國立臺灣大學工學院工程科學及海洋工程學系 碩士論文
Department of Engineering Science and Ocean Engineering College of Engineering
National Taiwan University Master Thesis
支援跨物種組織的整合性高通量序列分析 及功能註解參考系統
Integrated reference system for high-throughput sequence analytic and functional annotation on cross-species and
multiple-tissue
黃柏潤 Bo-Jun Huang
指導教授:黃乾綱 博士
Advisor: Chien-Kang Huang, Ph.D.
中華民國 97 年 6 月
June, 2008
i
ii
誌謝
兩年時光一扎眼便過了。記得剛進實驗室時,「研究」這兩個字對我而言仍處 於懵懵懂懂階段。然而,在參與指導教授的計畫與論文程式實作後,我開始對「做 研究」的精神與態度有了更深一層的認識,同時也發現自己在專業能力與心態上 的許多不足之處。
因此,首先最要感謝我的指導教授 黃乾綱博士,謝謝您在這兩年用心指導,
並分享許多經驗與想法,給我許多啟發與動力。再者,萬分感謝動物科學系的林 恩仲教授,謝謝您在系統設計上給予相當多寶貴意見,並耐心地解釋各種生化反 應關係,使非生物背景我能及早進入狀況。在此對以上兩位老師致上最由衷的謝 意。此外,要非常感謝我的口試委員洪振發教授、丁詩同教授以及蔡孟勳教授對 論文的指導與建議,讓論文更臻於完善。在此謹致最誠摯的謝意。
另外,特別感謝資工所的黃鈺峰學長在研究的方向與方法上,提供寶貴的建 議,使我研究過程遭遇的難題大多能迎刃而解。也謝謝動科所的慶儀與怡惠學姐 在學習過程中給予許多指教以及鼓勵。感激基安大哥在工作之餘尚需花費許多時 間幫我分析資料,使我的系統能順利完成。
還有跟我一起度過研究所生涯的家禎、家瑞、硯農、庭毓、逸偉、敦威、慎 清、書宇、耕維、基安、俊欽、佳憲、胤辰、濠欣、明翰,你們都是很有趣且優 秀的夥伴,跟你們相處的點點滴滴我永遠都不會忘記。欣慰這段時光大家都能平 安健康,在未來各奔前程的歲月裡,也祝福大家一路順風。
最後,我要感謝我的家人。謝謝我的父親黃治源先生適時的補貼生活費,不 定時的言語關心,讓我不必擔心民生問題,能夠更專注於學業上。謝謝我的母親 鄭雲霞女士在我回家之時準備豐富營養的餐點,讓我的身體總是維持很好的活 力。還有感謝在我面對挫折時,背後給予我鼓勵的親朋好友們,有了你們的陪伴,
讓我知道在這條路上並不孤單。
黃柏潤 謹誌 中華民國九十七年七月二十六日 于 國立臺灣大學工程科學及海洋工程系 Lab125A
iii
摘要
生物學家會利用較常研究的模式動物(model animal) ,透過蛋白質或基因序列 的來預測與非模式動物的同源關係。然而,從過去研究的文獻了解,跨物種組織 的整合平台與高通量的序列比對系統顯得相當重要。若是缺乏一個整合平台,則 研究人員需要先至網站下載欲研究的物種資料,並建構各資料間的關聯性。若尚 需考慮跨物種組織的序列比對,操作流程會更加複雜。此外,針對基因功能註解 的部分還可以透過 GO Term 說明。
本論文目的為建構一個跨物種組織分析的系統平台,提供研究人員選擇欲研 究的物種與組織進行高通量序列比對,進而協助研究人員對跨物種組織之間的同 源性有所了解,而這也是目前大部分的生物資訊系統所缺乏的功能。此外,我們 也將序列比對結果與 GO 功能註解資訊建立連結。最後,我們運用 Web2.0 的技術 提供友好的人機互動介面,並將查詢結果封裝為 XML (Extensible Markup Language) 格式,以利於未來的信息交流。
關鍵字 : 跨物種組織、序列比對工具、基因註解、蛋白質、模式動物
iv
Abstract
The biologists expect to use model animal to predict non-model animal when they want to deal with sequence alignment. However, the integrated platform on cross-species and multiple-tissue for gene-protein-function inference is critical according to literature surveys on related service. Without integrated platform, researches have to download files, link the relationships between databases, and develop programs to deal with dataset manually. With the consideration of different species and multiple tissues, the complexity of platform is incredible to deal with multiple data sources for biologists. In addition, function inference for gene sequences could be done by GO (Gene Ontology) term.
In this paper, we propose a framework of integrated platform with
cross-species and multiple-tissue data reference, and it provides high-throughput sequence analytic tool for homology mapping. In addition, we also develop a web service to integrate different data sources and functional annotation information of GO.
Computer science technology is also applied such as XML (Extensible Markup Language) for information exchange to simplify flow combination and dynamic web design and web 2.0 technology for friendly interactive interface to provide enriched information.
Keyword: DNA, cross-species, multiple-tissue, BLAST, model animal
v
目錄
口試委員會審定書………... i
誌謝………...ii
中文摘要………..iii
英文摘要………..iv
圖次 ... vii
表次 ... ix
Chapter 1 緒論 ...1
1.1 研究動機 ... 1
1.2 研究目的 ... 2
1.3 研究流程 ... 3
1.4 論文架構 ... 4
Chapter 2 文獻探討 ...5
2.1 相關研究網站 ... 5
2.1.1 NCBI ... 5
2.1.2 The Gene Ontology ... 5
2.2 資料來源介紹 ... 7
2.2.1 UniGene 資料集 ... 7
2.2.2 NR 資料集 ... 8
2.2.3 RefSeq 資料集 ... 8
2.2.4 GOA (Gene Ontology Annotation, GOA@EBI) ... 9
2.2.5 Gene Ontology 資料集 ... 9
2.3 序列比對工具 ... 10
2.3.1 BLAST 簡介 ... 10
2.3.2 FASTA 格式 ... 12
2.3.3 HTML4BLAST 工具 ... 12
2.3.4 Graphviz 結構關係圖產生器 ... 13
2.3.5 BlastSummary ... 13
2.4 相關系統研究 ... 14
2.4.1 COMPARE ... 14
2.4.2 ZooDDD ... 14
2.4.3 BioMOBY ... 15
2.4.4 Taverna ... 15
Chapter 3 資料倉儲建置 ...16
3.1 Framework ... 16
vi
3.2 資料倉儲內之物種與組織 ... 16
3.2.1 物種 ... 17
3.2.2 組織與發展時期 ... 18
3.3 正規化資料來源之表格設計 ... 18
3.3.1 正規化 UniGene 之欄位設計 ... 18
3.3.2 正規化 NR 之欄位設計 ... 19
3.3.3 正規化 RefSeq 之欄位設計 ... 19
3.3.4 正規化 GOA 之欄位設計 ... 20
3.3.5 正規化 Gene Ontology 之欄位設計 ... 20
3.3.6 整合各資料集之 ERD ... 22
Chapter 4 系統設計與建構 ...23
4.1 系統運作流程 ... 23
4.2 系統模組介紹 ... 29
4.2.1 模組一 : 物種及組織選擇 ... 29
4.2.2 模組二 : FASTA 格式之產生與進行 BLAST 演算 ... 30
4.2.3 模組三 : Gene Ontology 相關資訊之擷取 ... 34
4.2.4 模組四 :蛋白質序列資訊之擷取與封裝為 XML 文件形式 ... 35
4.2.5 模組五 : GO 樹狀圖形及階層路徑分析與呈現 ... 37
Chapter 5 討論 ...41
5.1 與 COMPAER 系統進行比較 ... 41
5.2 與 ZooDDD 系統進行比較 ... 43
Chapter 6 結論與未來工作 ...47
6.1 結論 ... 47
參考文獻 ...49
附錄 A : UniGene 物種版本及各組織與發展時期的序列數目………...51
vii
圖次
圖 1. 1 研究流程 ... 4
圖 2. 1 GO 階層架構 ... 6
圖 2. 2 GO 樹狀結構 ... 7
圖 2. 3 UNIGENE 資料集蒐集流程 ... 8
圖 2. 4 BLAST 演算流程 ... 12
圖 2. 5 FASTA 格式 ... 12
圖 2. 6 BLAST 結果圖型顯示 ... 13
圖 2. 7 DOT 格式 ... 13
圖 3. 1 資料倉儲架構 ... 16
圖 3. 2 UNIGENE 資料集之 ERD ... 19
圖 3. 3 NR 資料集之 ERD ... 19
圖 3. 4 REFSEQ 資料集之 ERD ... 20
圖 3. 5 GOA 資料集之 ERD ... 20
圖 3. 6 GENEONTOLOGY 資料集之 ERD ... 21
圖 3. 7 整合各資料集之 ERD ... 22
圖 4. 1 系統 USE CASE DIAGRAM ... 25
圖 4. 2 系統 ACTIVITY DIAGRA ... 28
圖 4. 3 系統功能 - 選擇 INPUT 的物種及組織 ... 29
圖 4. 4 系統功能 - 選擇 DATABASE 的物種及組織 ... 29
圖 4. 5 系統功能 - 輸入 BLAST 參數 ... 30
圖 4. 6 模組一:系統運作流程圖 ... 30
圖 4. 7 系統功能 – SUMMARYBLAST 程式輸出結果 ... 31
圖 4. 8 系統功能 – BLAST 演算完成後的輸出 ... 31
圖 4. 9 系統功能 – BLAST 演算完成後的圖形化展示 ... 32
圖 4. 10 模組二:系統運作流程圖 ... 33
圖 4. 11 系統功能 – 展示對映至 GENE ONTOLOGY 資料集資訊(1) ... 34
圖 4. 12 系統功能 – 展示對映至 GENE ONTOLOGY 資料集資訊(2) ... 34
圖 4. 13 模組三:系統運作流程圖 ... 35
viii
圖 4. 14 比對結果之 DATA SCHEMA ... 36
圖 4. 15 比對結果以 XML 格式封裝 ... 36
圖 4. 16 模組四:系統運作流程圖 ... 37
圖 4. 17 系統功能 – 顯示 GO 路徑、階層以及 TERM ... 38
圖 4. 18 系統功能 – 顯示 GO 所對映的 PROTEIN 資訊 ... 38
圖 4. 19 系統功能 – 顯示 GO 樹狀結構之路徑 ... 39
圖 4. 20 系統功能 – 以 FASTA 格式展示 PROTEIN 資訊 ... 39
圖 4. 21 系統功能 – 顯示 GO 樹狀結構圖 ... 39
圖 4. 22 模組五:系統運作流程圖 ... 40
圖 5. 1 系統 CMPARE - 顯示 BLAST 演算後的結果 ... 41
圖 5. 2 系統 CMPARE - 進一步的資訊擷取 ... 42
圖 5. 3 系統 CMPARE - 顯示擷取資訊 ... 42
圖 5. 4 系統 ZOODDD - 選擇物種組織及參數設定 ... 43
圖 5. 5 系統 ZOODDD – 顯示比對結果 ... 43
圖 5. 6 系統 ZOODDD – 顯示 GO 資訊 ... 44
圖 5. 7 ZOODDD 序列比對流程 ... 44
圖 5. 8 本系統序列比對流程 ... 45
圖 5. 9 系統 ZOODDD 之 TPM 公式 ... 45
圖 5. 10 系統 ZOODDD 之 SPECIFICITY 公式 ... 45
ix
表次
表 2. 1 REFSEQ 標示格式 ... 9
表 3. 1 系統所包含的物種 ... 17
表 4. 1 系統建置環境 ... 23
表 4. 2 UML 建模圖 ... 24
表 4. 3 系統 USE CASE DESCRIPTION ... 26
表 A. 1 物種 BOS TAURUS 於 UNIGENE 之資訊 ... 51
表 A. 2 物種 CANIS FAMILIARIS 於 UNIGENE 之資訊 ... 52
表 A. 3 物種 DROSOPHILA MELANOGASTER 於 UNIGENE 之資訊 ... 52
表 A. 4 物種 DANIO RERIO 於 UNIGENE 之資訊 ... 53
表 A. 5 物種 FUNDULUS HETEROCLITUS 於 UNIGENE 之資訊 ... 53
表 A. 6 物種 GASTEROSTEUS ACULEATUS 於 UNIGENE 之資訊 ... 54
表 A. 7 物種 GALLUS GALLUS 於 UNIGENE 之資訊 ... 54
表 A. 8 物種 HOMO SAPIENS 於 UNIGENE 之資訊 ... 55
表 A. 9 物種 MACACA FASCICULARIS 於 UNIGENE 之資訊 ... 56
表 A. 10 物種 MUS MUSCULUS 於 UNIGENE 之資訊 ... 57
表 A. 11 物種 MACACA MULATTA 於 UNIGENE 之資訊 ... 58
表 A. 12 物種 OVIS ARIES 於 UNIGENE 之資訊 ... 58
表 A. 13 物種 ORYCTOLAGUS CUNICULUS 於 UNIGENE 之資訊 ... 59
表 A. 14 物種 ORYZIAS LATIPES 於 UNIGENE 之資訊 ... 59
表 A. 15 物種 ONCORHYNCHUS MYKISS 於 UNIGENE 之資訊 ... 59
表 A. 16 物種 PIMEPHALES PROMELAS 於 UNIGENE 之資訊 ... 60
表 A. 17 物種 RATTUS NORVEGICUS 於 UNIGENE 之資訊 ... 60
表 A. 18 物種 SALMO SALAR 於 UNIGENE 之資訊 ... 61
表 A. 19 物種 SUS SCROFA 於 UNIGENE 之資訊 ... 61
表 A. 20 物種 XENOPUS TROPICALIS 於 UNIGENE 之資訊 ... 62
表 A. 21 物種 TAKIFUGU RUBRIPES 於 UNIGENE 之資訊 ... 63
表 A. 22 物種 TRICHOSURUS VULPECULA 於 UNIGENE 之資訊 ... 64
表 A. 23 物種 XENOPUS LAEVIS 於 UNIGENE 之資訊 ... 64
1
Chapter 1 緒論
目前生物學家進行大量 DNA 序列比對往往必須先至 National Center for Biotechnology Information (NCBI)網站下載最新版本的 BLAST 工具,再至其他網站 下載不同物種序列資料以及序列比對完成後需對映之資訊。上述步驟極為繁瑣耗 時。
本研究目的為將目前公開物種序列資料下載且正規化後整合至本系統建構的 資料倉儲,並將生物學家經常利用的序列比對工具 BLAST 也結合至本系統,提供 相關研究人員進行跨物種序列比對以及從 DNA 層次推演至蛋白質層次的公開使用 平台。
分析比對後的結果以 The Extensible Markup Language (XML)的格式輸出,期 利 用 XML 可 擴 充 性 及 結 構 化 的 優 點 使 分 析 比 對 的 結 果 檔 案 具 可 利 用 性 (reusable)。希冀此系統對相關研究人員有顯著正面的幫助。
1.1 研究動機
1950 年代以後科學家研究得知染色體是由去氧核糖核酸 (DNA)和蛋白質所 組成的雙螺旋結構,而基因就是DNA分子的一小段。到了1975年發明了分析及定 序DNA核甘酸序列的方法。1980 年代Walter Gilbert提議以眾多科學家的力量將人 類23對染色體總共約30億對的核甘酸序列予以解讀。因為科學家們認為能解讀製 造人類特徵的基因就能了解疾病與人類發育的過程。
1990 年美國政府正式地支持人類基因體計畫 (Human Genome Project),預計 耗資 30 億美元,透過國際實驗室間的合作,用 15 年時間完成解讀三十億鹼基的 工作。此計畫在美國設立四處的定序中心,另外在英國劍橋桑格中心 (Sanger Institute)、法國、中國大陸、日本、德國以及台灣等定序中心也都協力合作。
1990 年代以後,隨著電腦科技以及網際網路快速的發展。科學家運用超級電
2
腦於 2000 年 4 月提前完成人類基因的定序草圖,並將基因資訊公開在美國國家生 物技術資訊中心 (NCBI)的基因資料庫裡,提供所有科學家查詢的服務。
在短短的十年間,人類由數十個基因的解碼,進步到目前累積了百餘個物種 的基因序列資料庫,這是生物科學上重大的成就。專家預測,21 世紀將是基因體 學大放光彩的世紀。生物資訊學對基因體學與後基因體學研究皆為重要工具,無 論由 DNA 序列至 RNA 表現或者 RNA 至蛋白質功能的研究功能註解,資訊學家嘗 試由無秩序的 DNA 或胺基酸排列中,找出規律和生物意義,並經由生物學家的實 驗驗證,解決生物學上的問題,此即為生物資訊學的最大目標。此外,網際網路 的發展,更能促進研究成果的交流,對相關領域的進步有相當幫助。
生物資訊學發展的核心,在於各種資料庫的建立,如人類基因組資料庫、基 因表現資料庫、胺基酸排列的資料庫、蛋白質與蛋白質間交互作用資料庫、以及 人類遺傳疾病的資料庫和蛋白質變異的相關資料庫。另一方面,其他物種如老鼠、
酵母菌與各種致病菌等相關的資料庫,也提供基因體研究的重要參考依據。這些 資料庫統合起來加以運用,進行各種檢索和連結,對基因和蛋白質的關係,以及 各種蛋白質的機能等作充分而有效的解析和探討。
而跨物種序列比對可針對不同物種的表現基因進行同源性比較篩選,所得結 果將有助生物學家瞭解各物種的共同表現基因所具備的功能。因此,本研究蒐集 二十三種脊椎動物的 UniGene 序列,並將各物種的器官組織及發展時期予以分 類,期望提供一個服務平台,能夠給予生物學家任意選取物種與組織進行序列比 對分析,並提供 DNA 層次至蛋白質層次此一分析路徑下的相關資訊。
1.2 研究目的
目前在生物界每個物種在基因與蛋白質研究的速度都不盡相同,生物學家對 不同物種的專注程度也不一。有些物種如小鼠 (Mus musculus)、大鼠 (Rattus norvegicus)以及人類 (Homo sapiens)等模式動物 (model organism),生物學家已有 較長的研究歷史以及擁有較豐富的基因與蛋白質資訊,但如兔子 (Oryctolagus cuniculus)這個物種的基因與蛋白質的資訊較為稀少。因此,若能夠透過擁有豐富
3
的基因與蛋白質資訊之物種了解目前基因與蛋白質資訊較缺乏的物種之功能,則 可大幅降低研究人員的成本與連結物種間與組織間的序列關聯性。
瀏覽大部分的文獻,其系統較缺乏跨物種組織的整合平台。因此,研究人員 需要至網站下載欲研究的物種資訊,並自行對各物種組織之資訊予以分類方能開 始進行研究,且若該研究物種可以得到 Gene Ontology (GO)或者 GO 的樹狀結構 與路徑,還需要下載許多表格或進入不同的查詢網站才能得到資訊。如此的使用 流程對研究人員相當的不便利。
基於上述描述,本研究有下列項研究目的:
(1) 蒐集二十三種脊椎動物的基因與蛋白質序列資訊,並予以彙整。
(2) 建構一個跨物種系統平台,讓研究人員可以選擇欲研究的物種與組織進行序列 比對。
(3) 從 DNA 序列比對結果透過對映之蛋白質資料推衍至 GO。
(
4)
將 GO 的樹狀結構關係圖,予以層次概念詮釋之。(5) 將分析結果以圖形或表格方式呈現。
1.3 研究流程
本研究流程結構與順序如下圖 1.1 所示。
4 圖 1. 1 研究流程
1.4 論文架構
本論文共包含六個章節,第一章為介紹研究動機與目的,第二章則探討研究 中所參考的資料來源與相關文獻,第三章為資料倉儲(Data warehouse)的建置,在 此章將詳細介紹研究過程中各資料集是如何正規化,第四章為系統設計與建置,
其中將分為五大模組進行討論,第五章為討論與其他相似系統有何差異之處,第 六章為結論與未來工作。
研究動機
研究目的
文獻蒐集與分析
資料來源蒐集
系統實做 相關演算工具之研究
研究報告與相關文件 系統測試與維護
5
Chapter 2 文獻探討
2.1 相關研究網站
2.1.1 NCBI
The National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/)成立於 1988 年,原為 The National library of Medicine (NLM)的其中一部門。由於 NLM 在維護生醫資料庫擁有豐富的經驗,故由它主持 NCBI 計畫。
NCBI 內有許多生物資訊搜尋模組與流程自動化模組,幫助研究人員了解健康 與疾病之相關的遺傳基因及分生機制,並加速研究的效率。NCBI 也負責維護如 GeneBank、dbEST 與 UniGene 等序列相關資料庫,並將之與各地作者所提供資訊 進行連結,不定期更新資料庫內容。本研究所使用之 UniGene、RefSeq 與 NR 資 料集即是由 NCBI (http://www.ncbi.nlm.nih.gov/)取得。
2.1.2 The Gene Ontology
The Gene Ontology (GO)專案 (http://www.geneontology.org/)建構於 1998 年,一 開始僅統合 FlyBase external link (Drosophila)、the Saccharomyces Genome Database external link (SGD)與 the Mouse Genome Database external link (MGD) 此三個模式 有機體資料庫,但之後又納入更多有機體資訊。
GO 為統合各物種基因功能性名詞的強大工具,其可針對不同物種之基因或蛋 白質序列進行功能性解析[1]。GO 系統在進行功能性解析分為三種輸入方式,分別 敘述如下。
(1) 使用者可輸入基因或蛋白質名稱找出在不同物種或組織該基因或蛋白質所司 之功能。
(2) 使用者可輸入特定的 GO Term 或 GO ID,針對不同功能的名詞定義及有哪些
6
基因具有該生物功能進行了解。
(3) 使用者也可輸入以 FASTA 形式呈現的特定序列至 GO 系統進行分析,則可知 其序列可能的功能。
輸入 GO ID 進行功能分析後,GO 系統還會顯示 GO 的階層架構圖如下圖 2.1;
以及樹狀結構圖如下圖 2.2。
本系統關於 GO Term 與 GO ID 關連性檔案也是由 The Gene Ontology 網站取 得。
圖 2. 1 GO 階層架構 圖片來源: the Gene Ontology 網頁
7
圖 2. 2 GO 樹狀結構
2.2 資料來源介紹
2.2.1 UniGene 資料集
UniGene 是由 NCBI、clusters ESTs 以及 mRNA sequences 所建立的,其使用 coding sequences (CDSs)解釋 genomic DNA 分成各種相關序列的子集[2]。UniGene 的蒐集是以自動化的方式,對每一條新增至 GenBank 之 cDNA 序列會進行序列品 質的分析,諸如引子 (primers)、大腸桿菌、載體 (vector)和連接子 (linker)等外源 性的汙染序列以及具高重複性的低品質序列會在進行自動化分析前被排除在外;
之後還會檢測其序列長度,每條序列長度最少須包含 100 鹼基對以上;最後進行 序 列 相 似 性 分 析 , 若 能 經 由 分 析 找 到 可 能 是 來 自 於 同 一 個 基 因 的 序 列 群 組 (cluster),則將此序列歸入這一個序列群組,若找不到則成立一個新的序列群組。
流程如下圖 2.3 所示。
ESTs 序列約佔 GeneBank 的百分之六十二,故以分類方式所產生的
圖片來源: the Gene Ontology 網頁
8
UniGene 資料庫,每一群序列 (包含了 EST、及 mRNA 序列的基因組),共同代表 一種獨特的基因之 mRNA 產物。利用這種經過分類整理的資訊,便較直接使用數 量大而資訊含量少的個別 EST 資料要來得有效率。
圖 2. 3 UniGene 資料集蒐集流程
2.2.2 NR 資料集
NR (non-redundant) Protein Database 整合自 SwissProt、SwissProt updates、
PIR 與 PDB,目前由 NCBI 所維護。其特點為資料庫內的蛋白質序列皆不重複,且 擁有 Protein ID、Protein GI 與 Protein sequence 資訊,提供使用者進行蛋白質序列 比對的參照資訊。
2.2.3 RefSeq 資料集
NCBI 的參考序列計畫 (RefSeq)為中心法則中自然存在的分子,從染色體 至 mRNA 到蛋白質提供參考序列標準。RefSeq 標準為人類基因組的功能註解提供 一個基礎。RefSeq 會針對各種不同的分子類型提供不同的標號格式,如下表 2.1 所示。
序列品質分析
來源序列長度限制
基因相似性分析
UuiGene 資料集
9
表 2. 1 RefSeq 標示格式
分子 標號格式 基因組
完整基因體 NC_##### 原核生物、細菌、細胞菌、病毒與疫苗 完整染色體 NC_##### 真核生物
完整序列 NC_##### 質粒 基因體 Contig NT_##### 人類
mRNA NM_##### 有限的脊椎動物、人類、小鼠與大鼠 Protein NP_##### 所有以上
2.2.4 GOA (Gene Ontology Annotation, GOA@EBI)
GOA (GO Annotation@EBI)是由 EBI (European Bioinformatics Institute)所提供 且維護的專案,其目的是為蛋白質提供高品質的 GO (Gene Ontology)註解。另外 GOA 專案也為每一條蛋白質序列建立 IPI (International Protein Index),利用 IPI 可 以將 GO ID、GO Term 與其他資料庫的蛋白質 ID (如 Ensembl 與 NCBI)建立關聯對 映。
2.2.5 Gene Ontology 資料集
Gene Ontology (GO)創立目的為提供一組可對所有生物之基因與蛋白質在細 胞角色中的表述語彙 (Vocabulary),而 GO Term 描述其生物反應過程。GO 將生物 的功能性分析分成三大類:(1)生物作用 (biological process, P)、(2)分子功能 (molecular function, F)及(3)細胞組成 (cellular component, C)。生物作用牽涉化學或 生理的轉變,由一個或多個分子功能所集合而成。如:細胞生長與維持。分子功 能是指基因產物的生物化學活性,包含配體和受體的特殊鍵結。如:酵素和配合 體。而細胞組成指細胞中基因產生活化的位置。如:核甘體和核膜。同時 GO 也 可將基因所司具有之功能以單向環狀的樹狀圖示方式呈現,提供學者瞭解不同功 能分層間的關係,而樹狀結構的鏈結可分為”is-a”與”part of”兩種連接 GO Term 與 GO Term 間的關聯。
10
至 2008 年 4 月 12 日,共有 25036 個 GO Term,其中百分之九十八被定義。
其 中 biological_process 包 含 14696 個 、 cellular_component 包 含 2077 最 後 molecular_function 包含 8263 個
2.3 序列比對工具
序列比對工具為基因體範疇研究的重要工具,其可針對多條核甘酸或蛋白質 序列間進行序列相似度分析。目前主要針對 DNA 序列進行分析工具有兩種,一種 為 FAST[3],另一種為 BLAST[4]。由於本系統利用 BLAST 進行序列分析,故後 一小節將對此工具進行詳細敘述。
FAST 理念為給予兩序列 S、T 及一個參數值 k,一開始找出兩個序列長度大 於 k 值的相似片段,將這些片段稱為熱點 (hotspot),再將於同一對角線的熱點集 合起來,並將個熱點分數加總得到集合總分,找到分數最高的集合,將其中的熱 點連結,針對此對角線進行帶狀動態規劃,使其能包含其他相近之熱點,以取得 較佳解。FAST 運用於序列比對的靈敏度極高,但時間複雜度仍舊無法符合需求,
故大多數的研究人員使用 BLAST 進行序列分析的頻率較高。
2.3.1 BLAST 簡介
BLAST (Basic local alignment search tool) 為 區 域 相 似 比 對 採 用 啟 發 式 (Heuristic)演算實做之,其目的為花費越少的時間找到較為相似的序列片段,所以 結果並非最佳解,但卻能在有效的時間內完成,且區域序列片段的相似性有重要 的特性,又其可運做於 Unix 系統且執行效率高,使得大量的序列比對得以於短時 間內完成,故多數的生物資訊網站及研究人員皆採用此工具。
初期的 BLAST 不允許空缺 (gap)的片斷序列配對 (ungapped alignment),但 Altschul 等人於 1997 年提出修正版本,此時便克服上述的限制。BLAST 主要的基 本步驟為以下圖 2.4 所示。
BLAST 是採行 pair wise sequence alignment (成對序列比對),而一條序列為一
11
串英文字母所組成,又一個 alignment 為兩條序列成對排列,進行比對計分依據鹼 基 (base)或胺基酸 (Amino Acid)是否吻合 (match),若吻合則為正分,不吻合 (mismatch)與空缺 (gap)則為負分。
BLAST 目 前 是 由 NCBI 管 理 , 此 分 析 方 法 可 透 過 網 頁 介 面 (http://www.ncbi.nlm.nih.gov/BLAST/)或安裝於本端主機進行運作,NCBI 提供各種 不同型態及用途的 BLAST 種類如下:
(1) megablast : 用於基因體序列之比對,針對核苷酸序列利用連鎖查詢多條序 列以加速搜尋速度。
(2) blastn : 利用輸入核苷酸序列針對核苷酸序列之資料庫進行相似性比對。
(3) blastp : 利用輸入胺基酸序列針對胺基酸序列之資料庫進行相似性比對。
(4) blastx : 用於鑑別核苷酸序列之身分,其將核苷酸序列轉譯成六種形式的 胺基酸序列,並進一步在蛋白質資料庫內進行比對工作,以期找出此核苷 酸序列之潛在蛋白質產物。
(5) tblastn : 用於胺基酸序列之比對,但比對之胺基酸序列資料由核苷酸序列 資料庫中序列所轉譯成胺基酸序列為基礎。
(6) tblastx : 用於核苷酸序列之比對,將此核苷酸序列轉譯成六種形式的胺基 酸序列,並與由核苷酸序列資料庫中序列所轉譯出的胺基酸作序列相似性 比較。
本系統所使用的 BLAST 種類為 blastn,其版本為 2.2.17
12
圖 2. 4 BLAST 演算流程
2.3.2 FASTA 格式
當執行 BLAST 分析需使用的輸入格式為 FASTA 格式。可利用 NCBI Entrez 查詢網頁,輸入 Nucleotide Accession Number 或 Protein Accession Number 將可得 其 FASTA 格式。FASTA 格式之樣式,如下圖 2.5 所示。
2.3.3 HTML4BLAST 工具
HTML4BLAST 是一套能將 BLAST 結果格式轉換為網頁形式的工具,並將其 找出可能相似的區域並計算分數
將分數標準化
依照分數排序
轉算為期望值(E-VALUE)
>gnl|UG|Bt#S35034864 Bos taurus calpain 1, (mu/I) large subunit, mRNA (cDNA clone MGC:143348 IMAGE:8139008), complete cds /cds=p(68,2218) /clone=null /clone_end=null /gb=BC123635 /gi=115305057 /ug=Bt.252 /len=2965 GTCCTCAGTTGCCACCCGGGAAGCCAGAGCAGGGACCGCAGCGACCCCCCAACACTCCTCCCCCAGGATGGC CGAGGAGTTCATCACTCCGGTGTACTGCACCGGGGTGTCTGCACAAGTGCAGAAGCAGCGGGCCAAGGAGCT GGGCCTGGGCCGCCATGAAAATGCCATCAAGTACCTGGGCCAGGATTACGAGCAGCTGCGGGTTCACTGCCTG CAAAGAGGGGCCCTTTTCCGTGACGAGGCTTTCCCCCCAGTGCCCCAGAGCCTGGGCTTCAAGGAGCTGGGC CCCAACTCCTCCAAAACCTATGGCATCAAGTGGAAGCGTCCCACGGAGCTGTTCTCAAACCCCCAGTTCATCG TGGATGGAGCCACCCGCACGGACATCTGCCAGGGCGCACTGGGGGACTGTTGGCTCCTGGCTGCCATCGCCTC CCTTACCCTCAATGACACCCTCCTGCACCGAGTAGTTCCACATGGCCAAAGCTTCCAGGATGGCTACGCTGGCA TCTTCCATTTCCAGCTGTGGCAGTTTGGTGAGTGGGTGGATGTGGTGGTGGATGACCTGCTGCCCACCAAGGA CGGGAAGCTGGTGTTTGTGCACTCTGCCCAAGGCAACGAGTTCTGGAGCGCCCTGCTGGAGAAGGCCTATGCC AAGGTGAACGGCAGCTACGAGGCCCTCTCAGGAGGCAGCACGTCTGAGGGCTTTGAGGACTTCACCGGTGGA GTCACCGAGTGGTACGAGCTGCGCAAGGCGCCCAGCGACCTCTACAACATCATCCTCAAGGCCCTGGAGCGTG GCTCCCTGCTGGGCTGCTCCATCGATATCTCCAGCATTCTGGACATGGAGGCTGTCACCTTCAAGAAGCTGGTG AAGGGCCACGCCTACTCTGTGACCGGGGCCAAACAGGTGAACTACCAGGGCCAGATGGTGAACCTGATCCGG ATGCGGAACCCCTGGGGCGAGGTGGAGTGGACAGGAGCCTGGAGTGACGGCTCCTCGGAGTGGAACGGCGT
圖 2. 5 FASTA 格式
13
alignment 結果整合提供圖示顯示,如下圖 2.6 所示。本系統採用 HTML4BLAST 工具之版本為 1.6a。
圖 2. 6 BLAST 結果圖型顯示
2.3.4 Graphviz 結構關係圖產生器
Graphviz 為一套開放原始碼 (open source)的軟體工具,它能夠將抽象的圖形 網絡關係展示成為圖表,其產生圖表檔案類型可為 JPG、PNG 或 SVG 等。使用者 需先將其網絡關係封裝為文字型態的描述語言,其描述語言有固定格式,格式名 稱為 DOT,其格式如下圖 2.7;再由程式執行輸出為圖表形式。本研究需要利用其 工具展示 Go Term 的關連性。
圖 2. 7 Dot 格式
2.3.5 BlastSummary
digraph test {
graph [ratio=fill];
node [label="\N", color=black, fillcolor=white, fontcolor=blue, fontsize=10, shape=box, style=filled];
edge [fontsize=8];
graph [bb="0,0,382,610"];
accall [label="all\nall", fontname=Courier];
node1 [label="molecular_function\nGO:0003674", fontname=Courier];
node2 [label="binding\nGO:0005488", fontname=Courier];
node3 [label="ion binding\nGO:0043167", fontname=Courier];
node4 [label="metal ion binding\nGO:0046872", fontname=Courier];
node5 [label="calcium ion binding\nGO:0005509", fontname=Courier];
node6 [label="cation binding\nGO:0043169", fontname=Courier];
node1->accall[color=red, label=is_a];
node2->node1[color=red, label=is_a];
node3->node2[color=red, label=is_a];
node4->node3[color=red, label=is_a];
node5->node4[color=red, label=is_a];
node6->node3[color=red, label=is_a];
node5->node6[color=red, label=is_a];
}
14
此程式為國立台灣大學動物科學技術學系研發,其目的是將 BLAST 後的結果 予以表格化的統整呈現,使相關研究人員可更清晰明瞭比對結果,本研究會利用 此程式進行比對結果的彙整以及將彙整的資訊再對映至本研究所建構的資料倉儲 系統的其他資訊。
2.4 相關系統研究
近幾年生物資訊整合系統已有顯著的成長,即使各個系統的使用方式以及目 的皆有所差異,但進行本研究時仍涉獵多篇相關研究論文,最後選擇兩篇其想法 最為接近本系統之論文予以討論。以下兩節將進行系統簡介。
2.4.1 COMPARE
COMPARE 是一個利用 Web Service 技術整合分散且異質的資料,如 ZFIN [5]、FlyBase[6]與 ENSEMBL[7]等予以分析,而這些資料庫包含各種不同物種及組 織,並提供染色體結構 (genomic structure)、表示資料 (expression data)、註釋 (annotations)、反應路徑 (pathway)以及文獻鏈結 (literature link)等資訊。
使用者可將系統的回報資訊,透過選項設定而得到更精鍊 (refine)的結果,其 展示結果的部分與本研究有些許相似,將於第五章討論。
2.4.2 ZooDDD
ZooDDD 是由台灣中央研究院研究團隊於 2006 年發表的系統[8],其系統是從 UniGene 結合 EST 建構 ZooDDD 資料庫,藉由資料庫的資料進行跨物種跨組織比 對,得到可能同源相關資訊。
其資料庫包含有 human (Homo spiens)、mouse (Mus musculus)、rat (Rattus norvegicus)、dog (Canis familiaris)、chicken (Gallus gallus)、forg (Xenopus tropicalis)、
zebrafish (Danio rerio)與 tunicate (Ciona intestinalis)八個物種,以及各物種於 EST 所 擁有的組織。將於第五章進一步進行討論。
15
2.4.3 BioMOBY
BioMOBY[9]是一個牽涉生物資料主機 (biological data hosts)、生物資料服務 提供者 (biological data service providers)與程式編碼的國際性研究專案,其目的是 查詢與分配各種生物網路服務 ((biological web service)。此專案實質上是改造舊標 準的分佈式計算(例如 RPC、CORBA 與 DCOM),並提供一個標準介面讓使用者 方便呼叫服務項目。
2.4.4 Taverna
Taverna [10]是由 EBI((European Bioinformatics Institute)、IT Innovation、紐加 塞爾大學電腦科學系 (School of Computer Science, University of Newcastle)、曼徹斯 特大學電腦科學系 (School of Computer Science at the University of Manchester) 與 諾丁安大學混合真實實驗室 (Nottingham University Mixed Reality Lab)共同開發。
它是一個使用於規劃與執行工作流程的免費軟體工具,允許使用者整合許多的軟 體工具,例如整合由 NCBI 、EBI 、DDBJ (DNA Databank of Japan)、SoapLab、
BioMOBY 與 EMBOSS。
Taverna 提供一個桌面的編輯環境讓使用者可以建構欲執行工作流程,並會將 所編輯的工作流程封裝為 Scufl (Simple Conceptual Unified Flow language)格式,封 裝為此格式的好處是可以方便其他相容於 Scufl 格式的軟體使用。
16
Chapter 3 資料倉儲建置
3.1 Framework
本研究所需要用到的資料集有 UniGene、NR、RefSeq、GOA 與 Gene Ontology,
其詳細的說明在第二章提及。雖然某些網站如 DDBJ 有提供 Web Service 的查詢服 務元件,但考量網路狀態的不穩定,故本研究傾向將各資料集從來源網站擷取後 整合並建置於本地端的資料倉儲 (Data Warehouse)系統。
資 料 倉 儲 正 式 定 義 由 Inmon[11] 所 提 供 , 它 為 一 種 主 題 導 向 (subject-oriented) 、 整 合 的 (integrated) 、 隨 時 間 改 變 (time-variant) 與 非 揮 發 性 (nonvolatile)的資料集合。故本研究將擷取的資料集經過前置的篩選,再進行正規 化處理,最後輸入至本研究之資料倉儲,其概約的架構圖如下圖 3.1。
圖 3. 1 資料倉儲架構
3.2 資料倉儲內之物種與組織
UniGene GOA NR Gene
Ontology
正規化篩選後的資料 資料集過濾篩選
Data Warehouse
RefSeq
17
本研究的資料倉儲內含二十三種脊椎動物,而每項物種(species)又有個別擁有 的組織 (tissue),將於下兩小結介紹。
3.2.1 物種
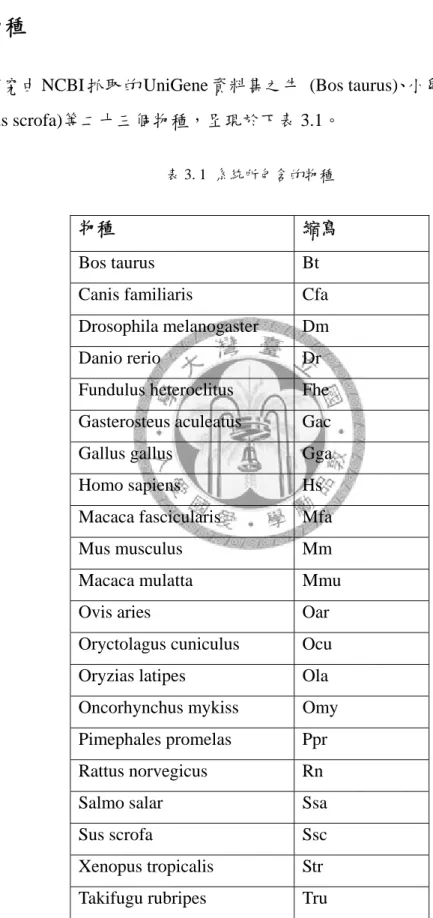
本研究由 NCBI 抓取的 UniGene 資料集之牛 (Bos taurus)、小鼠 (Mus musculus) 與猪 (Sus scrofa)等二十三個物種,呈現於下表 3.1。
表 3. 1 系統所包含的物種
物種 縮寫
Bos taurus Bt Canis familiaris Cfa Drosophila melanogaster Dm Danio rerio Dr Fundulus heteroclitus Fhe Gasterosteus aculeatus Gac Gallus gallus Gga Homo sapiens Hs Macaca fascicularis Mfa Mus musculus Mm Macaca mulatta Mmu Ovis aries Oar Oryctolagus cuniculus Ocu Oryzias latipes Ola Oncorhynchus mykiss Omy Pimephales promelas Ppr Rattus norvegicus Rn Salmo salar Ssa Sus scrofa Ssc Xenopus tropicalis Str Takifugu rubripes Tru
18
Trichosurus vulpecula Tvu Xenopus laevis Xl
3.2.2 組織與發展時期
本章節將描述系統中擷取各物種的 UniGene 版本以及所擁有的組織與發展時 期,如 Bos taurus 的 UniGene 版本為#90 而其擁有 boold、adult 與 brain 等組織與 發展時期。由於資料量過於龐大,故將其撰寫於附錄 A。
3.3 正規化資料來源之表格設計
傳統關聯式資料庫為了解決資料重複過高造成磁碟空間浪費問題,於是便對 資料庫中的關聯表進行正規化。1972 年 E.Codd 提出三項正規化階段 , 此 三 階 段 分別稱為第一正規化 (First Normal Form)、第二正規化 (Second Normal Form)及第 三正規化 (Third Normal Form),經此三階段處理的關聯表,不僅符合關聯綱要設 計時的兩項原則,也大幅減少更新後所產生的異常現象。
本研究將資料集予以正規化,使資料倉儲系統於效能與效率上有良好的表 現,以下將使用 Entity Relationship Diagram (ERD)表示其不同資料集所建構的表格 欄位以及關連性。
3.3.1 正規化 UniGene 之欄位設計
由於將 UniGene 資料集下載後為 FASTA 格式,故必須先進行程式剖析,產生 正規化後的檔案再輸入至資料倉儲系統。而在此建構的表格主要是建立物種、組 織與 DNA 序列的關連對映,其 ERD 呈現於下圖 3.2。其中紅色自體代表欄位為主 鍵,而藍色自體表欄位為索引鍵。
19 species
name varchar(50) sp_id varchar(10)
species_tissue sp_id varchar(10) express varchar(80)
data_express
id varchar(10)
express varchar(80) seq_uniq_gz
Seqhead varchar(3) sourcedb varchar(2) seqcies varchar(13) description text cds_start int cds_end int
clone varchar(42) clone_end varchar(5)
gb varchar(13)
gi varchar(10)
ug varchar(10)
len int
seq mediumtext
* 1
1
1
圖 3. 2 UniGene 資料集之 ERD
3.3.2 正規化 NR 之欄位設計
NR 的資料格式也為 FASTA,故必先經剖析,擷取本研究所預取得的資料。而 此資料集是欲建構蛋白質 GI 編碼與蛋白質序列的關連性,其 ERD 呈現於下圖 3.3。
圖 3. 3 NR 資料集之 ERD
3.3.3 正規化 RefSeq 之欄位設計
RefSeq 的資料集提供功能性 DNA 的 Accession 與 GI 編號對映至蛋白質的 Accession 與 GI 編號,此後方能再由蛋白質的 Accession 編號對映至 GOA 的資料 集欄位,其 ERD 呈現於下圖 3.4。
20
圖 3. 4 RefSeq 資料集之 ERD
3.3.4 正規化 GOA 之欄位設計
GOA 的資料集是提供蛋白質 ID 或 GI 對映至 GO ID 或 GO Term,而其中必須 先中介對映至 IPI 編碼,其 ERD 呈現於下圖 3.5。
圖 3. 5 GOA 資料集之 ERD
3.3.5 正規化 Gene Ontology 之欄位設計
Gene Ontology 建構的目的為建立其 GO Term 間的樹狀階層關係,與得知某階 層所包含的 GO Term,而欄位 kind 紀錄 GO Term 為”is a”或”part of ”,其 ERD 呈
21
現於下圖 3.6。
圖 3. 6 GeneOntology 資料集之 ERD
22
3.3.6 整合各資料集之 ERD
整合上述各資料集的表格設計以 ERD 表示,呈現於下圖 3.7。
species name varchar(50) sp_id varchar(10)
species_tissue sp_id varchar(10) express varchar(80)
data_express
id varchar(10)
express varchar(80) seq_uniq_gz
seqhead varchar(3) sourcedb varchar(2) seqcies varchar(13) description text cds_start int cds_end int clone varchar(42) clone_end varchar(5)
gb varchar(13)
gi varchar(10)
ug varchar(10)
len int
seq mediumtext
*
1 *
* 1
Xrefs_dgi_pgi rna_acc varchar(12) rna_gi varchar(9) prot_acc varchar(12) prot_gi varchar(9) gene_id varchar(9) unigene_id varchar(11) bases varchar(14) strain varchar(9) source_db varchar(12) define text 1
1 Xrefs_accession
IPI varchar(11) cod varchar(3) State varchar(14) Protein_Acc varchar(12)
GI varchar(9)
Xrefs_goid_ipi GO_ID varchar(7) IPI varchar(11) Aspect varchar(1)
Xrefs_Main IPI varchar(11) cod varchar(3) MDB_ID varchar(18) Main_DB varchar(18) UNIPARC varchar(13) 1
1
*
1
Xrefs_GO_Term GO_ID varchar(7) GO_TERM varchar(200) Ontology varchar(1)
1
*
1
*
Xrefs_accession IPI varchar(11) cod varchar(3) State varchar(14) Protein_Acc varchar(12)
GI varchar(9)
Xrefs_ensembl IPI varchar(11) ENSEMBL varchar(25)
Xrefs_unigene IPI varchar(11) UniGene varchar(9)
*
1
*
Xrefs_gi_seq * gi varchar(9) seq mediumtext 1
1 go_inheritance_relation
ID varchar(10)
is_a varchar(10) kind char(1) 1
*
*
圖 3. 7 整合各資料集之 ERD
23
Chapter 4 系統設計與建構
本系統於頁面呈現上使用 Java Server Page (JSP)技術,搭配 Java Bean 運作於 資料庫的擷取運算。系統的建置環境如下表 4.1 所示。
表 4. 1 系統建置環境
(1)硬體部分
中央處理器 2.8Ghz (Intel Pentium 4)
主記憶體容量 4 GB
硬碟容量 2 TB
(2)軟體部分
作業系統 Ubuntu (http://www.ubuntu.com/) Java 開發工具版本 Sun j2sdk1.5.0_08 (http://www.sun.com) 資料庫管理系統 MYSQL5.0.24a(http://www.mysql.com/)
4.1 系統運作流程
接下來介紹系統流程,將採用統一塑模語言 (Unified Modeling Language, UML)描述。UML 是軟體和系統開發的標準塑模語言,它可用規格化 (Specifying)、
視覺化 (Visualing)、文件化 (Documenting)及建構化 (Constructing)的方式塑模軟 體系統。[12] 認為 UML 是讓塑模者皆能夠使用的一種通用模組語言,及讓系統開 發者使用一個標準化的標記符號塑造不同模式的系統。
UML1.0 於 1997 年發佈,經過多個本版的演進,至 2003 年 6 月發佈 UML2.0。
UML2.0 為了符合模型驅動架構 (Model Driven Architecture)的需求做了大幅度的 修改,除了在圖形基礎擴充及變化部分的展示,也增加圖形標準元件,到 2.0 版本 時共有十三種圖形,分別於下表 4.2 呈現。
24
表 4. 2 UML 建模圖
中文名稱 英文名稱
使用個案圖 Use Case Diagram
類別圖 Class Diagram
套件圖 Package Diagram
物件圖 Object Diagram
循序圖 Sequence Diagram
通訊圖 Communicate Diagram
活動圖 Activity Diagram
狀態圖 State Diagram
部署圖 Deployment Diagram
元件圖 Component Diagram
複合結構圖 Composite Structure Diagram
時序圖 Timing Diagram
互動概觀圖 Interaction Diagram
本章將利用使用個案圖 (Use Case Diagram)以及活動圖 (Activity Diagram);以 使用者之觀點描述系統的行為者與系統間的互動行為與關係,另表達執行某一作 業行為中的活動流程。
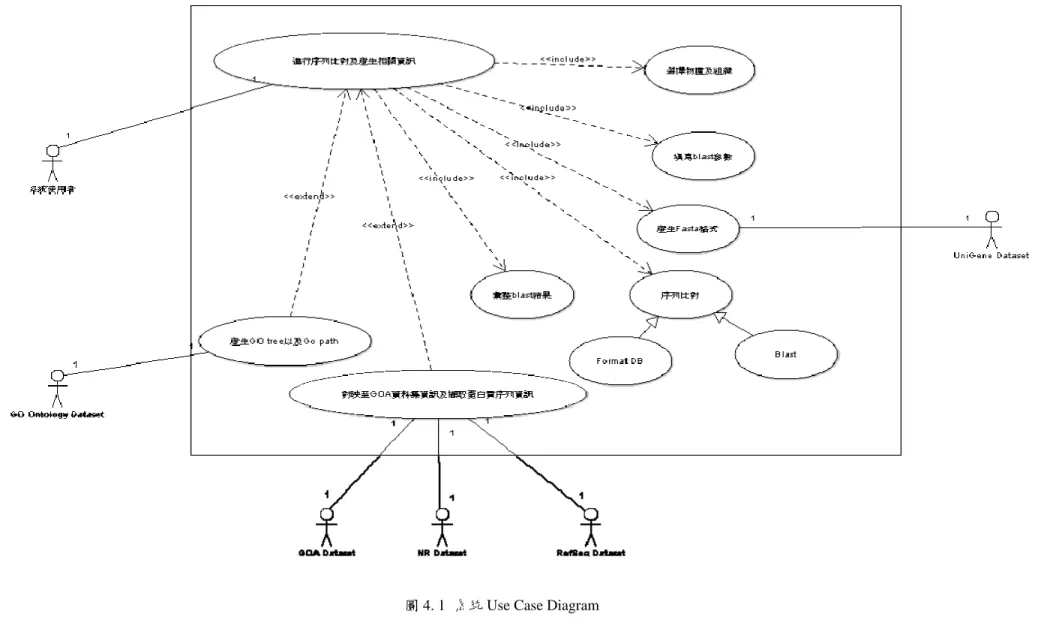
下圖 4.1 為本系統的使用個案圖,下表 4.3 將有對使用個案描述 (Use Case Description)。
25
圖 4. 1 系統 Use Case Diagram
26
表 4. 3 系統 Use Case Description
使用案例名稱: DNA 序列比對分析及整合資訊提供
情境目標 使用者選擇物種以及組織,欲得到序列比對後的資訊以
及整合蛋白質與 GO Ontology 的資訊。
前置條件 使用者須勾選 Database 的物種組織及 Input 的物種組織並 設定 BLAST 參數。
成功的結束狀態 將整合後的資訊提供予使用者下載。
失敗的結束狀態 導回至首頁重新進行設定。
主要行為者 生物相關系統使用者。
次要使用者 UniGene Dataset、GOA Dataset、NR Dataset、RefSeq Dataset 以及 GO Ontology Database。
觸發器 使用者選好完成後送出請求
主要流程 步驟 動作
1 使用者選擇物種及組織 2 BLAST 參數設定 3 產生 FASTA 格式
4 序列比對
5 產生序列比對結果 6 彙整序列比對結果
7 對映 GOA 資料集擷取 Gene Ontology(GO)資訊 8 對映 NR 資料集擷取蛋白質序列
9 產生 GO ID 與蛋白質序列想關結果檔案 10 展示 GO Tree 以及 GO ID 的路徑及皆階層分析
延伸步驟 步驟 分支動作
1.1 使用者未進行選取。
27
1.2 導向系統首頁。
2.1 使用者未進行參數設定。
2.2 使用系統預設參數。
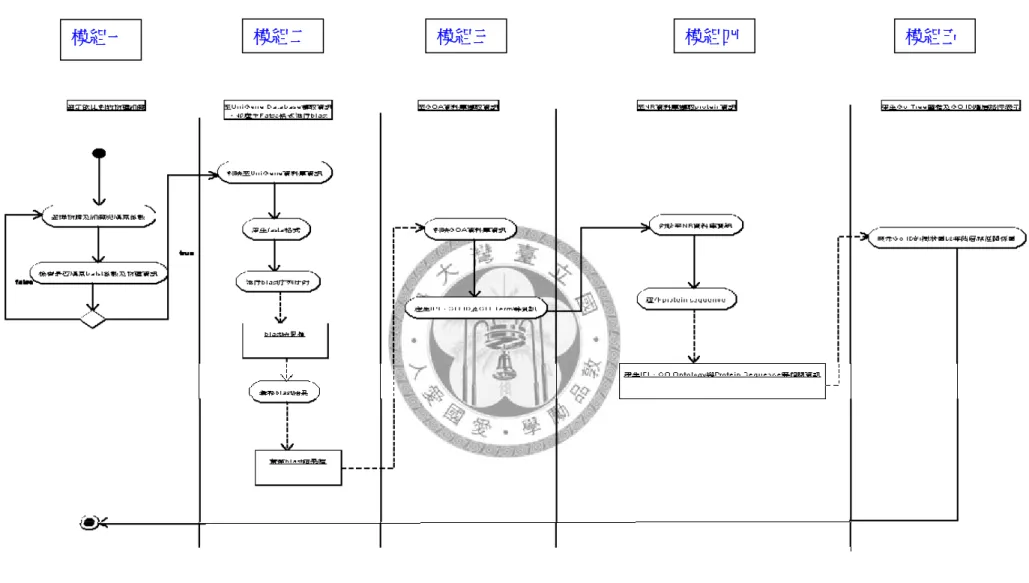
下圖 4.2 為系統的活動圖。本系統共分為五個模組,為了詳細解釋之,下一小 節將分別為這五個模組進行介紹。
28
圖 4. 2 系統 Activity Diagra
模組一 模組二 模組三 模組四 模組五
29
4.2 系統模組介紹
本節將更細部分為分為五小節,分別介紹本系統的五個模組,其五個模組分 別為: (模組一)物種組織選擇 (模組二)FASTA 格式之產生與進行 BLAST (模組 三)Gene Ontology 相關資訊之擷取 (模組四)蛋白質序列資訊之擷取 (模組五)GO 樹狀圖形及階層路徑分析與呈現。
4.2.1 模組一 : 物種及組織選擇
首先,使用者需選擇欲比對的物種,而在本系統由於考量效能因素,故使用 者目前僅能選擇某一物種中的某一個組織當成 Input 或 Query,於下圖 4.3 所示。
圖 4. 3 系統功能 - 選擇 Input 的物種及組織
至多選擇三項其中一物種的其中一組織當成 Database,於下圖 4.4 所示。
圖 4. 4 系統功能 - 選擇 Database 的物種及組織
選擇完畢後設定 BLAST 演算之參數設定,BLAST 演算之參數設定分別為(1) e-value (期望值) (2) show sequence number (至多比對結果的展示數目) (3)
30
alignment result number (至多比對序列的數目),於下圖 4.5 所示。
圖 4. 5 系統功能 - 輸入 BLAST 參數
若使用者無選擇 Input 的物種及組織與至少一個 Database 的物種與組織則無法 進行接下來的步驟,以下圖 4.6 為本模組的結合後端資料庫的概念圖。
圖 4. 6 模組一:系統運作流程圖
4.2.2 模組二 : FASTA 格式之產生與進行 BLAST 演算
當使用者選擇物種組織及填寫 BLAST 演算之參數完畢後,系統即會產生相對 應 FASTA 格式的 DB 以及 Input,並接著進行格式化資料庫 (fornatdb)以及執行
選擇欲比對 Input 及 Database 的物種及組織
檢驗是否填寫系統要求 的資料及參數
模組二 儲存系統目前
所擁有的物種
31
BLAST 演算。
BLAST 演算執行完畢後將執行 SummaryBlast 程式,於上述文獻探討曾提及,
此程式將彙整 BLAST 結果,提供使用者更簡潔的報表如下圖 4.7 所示。
圖 4. 7 系統功能 – SummaryBlast 程式輸出結果
SummaryBlast 程式執行完成後將執行 HTML4BLAST 程式,此程式的目的為 提供圖形化介面的 BLAST 比對結果。下圖 4.8 為 BLAST 比對完成的頁面,下圖 4.9 為點擊超鏈結後所呈現的頁面。
圖 4. 8 系統功能 – BLAST 演算完成後的輸出
32
圖 4. 9 系統功能 – BLAST 演算完成後的圖形化展示
本模組使用到 UniGene 資料集,產生 FASTA 格式、BLAST 比對結果、BLAST 彙整以及 BLAST 圖形表示輸出檔案,下圖 4.10 呈現此模組的概約的流程圖。
33
圖 4. 10 模組二:系統運作流程圖
FASTA 格式
產生器UniGene 資料集
DB 的 Fasta 格式檔案 Input 的 Fasta
格式檔案
格式化 DB
BLAST 演算
Blast 比對結果
SummaryBlast
Blast 彙整檔案
HTML4BLAST
Blast 圖形畫頁面
模組三
34
4.2.3 模組三 : Gene Ontology 相關資訊之擷取
完成模組二結果,得到 BLAST 比對後的結果檔,即可利用 RefSeq 的資料集,
將 DNA 序列的資訊對映至蛋白質序列的資訊。再利用蛋白質序列的 GI (GeneBank ID)透過 GOA 的資料集得到 IPI,接著對映至 Gene Ontology 的資訊。所謂的 Gene Ontology 資訊為 GO ID、Term 及 Ontology。產生比對後的結果檔,於下圖 4.11 及 圖 4.12 所示。
圖 4. 11 系統功能 – 展示對映至 Gene Ontology 資料集資訊(1)
圖 4. 12 系統功能 – 展示對映至 Gene Ontology 資料集資訊(2)
本模組會使用 RefSeq 以及 GOA 資料集,產生 Gene Ontology 的輸出檔案,下 圖 4.13 呈現此模組的概約的流程圖。
35
圖 4. 13 模組三:系統運作流程圖
4.2.4 模組四 :蛋白質序列資訊之擷取與封裝為 XML 文件形式
與模組三同樣使用 BLAST 比對後的結果檔,利用 RefSeq 的資料集,將 DNA 序列的資訊對映至蛋白質序列的資訊。接著使用 NR 的資料集,利用蛋白質序列的 GI 即可得知其原始的蛋白質序列。
除了得到蛋白質序列資訊外,系統在模組四將模組三得到的資訊加上蛋白質 序列資訊封裝為 XML (eXtensible Markup Language)的文件形式,目的為增加資訊 的可重複利用性。其 XML 的 Data Schema 於下圖 4.14 所示,而部份封裝內容,如 下圖 4.15 所示。
DNA 資訊對映 蛋白質資訊
BLAST 比對結果
RefSeq 資料集
蛋白質資訊對映 至 IPI 編號
IPI 編號對映至 Gene Ontology 資訊
Gene Ontology 資訊
GOA 資料集
模組四
36
<record number="2">
<clone_id>Bt.252</clone_id>
<dna_id>XM_864105</dna_id>
<dna_gi>119908207</dna_gi>
<source_db>UG</source_db>
<description>PREDICTED: Bos taurus calpain, large polypeptide L2</description>
<species>Bos taurus</species>
<unigene>Bt.60825</unigene>
<protein_id>XP_869198</protein_id>
<protein_gi>119908208</protein_gi>
<protein_seq>MKPRPARFVDNKLKQRVIQVCILHGQLSEWSAFAA LHGQLSEWSAFAA L</protein_seq>
<ipi_num>IPI00695673</ipi_num>
<mdb_id>A4IFD3</mdb_id>
<ensembl number="1">ENSBTAP00000044733</ensembl>
<go number="0004198">
<term>calpain activity</term>
<ontology>F</ontology>
</go>
</record>
<?xml version="1.0" encoding="UTF-8"?>
<xs:schema xmlns:xs="http://www.w3.org/2001/XMLSchema" elementFormDefault="qualified">
<xs:element name="result_file" minOccurs="1" maxOccurs="1">
<xs:complexType>
<xs:sequence>
<xs:element name="record" minOccurs="1" maxOccurs="unbounded">
<xs:complexType>
<xs:attribute name="number" use="required"/>
<xs:sequence>
<xs:element name="clone_id" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="dna_id" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="dna_gi" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="source_db" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="description" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="species" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="unigene" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="protein_id" type="xs:string" minOccurs="0" maxOccurs="1"/>
<xs:element name="protein_gi" type="xs:string" minOccurs="0" maxOccurs="1"/>
<xs:element name="protein_seq" type="xs:string" minOccurs="0" maxOccurs="1"/>
<xs:element name="ipi_num" type="xs:string" minOccurs="0" maxOccurs="1"/>
<xs:element name="mdb_id" type="xs:string" minOccurs="0" maxOccurs="1"/>
<xs:element name="ensembl" type="xs:string" minOccurs="0" maxOccurs="unbounded">
<xs:complexType>
<xs:attribute name="number" use="required"/>
</xs:complexType>
</xs:element>
<xs:element name="go" type="xs:string" minOccurs="0" maxOccurs="unbounded">
<xs:complexType>
<xs:attribute name="number" use="required"/>
<xs:sequence>
<xs:element name="term" type="xs:string" minOccurs="1" maxOccurs="1"/>
<xs:element name="ontology" type="xs:string" minOccurs="1" maxOccurs="1"/>
</xs:sequence>
</xs:complexType>
</xs:element>
</xs:sequence>
</xs:complexType>
</xs:element>
</xs:sequence>
</xs:complexType>
</xs:element>
</xs:schema>
圖 4. 14 比對結果之 Data Schema
圖 4. 15 比對結果以 XML 格式封裝
37
XML 是開放標準用於建立描述結構化資料標示語言的語言。其中資料交換的 便利性是它的優勢之ㄧ。早期使用於電子商務大量資訊的自動化處理以取代以往 的電子資料交換 (Electronic Data Interchange, EDI)。當今,生物資訊系統需處理的 資料量亦是相當龐大。雖然管理基因體與生物的研究資料一直被認為是一個大問 題,但似乎並沒有令人滿意的解決方案。這目前在許多的生物專案仍為一個瓶頸 [13]。Achard[14]提出以 XML 整合生物資訊的想法。自此後便有許多相關系統使 用 XML 包裝資訊內容,如 JXP4BIGI[15]。
本模組會使用 NR 資料集以及 JDOM 技術插入內容結點,產生 XML 格式的輸 出檔案,下圖 4.16 呈現此模組的概約的流程圖。
圖 4. 16 模組四:系統運作流程圖
4.2.5 模組五 : GO 樹狀圖形及階層路徑分析與呈現
模組五顯示 GO 的樹狀結構。在樹狀結構方面以兩種模式顯示 : (1)表格模式 蛋白質 GI 對映至
蛋白質序列
Gene Ontology 資訊
NR 資料集
整合為 XML 格式 的輸出格式
JDOM
XML 格式整合 資料出書檔
資訊
38
(2)點陣圖模式。
表格模式中,移動至某個 GO ID 欄位可得知其 Term 資訊,如下圖 4.17。使用 者尚可選取欲得知 GO 路徑 (如下圖 4.18)以及階層資訊,如下圖 4.19。顯示階層 資訊方面,此模組參照模組四的 XML 格式整合資料輸出檔,再將 GO ID 對映至 蛋白質序列、GI 與 Accession number 以 FASTA 格式顯示,如下圖 4.20。
圖 4. 17 系統功能 – 顯示 GO 路徑、階層以及 Term
圖 4. 18 系統功能 – 顯示 GO 所對映的 Protein 資訊
39
圖 4. 19 系統功能 – 顯示 GO 樹狀結構之路徑
圖 4. 20 系統功能 – 以 FASTA 格式展示 Protein 資訊
點陣圖模式中 (如下圖 4.21),系統先將 GO 的樹狀結構封裝為 DOT 格式,再 呼叫 Graphviz 套件輸出為 PNG 的點陣圖形式。
圖 4. 21 系統功能 – 顯示 GO 樹狀結構圖
40
本模組會使用 Gene Ontology 資料集建構 GO 樹狀構造以及 DOM 技術擷取 XML 蛋白質結點資訊,下圖 4.22 呈現此模組的概約的流程圖。
圖 4. 22 模組五:系統運作流程圖
建構 GO 樹狀 結構
Gene Ontology
資料集
DOM
XML 格式整合 資料輸出檔資訊 顯示 GO 樹狀
表格
將 GO 樹狀結構 封裝為 DOT 格式
產生點陣圖
GO 樹狀結構 點陣圖
資訊 顯示 GO 階層 顯示 GO 路徑
顯示 GO 對映 蛋白質資訊
41
Chapter 5 討論
本章將針對兩個與本研究相似的系統,ZooDDD 系統[8]以及 COMPARE 系統 [16]進行比較討論。
5.1 與 COMPAER 系統進行比較
COMPAER 系統與本研究較為相似的功能為 BLAST Search。使用者可輸入一 條或多條 DNA 序列或蛋白質序列進行 BLAST 演算,BLAST 演算完成後點選其 Gene ID 的超鏈結 (呈現於下圖 5.1),即可獲得 Gene ID 的相關資訊,之後點選 Refine 再選擇欲比對的物種與欲擷取的資訊 (呈現於下圖 5.2),即可獲得跨物種比 對的 Orthologues 訊息、GO 註解 (annotation)以及反應路徑 (pathway)等資訊,呈 現於下圖 5.3。
圖 5. 1 系統 CMPARE - 顯示 BLAST 演算後的結果
圖片來源: COMPARE 系統網頁
42
圖 5. 2 系統 CMPARE - 進一步的資訊擷取
圖 5. 3 系統 CMPARE - 顯示擷取資訊
COMPARE 系統進行比對結果後,提供的資訊相對於本系統更甚豐富。但 COMPARE (1)系統提供的物種資訊較少(4 種), (2)無法進行跨組織器官的比對,
且(3)無整體彙整的功能(使用者一次僅能瀏覽一個 Gene ID 關聯的 GO 註解及 Orthologues 等資訊)。而本系統提供二十三個物種的資訊供使用者進行跨物種跨組 織的比對,比對完成後整理成一份彙整報告。上述為本系統略佔優勢之處。
圖片來源: COMPARE 系統網頁
圖片來源: COMPARE 系統網頁
43
5.2 與 ZooDDD 系統進行比較
本研究與 ZooDDD 系統擷取自 UniGene 的資料集並將其物種及組織予以抽離 並建置於本端資料庫。使用者至多可選擇兩種欲比對的物種及組織發展時期進行 程式演算,呈現於下圖 5.4。比對完成後將產生一份彙整報告 (呈現於下圖 5.5),
並結合另一套系統 GOBU 展示 GO 註解資訊,呈現於下圖 5.6。
圖 5. 4 系統 ZooDDD - 選擇物種組織及參數設定
圖 5. 5 系統 ZooDDD – 顯示比對結果
圖片來源: ZooDDD 系統網頁
圖片來源: ZooDDD 系統網頁
44
圖 5. 6 系統 ZooDDD – 顯示 GO 資訊
雖然 ZooDDD 系統與本系統同樣皆進行跨物種跨組織的比對,但比對的方式 有所不同。ZooDDD 是將選取的不同物種組織序列先透過 Ensembl 資料庫進行 BLAST 演算比對,完成後再判斷是否有同源關係,於下圖 5.7 所示。而本系統直 接進行 UniGene 與 UniGene 間的比對,於下圖 5.8 所示。
圖 5. 7 ZooDDD 序列比對流程
選取 DB1 物種 及組織
選取 DB2 物種 及組織
與 Ensembl 資料庫進行 Blast 演算比對
判斷其同源關係
圖片來源: ZooDDD 系統網頁
45
圖 5. 8 本系統序列比對流程
另外 ZooDDD 為了使 UniGene 資料較具代表性使用了兩個過濾方程式,呈現 於下圖 5.9 及圖 5.10。
圖 5. 9 系統 ZooDDD 之 TPM 公式
圖 5. 10 系統 ZooDDD 之 Specificity 公式
公式 TPM 欲表示一個 EST 序列在其物種組織的所佔的比值,當 TPM 值越大 則代表其 EST 序列更具代表性。
公式 Specificity 則欲表示一個 EST 序列其本身於某組織所佔的比例,當 Specificity 值越大則代表其 EST 序列更具代表性。
選取 Input 物種及組織
選取 DB1 物種及組織
選取 DB2 物種及組織
選取 DB3 物種及組織
Input 序列與 DB 序列進行 Blast 演算比對
判斷其同源關係
46
但本研究認為此方式的篩選機制有仍可能摒棄具代表性的資訊,況使用者尚 需了解其公式的意涵,故本系統實做上傾向提供全面的資訊予使用者。
47
Chapter 6 結論與未來工作
本章節將對花費近二年的研究給予些許的看法以及提供幾項觀點,希冀本研 究對生物資訊界有微薄貢獻。
6.1 結論
目前許多生資網站提供相當多 Web Base 或者 Web Service 的分析工具,但尚 無從基因層次推衍至蛋白質層次的工具。本系統提供此一套高通量序列整合流程 系統,並藉由模式動物 (Model Animal)預測非模式動物間的同源關係,並延伸提 供 GO Term 與 GO Term 之間的關聯性、階層與路徑顯示。且以使用者的觀點而言,
所有的細部流程一氣呵成完成,以縮短生物學家於實驗上的時間。
本人於蒐集各資料集中發覺,各資料集其資料呈現上皆有些微的凌亂,導致 系統開發者在剖析文件相當的不容易,故本系統於彙整比對的報表中使用 XML 方 式將結果封裝 (package)。若未來研究人員欲針對比對結果進行細部剖析,即可非 常方便地使用各種剖析 XML 的套件予以開發。
6.2 未來可能延伸之研究方向
於生物的觀點方面,本系統 DNA 資料庫僅擁有 UniGene 的資料,希望未來能 將 TIGR 的資料集也包含至本系統,提供相關研究人員更多元的選擇。而目前 GOA 也 只 提 供 人 類 (Human) 、 鼠 (Mouse) 、 阿 拉 伯 芥 (Arabidopsis) 、 斑 馬 魚 (Zebrafish)、雞 (Chicken)與牛 (Cow),故使用者必須於首頁的 DB 選項選擇上述 物種,報表結果才能將 GO Ontology 的資訊彙整呈現予使用者。若後續找到其他 物種的 GOA 資料集,則可新增至資料倉儲,使資訊於呈現上更為完整。
另 外 目 前 的 結 果 也 僅 提 供 至 蛋 白 質 序 列 的 層 次 , 而 蛋 白 質 的 交 互 作 用 (protein-protein interaction)於進行基因研究時亦扮演相當重要的角色,系統仍可提 供蛋白質的交互作用的資訊,使研究人員比對的結果資訊更加具備生物意義。
48
於資訊的觀點方面,本系統可增加一套代理程式 (Agent),負責各資料集的資 料更新,保證本端資料庫的資料處於最新版本的狀態。另外本系統可以將各個模 組的功能程式發展為 Web Service 形式,並與 BioMOBY[9]和 Taverna[10]等工具進 行結合,使未來的遠端開發人員能直接利用 Web Service 呼叫而不需再花時間重新 撰寫。