube then mixed
with equal volume of isopropanol and 20 mg glycogen to let the pellet could be seen in
the following centrifugation step. After 10 minutes incubation at room temperature, it
was centrifuged at 12000 g for 20 minute at 4 °C to precipitate RNAs. The supernatant
was removed and the pellet was washed by 70% ethanol once at 13200 rpm for 5
minutes at 4 °C. Finally, the pellet was air-dried and dissolved in appropriate volume of
nuclease free water. The purified RNA samples were then subjected into northern blot
or quantitative RT-PCR analysis.
2.11 In vitro transcription of biotinylated RNA oligo
To PCR amplify the T7 promoter fused DNA templates for in vitro