國立臺灣大學醫學院毒理學研究所 碩士論文
Graduate Institute of Toxicology College of Medicine
National Taiwan University Master Thesis
柴油引擎懸浮微粒對心肌細胞致毒機制之探討 Toxicological Effects of Diesel Exhaust Particles on
Cardiomyocytes
謝媺其 Mei-Chi Hsieh
指導教授:劉興華 博士 Advisor: Shing-Hwa Liu, Ph.D.
中華民國一百年七月
July, 2011
致謝
轉眼之間,碩士班的生活就接近尾聲了。從準備考研究所、進入實驗室參與 每一次的 meeting、大 se 與小 se。直到今天,所有的生活點滴都彷彿昨日般地歷 歷在目。這兩年的生活就像是在登山,崎嶇的道路上充滿了波折,也爬得滿身汗,
雖然常常跌得滿身傷,總是有人向我伸出援手以及鼓勵我,而我也學會要努力地 撐過去。現在,就像是站在山頂,回味著來時路的艱辛,俯瞰著美景。無論是在 學術上、人際關係上,我都學到了很多。
首先,感謝我的指導教授劉興華老師,提供機會在我感興趣的題目上嘗試研 究以及給予指導,幫助我得以順利完成論文。您嚴格的要求,讓我了解遭遇挫折 仍須以努力不懈的態度來學習,在此獻上萬分的感謝。接著要感謝楊榮森醫師,
謝謝您常常給予我建議與關心,並且指引我許多方向。謝謝蕭水銀老師與姜至剛 醫師給予我論文上許多指導與建議,讓我的論文得以更完整。在此,也謝謝翁祖 輝老師與彭福佐老師,不斷地在研究上給予指導與建議,及協助提供設備以順利 進行我的實驗研究。
另外,特別感謝陳錦澤醫師與鄭志鴻博士,曾經提供機會讓我學習基礎,並 給予學術上的指導與人生方向的指引。除此之外,更是無私地給予學生的實驗研 究之建議與幫助。
陪伴我走過兩年的 541 實驗室,是一個非常溫暖的大家庭。衷心感謝元鵬學 長和鎮天學長總是慷慨地分享經驗、提供建議與協助。麗婷和振源兩位這麼好的 同學,謝謝你們的支持、鼓勵與不斷的為我著想。總是帶著溫暖笑容的瀅如學姊、
柏霖學長與常陪伴我的佳薇學姊,非常謝謝有你們的關心、鼓勵與協助解決難題 以順利度過難關。同時也要謝謝國棟學長、靜欽學長、曉怡學姊、瓊文學姊、孝
勳學長、天鳳、雅銨和何豐名學長的關心與鼓勵。
奕伸學長、惠潔學姊、阿偉學長和青澔學長,感謝你們協助我度過實驗的難 關。謝謝帝言學長的全力幫助與建言;師儀的傾聽與陪伴;幸宜學姊和佑民的關 心與鼓勵。謝謝班上的同學的互相支持與打氣,大家將要一起邁入下個旅程,也 要一起加油!謝謝小孟、馬立學姊、燕玲學姊、允君、淑君、楊小白、黃小白和柔 慧,你們陪我度過難關與挫折,總是鼓勵我,讓我知道人生道路必然曲折,但努 力過後將是一片遼闊的天空。
最後,我要感謝我的家人,爸爸、媽媽、姊姊和弟弟,有你們的鼓勵與支持,
我才能走到今天。也謝謝小芳默默的支持與鼓勵。希望大家都健康、平安、快樂,
並在未來找到屬於自己的一片天空。
謝媺其 2011.07.29
Contents
Contents……….i
中文摘要……….………...……..iii
Abstract………....v
Abbreviations……….…………...………….vii
CHAPTER I Introduction……….………1-13 1. Diesel exhaust particles (DEPs)………...………..1
2. Cardiac hypertrophy………...………...5
3. Endoplasmic reticulum stress………7
4. Specific aims………...………13
CHAPTER II Materials and Methods………..………….…….14-21 CHAPTER III Results………...…..………22-27 Part I: Effects of DEPEs on neonatal rat cardiomyocytes………...….…22-24 1.1 Effects of DEPEs on neonatal rat cardiomyocyte viability………...………..….22
1.2 DEPEs induced cardiomyocyte hypertrophy………....…22
1.3 DEPEs induced upregulation of pathologic hypertrophy marker BNP and β-MHC but not ANP in neonatal rat cardiomyocytes………...…….……..23
1.4 DEPEs induced activation of PKC, p38, ERK, and JNK in cardiomyocytes..…23
Part II: Effects of DEPEs on H9c2 cells…………...……….….25-27 2.1 DEPEs reduced H9c2 cell viability……….……….………25
2.2 DEPEs induced apoptsis in H9c2 cells………..………..25
2.3 Exposure to DEPEs increased activation of JNK, ER chaperone GRP78, and pro-apoptotic factor CHOP……….…25 2.4 Phospho-JNK mediated DEPEs-induced upregulation of GRP78 and CHOP…26
2.5 Necrosis is also involved in DEPEs-induced cytotoxicity………….…….……26 CHAPTER IV Discussion………..……….…….28-32 Part II: Effects of DEPEs on neonatal rat cardiomyocytes…………...……..….28-30 Part II: Effects of DEPEs on H9c2 cells………..………...……31-32 CHAPTER V Conclusions and future applications……….………..33 Figures and Tables……….……….………….34-51 References……….52-65
中文摘要
近年來,流行病學研究顯示空氣懸浮微粒的增加及其所含之化學成分與心血
管疾病致死率有關。粒徑小於 2.5 微米的懸浮微粒(particulate matter; PM2.5)
可深入肺部並累積於體內,與血栓的形成和發炎反應具高度相關性。柴油引擎懸 浮微粒(diesel exhaust particles, DEPs)的平均直徑約為 0.2 微米,且為 PM2.5 之主要成分。柴油引擎車的使用量逐年漸增,因柴油引擎的效率較汽油引擎高,
因而降低石油消耗速度及二氧化碳的排放量,較不易加劇溫室效應,但氮氧化物 與懸浮微粒的排放量較汽油引擎相對得多。過去研究顯示柴油引擎懸浮微粒萃取 物(DEP extracts, DEPEs)可降低心肌細胞存活率,且可能導因於活性氧分子的生 成,但其造成的心血管系統毒性的確切機制尚未清楚。因此,本研究主要探討 DEPEs 對心肌細胞的毒性及參與的機制。
首先,利用初生大鼠心肌細胞探討 DEPEs 對心肌細胞的毒性。將初生大鼠心 肌細胞處理 DEPEs (1-15 μg/mL) 24 小時,發現對細胞存活率沒有顯著的影響,
但細胞大小與細胞中蛋白質含量則有增加,且細胞肥大指標因子[腦鈉素(BNP)及 肌凝蛋白質重鏈(β-MHC)]的基因表現量上升,顯示 DEPEs 具引起心肌細胞肥大的 作用。另外,DEPEs 亦可活化 mitogen-activated protein kinases (MAPKs),即 extracellular signal-regulated kinase、c-Jun-N-terminal kinase (JNK)和 p38,及其上游調控分子 protein kinase C (PKC)的磷酸化程度也增加。結果顯示 DEPEs 可能經由 PKC 調控之 MAPKs 路徑導致初生大鼠心肌細胞肥大。
接著,利用 H9c2 心肌細胞株探討 DEPEs (3-25 μg/mL)造成的心肌細胞毒性。
將細胞處理 25 μg/mL DEPEs 24 小時後,細胞存活率降低,並且利用流式細胞儀 技術發現,細胞會走向凋亡與壞死。同時,細胞內 ATP 降低,乳酸脫氫酶釋放情 形也增加。利用西方墨點法顯示處理 DEPEs 於短時間內可使 JNK 磷酸化增加。內 質 網 壓 力 恆 定 相 關 蛋 白 glucose-regulated protein 78 (GRP78) 和 C/EBP-homologous protein (CHOP)的表現量亦會上升,並且可被 JNK 抑制劑回復。
結果指出 DEPEs 可使心肌細胞凋亡與壞死,並且可經 JNK 調控引起內質網壓力,
但此內質網壓力在 DEPEs 造成的心肌細胞死亡中所扮演的角色仍須進一步探討。
綜合上述結果顯示,DEPEs 可造成初生大鼠心肌細胞肥大,亦可使 H9c2 心肌
細胞走向凋亡與壞死。此研究有助於進一步地了解 DEPs 造成的心血管毒性作用及 參與之機制。
關鍵詞:柴油引擎懸浮微粒,心肌細胞,細胞肥大,細胞凋亡,細胞壞死
Abstract
Evidences from epidemiological studies indicate that exposure to particulate matter (PM) air pollution contributes to cardiovascular morbidity and mortality. Fine particles with a diameter <2.5 μm (PM2.5) have an important role in triggering biological responses such as thrombosis and inflammation. Diesel exhaust particles (DEPs) with a mean size of about 0.2 µm are a major component of ambient PM2.5. Moreover, the use of diesel engine powered cars has recently been increasing in the world, because diesel engines offer better fuel efficiency and lower emissions of carbon dioxide than gasoline engines, which means that vehicles running on diesel are less responsible for global warming. However, diesel engines emit more nitrogen oxides and particles than do gasoline engines. It was reported that DEP extracts (DEPEs) decreased cardiomyocytes viability and it might be due mainly to reactive oxygen species formation. However, the mechanisms by which DEPs produce adverse cardiovascular effects at the cellular level are not fully understood. In this study, we used rat neonatal cardiomyocytes and H9c2 cardiac myoblasts to investigate the effects of DEPEs.
First, we used neonatal rat cardiomyocytes to determine the effects of DEPEs on cardiomyocyte hypertrophic response. Exposure to DEPEs (1-15 μg/mL) had no effects on cardiomyocyte viability. DEPEs treatment was found to elevate not only the cell size but also the ratio of total protein/cell number; additionally, the gene expression of hypertrophy markers including brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC) but not atrial natriuretic peptide (ANP) in neonatal rat cardiomyocytes was also increased, which indicated that DEPEs might induce cardiomyocyte hypertrophy. Besides, DEPEs also caused an increase in phosphorylation of extracellular signal-regulated kinases, c-Jun-N-terminal kinase (JNK) and p38 mitogen-activated
protein kinases (MAPKs), upstream regulators of ANP, BNP and β-MHC. And protein kinase C (PKC), one of the upstream regulators of these MAPKs, was also activated.
These results suggested that DEPEs was capable of inducing cardiomyocyte hypertrophy and implied that a PKC-regulated MAPK pathway may be involved.
On the other hand, we also investigate the effects of DEPEs on H9c2 myoblast death. After DEPEs (3-25 μg/mL) treatment for 24 hours, cell viability significantly decreased. DEPEs also increased annexin-V and propidium iodide binding, indicating that DEPEs could induce apoptosis and necrosis in H9c2 cells. Exposure to DEPEs also caused an increase in phosphorylation of JNK. Expression of endoplasmic reticulum (ER) chaperone protein glucose-regulated protein 78 and pro-apoptotic factor C/EBP-homologous protein was augmented after exposure to DEPEs, which could be reversed by JNK inhibitor. Meanwhile, DEPEs triggered the depletion of intracellular ATP and increased lactate dehydrogenase release. Collectively, these data showed that DEPEs could lead to not only apoptosis but also necrosis and induce ER stress through JNK pathway.
The present study demonstrated that DEPEs prompted neonatal rat cardiomyocyte hypertrophy and also induced apoptosis as well as necrosis in H9c2 myoblasts. These data provide the better understanding of potential mechanisms that link DEPs to toxicological regulation of cardiomyocytes.
Keywords: diesel exhaust particles, cardiomyocyte, H9c2 cell, hypertrophy, apoptosis, necrosis
Abbreviations
ANP atrial natriuretic peptide BNP brain natriuretic peptide CHOP C/EBP-homologous protein DEPs diesel exhaust particles DEPEs DEP extracts
ER endoplasmic reticulum
ERK extracellular signal-regulated kinase GRP78 glucose-regulated protein 78 JNK c-Jun-N-terminal kinase β-MHC β-myosin heavy chain PKC protein kinase C PM particulate matter
CHAPTER I Introduction
1. Diesel exhaust particles (DEPs) 1.1 Sources and exposure
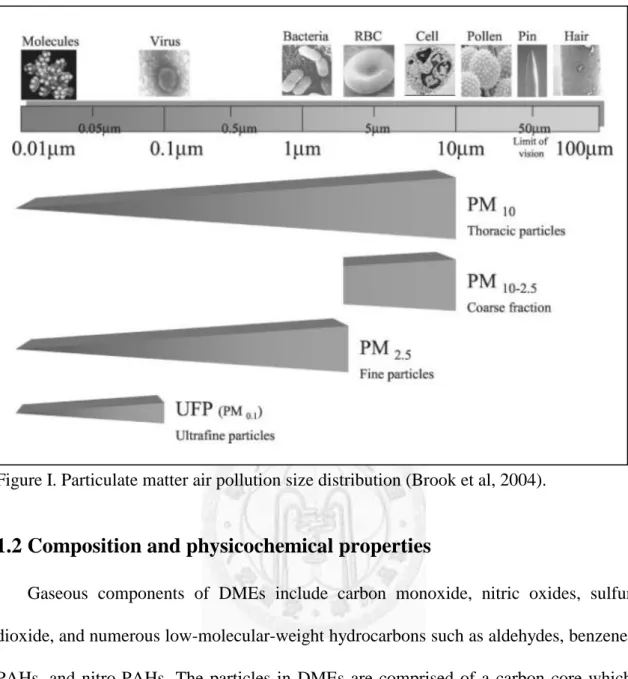
Airborne particulate matter (PM) consists of a heterogeneous mixture of solid and liquid particles suspended in air, continually varying in size and chemical composition in space and time (Figure I). The major source of PM2.5 throughout the world today is the human combustion of fossil fuel from industry or traffic, biomass burning, heating, cooking, indoor activities, and nonhuman sources like fires. The primary and secondary National Ambient Air Quality Standards for PM2.5 are 15.0 μg/m3 annual arithmetic mean concentrations (Environmental Protection Agency, U.S.A., 2002). Of the motor vehicle generated air pollutants, diesel motor emissions (DMEs) account for a highly significant percentage. The diesel engine’s popularity expanded because of its excellent fuel economy and durability, and it requires less maintenance. Diesel is the fuel used in mass transportation vehicles such as trucks, buses, and trains. Because of their lower per-mile fuel consumption, diesel engines generally release less carbon dioxide from the tailpipe, which means that vehicles running on diesel put less global-warming pollution into the air. However, on an equal horsepower basis, diesel exhaust is 100 times more toxic than gasoline exhaust. Diesel engines generate up to 100 times more fine particles than gasoline engines of a similar size (Zielinska et al, 2004). DMEs are a complex mixture composed of hundreds of constituents in either gas or particulate form, and DEPs are a major component of ambient PM2.5 (Yan et al, 2008).
Figure I. Particulate matter air pollution size distribution (Brook et al, 2004).
1.2 Composition and physicochemical properties
Gaseous components of DMEs include carbon monoxide, nitric oxides, sulfur dioxide, and numerous low-molecular-weight hydrocarbons such as aldehydes, benzene, PAHs, and nitro-PAHs. The particles in DMEs are comprised of a carbon core which adsorbs organic compounds including PAHs and nitro-PAHs, as well as small amounts of sulfate, nitrate, and metals. Particles with a diameter <5 μm can be deposited at the alveoli, but larger particles only reach the proximal airways and are eliminated by mucociliary clearance (Stuart, 1984). Recent electron microscopy studies suggest that over 80% of DEPs are ≦0.1 μm; PM0.1 can penetrate the epithelium and vascular walls, and enter the bloodstream (Sydbom et al, 2001). Therefore, the small size of DEPs allows them to penetrate deep into the lung, and even reach the blood circulation. Rats injected systemically with DEPs showed the presence of particles deposition in the
interalveolar interstitial tissue, myocardial interstitium, renal glomeruli, tubules and interstitium (Nemmar et al, 2010). Additionally, the large surface area of DEPs makes them an excellent medium for absorbing organics, which are known to have mutagenic and carcinogenic properties.
1.3 DEPs’ cardiopulmonary effects
Epidemiological studies have reported that ambient air pollution affects the respiratory tract, and it has systemic and cardiovascular effects as well (de Hartog et al, 2003; Künzli et al, 2010; Künzli et al, 2004; Rückerl et al, 2006). Ambient PM air pollution has been observed to be associated with increased risk of hospital admissions and deaths attributed to cardiovascular causes (Wellenius et al, 2006; Zanobetti &
Schwartz, 2005). Short-term exposure to particulate air pollution results in an increase in the susceptibility to ischemia (Pekkanen et al, 2002) and myocardial infarction (Peters et al, 2001). Risk estimates provided by several cohort studies demonstrate that a 10-μg/m3 increase in PM2.5 exposure is in general positively associated with excess mortality, largely driven by increases in cardiopulmonary or cardiovascular deaths (Brook et al, 2010). In previous studies, systemic administration of DEPs not only induced a significant decrease of heart rate and blood pressure but also triggered pulmonary inflammation in rats (Nemmar et al, 2007). Spontaneously hypertensive rats exposed to DEPs exhibited a dose-dependent increase in systolic blood pressure and heart rate, and it also showed pulmonary inflammatory reaction involving interleukin (IL)-6, leukotriene B4 and oxidative stress (Nemmar et al, 2009). Besides, there were more significant elevations of cytokines and albumin-specific immunoglobulin, which are biomarkers associated with allergy-related changes in the airways, observed among sensitized mice that were exposed to concentrated fine and ultrafine particles at
50 meters downwind than 150 meters downwind of a heavily trafficked freeway system (Kleinman et al, 2007). Rats instilled with DEPs intra-tracheally showed acute cardiac dysfunction, including the left ventricular systolic and diastolic dysfunction at 1 day after the exposure (Huang et al, 2010a). In addition to the circulating cytokines, cardiac IL-1β expression increased, which might be one of the mechanisms inducing acute cardiac dysfunction after short-term DEPs exposure. Functional changes and the increased circulating cardiac troponin I levels indicated that short-term DEPs exposure led to myocardial damage, and necrosis happened. In another study, exposure to diesel exhaust for 1 month produces a hypertensive-like cardiac gene expression pattern associated with mitochondrial oxidative stress in healthy rats (Kodavanti et al, 2009). In cellular level, treatment of DEP extracts (DEPEs) was reported to decrease cardiomyocyte viability and it might be due mainly to reactive oxygen species (ROS) formation (Okayama et al, 2006). Relative to participants living more than 150 meters from a major roadway, participants living within 50 meters of a major roadway had higher left ventricular mass (Van Hee et al, 2009). A model of isolated atria from guinea pigs was used to examine the direct toxic action of DEPs (Sakakibara et al, 1994). In this model, DEPs concentration ranged from 10 to 500 μg/mL induced a transient but dose-dependent increase in contractile force; DEPs in doses >500 μg/mL, however, decreased contractile force and induced cardiac arrest. It was concluded that cardiac toxicity contributes to the lung edema which is known as one prominent cause of death in DEPs-exposed animal.
2. Cardiac hypertrophy 2.1 Definition
Hypertrophy is used to describe cardiac response to stress under one or a few specific pathophysiological conditions, in which cardiac enlargement was largely assumed to be the result of increased cardiomyocyte size (Dorn et al, 2003). It is often associated with disease, including ischemic heart disease, hypertension and heart failure.
At the cellular level, cardiomyocyte hypertrophy is characterizedby an increment in cell size, increased protein synthesis, andchanges in the organization of sarcomeres. In contrast to developmental growth of heart through hyperplasia, growth of the postnatal heart occurs primarily through hypertrophy due to significant reduction in cardiac myoctes proliferation capacity soon after birth.
2.2 Classification
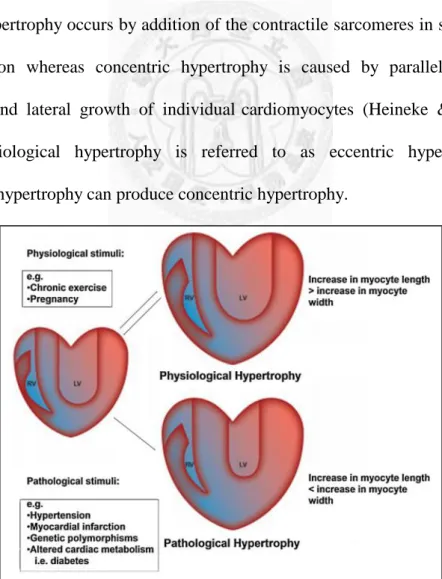
Cardiac hypertrophy is not all detrimental, as extensive aerobic conditioning through exercise induces a state of physiological growth regarded as adaption in the long term. Hypertrophy can be classified into two types. One is ‘physiological’, which is the normal response to healthy exercise or pregnancy, and the other is ‘pathological’, which is the response to stress or disease (Figure II) (Barry et al, 2008). Physiological hypertrophy is reversible and occurs without morbid effect on cardiac function. This normal growth of heart or in conditioned athletes enhances cardiac output to meet increased metabolic demands. Pathological hypertrophy occurs in response to pathological stress signals, such as neurohormonal activation, inflammation or cardiac injury. It has traditionally been considered to be adaptive initially, allowing the heart to increase cardiac output and compensate for adverse hemodynamics. In animal models,
inhibition of cardiac hypertrophy with cyclosporine A was demonstrated to result in increased mortality because of heart failure (Meguro et al, 1999). Prolonged hypertrophy is, however, associated with a significant increase in the risk for sudden death or progression to heart failure. At the molecular level, pathological hypertrophy is characterized by the activation of fetal genes, for example ANP, BNP, and β-MHC, which are considered and employed as hypertrophic markers (Cameron & Ellmers, 2003;
Rohini et al, 2010). Classically, hypertrophic growth develops in two ways: (1) concentric hypertrophy due to chronic pressure overload leading to reduced left ventricular volume and increased wall thickness, and (2) eccentric hypertrophy due to volumeoverload or prior infarction causing dilation and thinning of the heart wall.
Eccentric hypertrophy occurs by addition of the contractile sarcomeres in series causing cell elongation whereas concentric hypertrophy is caused by parallel addition of sarcomeres and lateral growth of individual cardiomyocytes (Heineke & Molkentin, 2006). Physiological hypertrophy is referred to as eccentric hypertrophy, and pathological hypertrophy can produce concentric hypertrophy.
Figure II. Pathological and physiological hypertrophic response to stimuli (Barry et al, 2008).
3. Endoplasmic reticulum (ER) stress 3.1 Definition
The ER is the first compartment in an ordered membranous network called the secretory pathway. Two primary functions of the ER are to (1) process nascent polypeptide chains so that properly modified proteins can be transported to the Golgi apparatus and (2) maintain internal stores of Ca2+ (Harr & Distelhorst, 2009). For example, GRP78, which is the ER-located member of the family of heat shock protein 70 molecular chaperones, promotes the folding of hydrophobic regions in polypeptides to the interior in a Ca2+-dependent manner (Gething, 1999). If the influx of nascent, unfolded polypeptides exceeds the folding and/or processing capacity of the ER, the ER homeostasis is compromised. Perturbation of ER-associated functions leads to ER stress via the activation of complex cytoplasmic and nuclear signaling pathways, collectively termed the unfolded protein response (UPR), also known as misfolded protein response, resulting in upregulation of expression of ER resident chaperones, facilitating the degradation of misfolded proteins and inhibiting protein synthesis to return the ER to its normal physiological state. When these adaptive responses are not sufficient to relieve ER stress, cells subjected to sustained and irreversible stress undergo programmed cell death.
3.2 UPR signaling
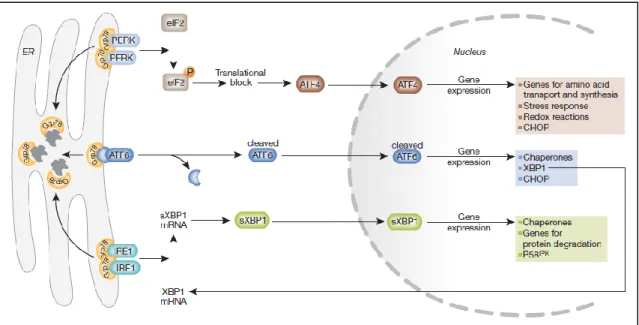
Three steps are involved in UPR activation. First, translation is attenuated to avoid further accumulation of misfolded proteins in the ER; second, chaperone and protein folding genes are activated transcriptionally; and the last, ER-associated degradation is activated in an attempt to rectify the accumulation of misfolded protein (Schröder &
Kaufman, 2005b). The UPR to ER stress can be initiated by activating three ER membrane receptors to initiate adaptive responses, including protein kinase RNA (PKR)-like ER kinase (PERK), inositol-requiring protein-1 (IRE1), and the transcriptional factor activating transcription factor-6 (ATF6) (Kaufman, 2002; Kim et al, 2008; Ron & Walter, 2007). These receptors are located with their N-terminus inside the lumen of the ER and their C-terminus in the cytosol, thereby connecting the ER with the cytosol. All three receptors are maintained in an inactive state through the interaction of their N-terminus with GRP78. When unfolded proteins accumulate in the ER, GRP78 releases these receptors to allow their activation (Figure III). PERK is activated first, rapidly followed by ATF6, whereas IRE1 is the last. Activated PERK phosphorylates eukaryotic translation initiation factor 2 (eIF2α) and consequently blocks its translation. However, eIF2α phosphorylation enables translation of ATF4, which occurs through an eIF2α-independent translation pathway. ATF4 translocates to the nucleus and induces the UPR-related genes. ATF6 is activated by limited proteolysis after its translocation from the ER to the Golgi apparatus. ATF6 is also a transcription factor and activates a subset of the UPR-related genes, including X-box binding protein 1 (XBP1). To achieve its active form, XBP1 must undergo mRNA splicing, which is carried out by IRE1. The three arms of the UPR, including ATF4, XBP1 and ATF6, all of which can induce transcription of CHOP, coordinately regulate the transcription of UPR-related genes encoding ER chaperones and protein folding enzymes to reduce the accumulation of unfolded proteins.
Figure III. The unfolded protein response (Szegezdi et al, 2006).
3.3 The relationship of DEPs and ER stress
A recent study on the molecular basis of PM2.5-induced intracellular events has highlighted the activation of a pathophysiological ER stress response upon PM2.5 exposure (Laing et al, 2010). It suggested that PM2.5 exposure differentially activates the UPR branches in C57BL/6 male mice, leading to ER stress-induced apoptosis through the PERK-eIF2α-CHOP UPR branch. PM2.5 also caused the upregulation and activation of caspase-12, and also induced ER stress in human bronchial epithelial cells (Watterson et al, 2009; Watterson et al, 2007). Similarly, the human bronchial epithelial cells treated with organic DEP extracts were showed an induction in a proinflammatory response accompanied by the UPR, such as Hsp70, ATF4, IL-6, and IL-8 (Jung et al, 2007). In addition, 1-nitropyrene (1-NP) is one of the most abundant nitro-PAHs in diesel exhaust and a major contributor to the mutagenicity of diesel exhaust particulate matter. And previous study has demonstrated that low levels of 1-NP (≦10 μM) induced DNA damage, increased intracellular ROS levels and increased protein
expression of the ER stress chaperone GRP78 in human umbilical vein endothelial cells (Andersson et al, 2009).
3.4 ER in the heart
As other cells, cardiac cells require ER membrane for housekeeping functions like protein turnover, modification and folding. ER in cardiomyocytes is involved in continuous turnover and synthesis of many membrane proteins including ion channels gap junction components and cell surface receptors (Mesaeli et al, 2001). The ER in non-muscle cells and SR in cardiac and skeletal muscle cells are also considered one of the most important and metabolically relevant sources of cellular Ca2+ for variety of functions including secretion, contraction-relaxation, cell motility, cytoplasmic and mitochondrial metabolism, protein synthesis, modification and folding, gene expression, cell cycle progression and apoptosis (Clapham, 1995; Pozzan et al, 1994). In the myocardium, the ER involves in maintenance of cellular Ca2+ homeostasis and synthesis of secretory proteins such as ANP, BNP (Forssmann et al, 1989), and vascular endothelial growth factor (Ozawa et al, 2001). Thus, dysfunction of the ER might contribute to heart diseases.
3.5 ER stress in the heart
A concept of the ER stress response was first described more than 2 decades ago (Kozutsumi et al, 1988), but only recently has it received significant attention by the cardiovascular community. The UPR and/or ER-initiated apoptosis have been implicated in the pathophysiology of various cardiovascular diseases such as cardiac hypertrophy, heart failure, atherosclerosis, and ischemic heart disease (Table I). Heat, hypoxia, ischemia, disease, glucose and metabolic starvation are potent inducers of the
ER stress. The response to ER stress occurs in two phases (Groenendyk et al, 2010).
First, the pathway is turned on in an attempt to address one consequence of the insult, the accumulation of protein in the ER. The fetal gene program is also activated in attempt to remodel the diseased tissue. If the heart continues being stressed, the UPR triggers autophagy and apoptosis to deal with the problem. This may ultimately result in heart failure and death. Therefore, the response to insult or injury can be classified into a pathologically relevant ER stress response and physiologically relevant ER stress response. XBP1 and ATF6 mediate induction of ER chaperones, protecting the heart from ischemia/reperfusion injury (Martindale et al, 2006), whereas activation of the PERK/ATF4/CHOP branch of the UPR triggers the pro-apoptotic signals. Recent observations indicate that in the heart, the UPR is activated during acute stresses, including ischemia/reperfusion, as well as upon longer term stresses that lead to cardiac hypertrophy and heart failure. For example, GRP78, CHOP, caspase-12, and phospho-JNK were significantly increased in rats with abdominal aortic coarctation (Guan et al, 2011). ATF6 was activated by ischemia but inactivated upon reperfusion in a cultured cardiomyocyte model system of simulated ischemia/reperfusion, suggesting that it may play a role in the induction of ER stress response genes during ischemia that could have a preconditioning effect on cell survival during reperfusion (Doroudgar et al, 2009). Mice subjected to transverse aortic constriction had cardiac hypertrophy and failure and showed an induction in prolonged ER stress, which might contribute to cardiomyocyte apoptosis during progression from cardiac hypertrophy to failure (Kitakaze & Tsukamoto, 2010; Okada et al, 2004). Besides, ER Ca2+ depletion occurs in the presence of ER stress inducer thapsigargin. Increases in cytosolic Ca2+ through changes in excitation-contraction coupling in cardiomyocytes such as that demonstrated for angiotensin II (Gusev et al, 2009) may drive the hypertrophic pathway and lead to
ER stress induction because of ER Ca2+ depletion. On the other hand, CHOP has shown to be responsible for ER stress-induced Puma, p53-upregulated modulator of apoptosis, activation during neonatal cardiomyocyte apoptosis (Nickson et al, 2007).
Cardiomyocytes treated with imatinib showed activation of the ER stress response, collapse of the mitochondrial membrane potential, release of cytochrome c into the cytosol, reduction in cellular ATP content and cell death (Kerkelä et al, 2006). In cardiomyocytes, activation of AMP-activated protein kinase (AMPK) contributes to protection of the heart against hypoxic injury through down regulation of ER stress (Terai et al, 2005). And inhibition of protein synthesis via eukaryotic elongation factor 2 inactivation may be the mechanism of cardioprotection by AMPK. Overexpression of ATF6, GRP78 and Derlin3 protects cardiomyocytes from ischemia/reperfusion damage (Belmont et al, 2009; Fu et al, 2008; Martindale et al, 2006)
Table I. ER stress and cardiovascular disease (Minamino & Kitakaze, 2010).
4. Specific aims
Numerous Epidemiological Studies have reported consistent associations between exposures to particulate air pollution and cardiovascular mortality and morbidity. In particulate air pollution, DEPs with a mean size of about 0.2 μm are a major component of PM2.5. Previous study demonstrated that traffic exposure is associated with higher left ventricular mass in adults. In cellular level, it was reported that DEPEs decreased cardiomyocytes viability and it might be due mainly to reactive oxygen species formation. However, the mechanisms by which DEPs produce adverse cardiovascular effects at the cellular level are not fully understood. The present study is designed to examine the actions and possible mechanisms of DEPs on growth and function of cardiomyocytes.
CHAPTER II
Materials and Methods
1. Reagents and Antibodies
Standard diesel particulate matter (DPM, SRM 2975) was purchased from National Institute of Standards and Technology (NIST, Gaithersburg, MD, USA). Anti-phospho (P)-p38, anti-P-JNK, anti-GRP78, anti-CHOP, secondary horseradish peroxidase (HRP)-conjugated antibodies, and polyclonal rabbit anti-desmin antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-PKC was from Cell Signaling Technology (Danvers, MA, USA). FITC-goat anti-rabbit IgG secondary antibodies were purchased from Zymed Laboratories (South San Francisco, CA, USA). Hoechst 33258 nuclear dye and SP600125 (JNK inhibitor) were obtained from Sigma–Aldrich Corporation (St. Louis, MO, USA)
2. Preparation of DEPEs
This method was carried out using a protocol modified from Tzeng et al.
(2003). Certified values for concentratio ns are provided for 11 PAHs in Table 1 (National Institute of Standards and Technology, 2009). Reference values for concentrations are provided for 28 additional PAHs in Table 2 and for 17 nitro-substituted PAHs in Table 3 (National Institute of Standards and Technology, 2009). In brief, DEPs were immersed in solvent (dichloromethane : n-hexane [1:1, v/v]) in the dark for 18 hours (Escobal et al, 1997).
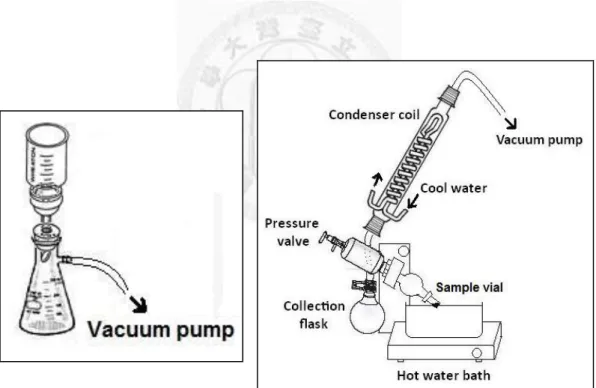
And the DEPs were then sonicated for 1 hour and centrifuged at 6,000 rpm for 15 min at
4℃. The supernatant was filtered by HPLC filter to obtain the DEPEs solution (Figure IV). The mixture was resuspended in solvent, sonicated and centrifuged at 6,000 rpm for 15 min at 4℃. The three steps were repeated for 4 times. The DEPEs solution was concentrated by a vacuum evaporator to get DEPEs (Figure V). Vacuum evaporation is the process of causing the pressure in a liquid-filled container to be reduced below the vapor pressure of the liquid, causing the liquid to evaporate at a lower temperature than normal. The crude DEPEs was dried by air flow, weighed, dissolved in dimethyl sulfoxide (DMSO) at a desired concentration, and stored at -20℃. After DEPEs were diluted by a certain medium, the final concentration of DMSO was 0.05 % (v/v).
Figure IV. Filtration system. Figure V. Vacuum evaporator.
3. Preperation of Neonatal Rat Cardiomyocytes
This method was carried out using a protocol modified from Cheng et al.
(1999). Primary cultures of neonatal rat cardiomyocytes were prepared from ventricles
of 1- to 3-day-old neonatal Wistar rats. Briefly, hearts were cut into chunks of approximately 1 mm3 using scissors and treated with pancrease (Sigma–Aldrich Corporation) at 37 ℃ for 15 min. Pancrease digested cells were collected by centrifugation at 1,600 rpm for 3 min. The cell pellets were resuspended in F10 medium containing 20% (v/v) fetal bovine serum (FBS) and plated into a Petri dish for 2 hours.
The nonattached myocytes in the F10 medium (Sigma–Aldrich Corporation) were collected and centrifuged at 1,600 rpm for 3 min. The cell pellets were resuspended in Dulbecco’s modified Eagle medium (DMEM; Gibco) containing 0.1 mM 5-bromo-2'-deoxyuridine (BrdU; Sigma–Aldrich Corporation) and 10% (v/v) FBS. The cells were then seeded on culture dishes at an appropriate cell density and cultured at 37°C in 95% air-5% CO2. After 2 days, cardiomyocytes obtained were 80% pure as revealed by their contractile characteristics under light microscopy. The culture medium was changed to fresh DMEM containing BrdU and 0.2% (v/v) FBS for 12 hours before exposure to DEPEs.
4. H9c2 Cell Cultures
H9c2 cells derived from embryonic rat heart myocardium were obtained from American Type Tissue Collection (ATCC, Rockville, MD, USA). The cells were grown in DMEM supplemented with 10% FBS at 37°C in 95% air-5% CO2. When they reached 70–85% confluency, the cells were washed with phosphate-buffered saline (PBS) (pH 7.4), trypsinized, and centrifuged at 1,000 rpm for 5 min. the cell pellets were resuspended in DMEM containing 10% (v/v) FBS and further passaged or appropriately treated. The culture medium was changed to fresh DMEM 1% (v/v) FBS for 12 hours before exposure to DEPEs.
5. Preparation of Total Cell Lysates
Cells were washed with ice-cold PBS and lysed with RIPA buffer (20 mM Tris-HCl (pH7.4), 150 mM NaCl, 1 mM EDTA, 1 mM ethylene glycol tetraacetic acid, 0.1%
Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM sodium fluoride (NaF), 1 μg/ml leupeptin, and 1 μg/ml aprotinin). PMSF and NaF in the buffer were the serine protease inhibitor and phosphatase inhibitor, respectively. The lysates were left on ice for 10 min, and centrifuged at 10,000 rpm for 10 min at 4℃. The supernatants were normalized for protein concentration by BCATM Protein Assay Kit (PIERCE) with bovine serum albumin as standard.
6. Western Blotting
Equal amounts of proteins (50 μg per lane) were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by electrotransfer to polyvinylidene difluoride membranes (Millipore Corporation, Bedford, MA, USA). The membranes were blocked with 5% nonfat powder milk in 0.1% Tris-buffered saline + Tween 20 (TBST) for 1 hour and probed with various primary antibodies at 4℃. Subsequently, membranes were washed three times with 0.1% TBST and incubated with secondary HRP-conjugated antibodies at room temperature for 1 hour. After three washes, the signals were visualized by an enhanced chemiluminescence reagent detection system according to the manufacturer’s protocol (Millipore Corporation).
7. Measurement of Cell Viability
The yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium salt (MTT;
Sigma-Aldrich Corporation) is reduced by mitochondrial succinate dehydrogenase in
viable cells to form insoluble purple formazan crystals, which are solubilized by DMSO.
In brief, cells seeded in 96-well plates were treated with DEPEs for 24 hours and then stained with MTT (0.5 mg/ml) for 4 hours. The media were removed, and formazan crystals produced were dissolved in 100 μL DMSO (Sigma–Aldrich Corporation). The absorbance was measured at 570 nm.
8. Lactate Dehydrogenase (LDH) Release Assay
After exposure to DEPEs for 24 hours, the amount of LDH leaked from the cytosol of damaged cells into the medium was detected. The released LDH was quantified by Cyto 96® Non-Radioactive Cytotoxicity Assay (Promega Corporation, Madison, WI, USA) according to the manufacturer's instructions. The absorbance was measured at 492 nm.
9. Analysis of Intracelluler ATP Levels
The intracellular ATP content was measured by Adenosine 5’-triphosphate bioluminescent assay kit (FL-AA, Sigma-Aldrich Corporation). After exposure to different concentrations of DEPEs for 24 hours, cells were washed two times with PBS and lysed with RIPA buffer. The lysates were centrifuged at 10,000 rpm for 10 min at 4℃. Subsequently, 100 μL supernatant and 100 μL ATP Assay Mix solution were mixed and immediately measured the amount of light produced with Orion L Microplate Luminometer (Berthold Detection Systems, Bad Wildbad, Germany).
10. Annexin V-FITC apoptosis detection
H9c2 cells were cultured in 6-well plates. Cells were treated with DEPEs for 24
hours, and then apoptosis was assessed by using an annexin V-FITC apoptosis detection kit (Becton Dickinson, Franklin Lakes, NJ, USA). In brief, cells were dissociated by 0.05% trypsin/EDTA for 3 min, and then centrifuged at 1,000 rpm for 5 min and re-suspended in 100 μL 1X binding buffer, and transferred into a 5 mL FACS tube, and added 5 μL annexin V-FITC (conjugated with fluorescein isothiocyanate) and 10 μL propidium iodide (PI). After incubation for 30 min at room temperature in dark, 400 μL of 1X binding buffer was added to each tube and the samples were immediately analyzed using a FACS flow cytometer.
11. Total RNA Extraction
Total RNA was isolated from cells with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) (1 mL to 1 × 106 cells), followed by chloroform extraction and isopropanol precipitation. The RNA was washed with 75% ethanol, resuspended in RNase-free water and, incubated at 65℃ for 5 min.
12. Real-time Reverse Transcription-Polymerase Chain Reaction
1 μg RNA was reverse transcribed using SuperScriptTM III First-Strand Synthesis System (Invitrogen). The RT reaction products were diluted to the volumes of 256 μL, and 1-μL aliquots were used as template. The mRNA expression levels were quantified by StepOne Real-Time PCR Detection Systems (Applied Biosystems, Warrington, UK) using SYBR® GreenERTM qPCR SuperMix (Invitrogen). Thermal cycler conditions were initiated at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15
s, and annealing/extension at 60 °C for 1 min. Polymerase chain reaction products were confirmed by melting curve analysis to ascertain the specificity of the primers and the purity of the final PCR product. The sequences of the primers used are shown in table 4.
The mRNA expression was normalized to expression of the housekeeping gene GAPDH amplified in a separate reaction.
13. Measurement of Protein Content Per Cell
After treatment with DEPEs for 24 hours, cells were trypsinized and washed twice with PBS. Cells were then collected via centrifugation at 1,000 rpm for 5 min, stained with trypan blue, and the viable cells were counted by microscopic examination. Cells were lysed with RIPA buffer as described above, and the lysates was centrifuged at 10,000 rpm for 10 min at 4℃. Protein concentration in the lysates was measured using BCATM Protein Assay Kit (PIERCE). Protein content per cell was determined by dividing the total amount of protein by the cell number.
14. Immunofluorescence
This method was carried out using a protocol modified from Liu et al.
(2009). To measure the cell size of cardiomyocytes, cells cultured in 12-well plates were washed with PBS followed by fixation with 4% paraformaldehyde in PBS at room temperature for 10 min. After three washes, the cells were permeabilized with 0.2%
Triton-100 for 5 min at room temperature. Non-specific binding of the fixed cells was blocked with PBS containing 2% bovine serum albumin for 1 hour, and cells were incubated overnight at 4℃ with polyclonal rabbit anti-desmin antibody diluted with blocking solution to 1:200. After three washes with PBS, cells were incubated for 1
hour at room temperature in the dark with FITC-goat anti rabbit IgG secondary antibodies diluted in blocking solution to 1:1,000. Finally, Hoechst 33258 nuclear dye was added with secondary antibodies at a final concentration of 1 μg/mL for 15 min, and cells were washed three times in PBS. Fluorescent images were captured by a Leica DMIL inverted microscope equipped with a charge-coupled device camera and SPOT software, version 4.6 (Diagnostic Instruments, Sterling Heights, MI, USA). Cell surface areas were measured by morphometric analysis of desmin-stained cardiomyocytes using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA).
15. Statistical Analysis
Results are expressed as means ± S.E.M. for at least three independent experiments.
Differences between two groups were evaluated by using the Student’s t-test. P<0.05 was considered statistically significant.
CHAPTER III Results
Part I: Effects of DEPEs on neonatal rat cardiomyocytes
1.1 Effects of DEPEs on neonatal rat cardiomyocyte viability
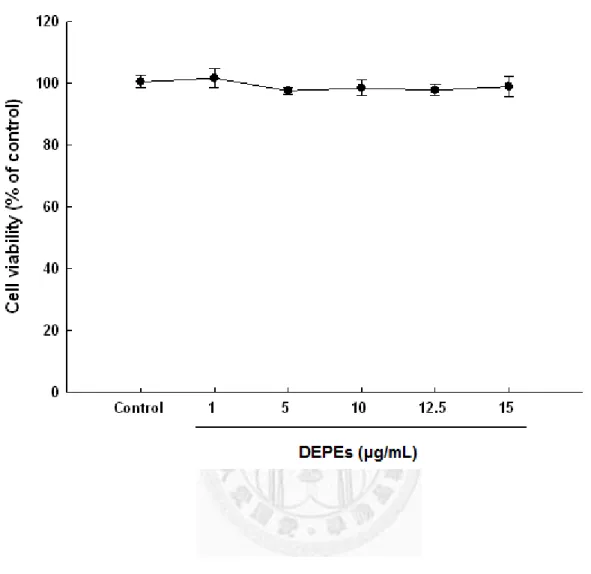
After incubation with various concentrations of DEPEs (1, 5, 10, 12.5, and 15 μg/mL) for 24 hours, there were no significant changes on neonatal rat cardiomyocyte viability (Figure 1).
1.2 DEPEs induced cardiomyocyte hypertrophy.
To evaluate the effects of DEPEs on cellular hypertrophy in cardiomyocytes, neonatal cardiomyocytes was treated with DEPEs and subjected to immunofluorescence analysis for measuring cellular area size (Figure 2A). After incubation with various concentrations of DEPEs (0.1, 0.5, 1, and 5 μg/mL) for 24 hours, a marked increase in cell size was observed at 5 μg/mL DEPEs. The quantitative results showed that the size area of neonatal cardiomyocytes treated with 5 μg/mL DEPEs for 24 hours significantly increased by approximately 2.4 fold. And hypertrophy determination showed that the total protein/cell numbers were increased to 112%, 126% and 161% by treatment of 0.5, 1 and 5μg/mL DEPEs for 24 hours, respectively. These results suggested that DEPEs induces cardiomyocyte hypertrophy.
1.3 DEPEs induced upregulation of pathologic hypertrophy marker BNP and β-MHC but not ANP in neonatal rat cardiomyocytes.
To further understand whether the expression of genes associated with myocardial pathologic hypertrophy was induced by DEPEs treatment, the mRNA expression levels of the fetal genes ANP, BNP, and β-MHC were analyzed by real-time RT-PCR following the administration of DEPEs (Figure 2C). Cultured cardiomyocytes were treated with various concentrations of DEPEs (0.1, 0.5, 1 and 5 μg/mL) for 24 hours and subsequently subjected to real-time RT-PCR analysis. The mRNA levels of BNP and β-MHC were elevated. The results suggested that DEPEs statistically lead to the increases in gene expression of hypertrophic markers BNP and β-MHC but not in ANP.
1.4 DEPEs induced activation of PKC, p38, ERK, and JNK in cardiomyocytes.
The results in Figure 2 indicated that DEPEs treatment markedly promoted the development of myocardial hypertrophy; therefore, the molecular events involved in cardiac hypertrophic process were further examined. Activation of p38 and JNK signaling pathways has been reported to enhance hypertrophic markers (ANP, BNP, and β-MHC) expression, and subsequently lead to cardiac hypertrophy (Huang et al, 2010b).
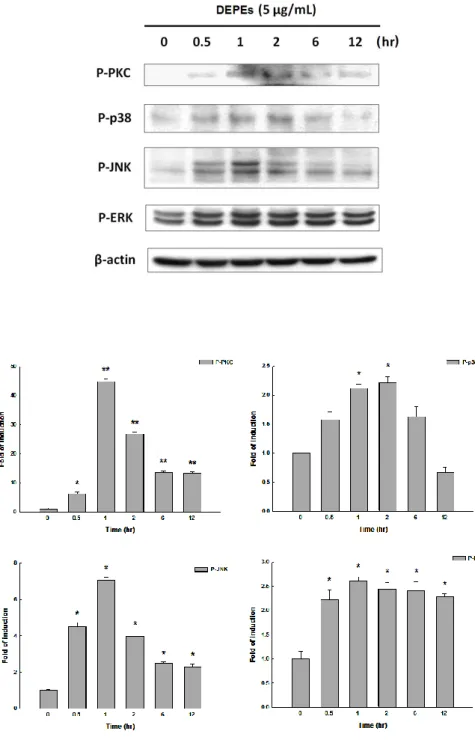
To examine if phosphorylation of p38 and JNK was induced by DEPEs, neonatal rat cardiomyocytes were treated with 5 μg/mL DEPEs for various time periods (0.5, 1, 2, 6, and 12 hours) followed by western blot assay (Figure 3). DEPEs significantly induced the phosphorylation of p38 and JNK within 1 hour. Activation of ERK, which is implicated in the development of cardiac hypertrophy (Muslin, 2008; Zou et al, 2001), was also increased within 1 hour (Figure 3). The results showed that DEPEs elevated the expressions of MAPKs (p38, JNK, and ERK). Besides, PKC is one of upstream regulators of these MAPKs (Frey & Olson, 2003), and its expression level was
determined by western blot assay. DEPEs treatment also resulted in activation of PKC (Figure 3). These results suggested that DEPEs induces myocardial hypertrophy through PKC-mediated MAPKs pathway.
Part II: Effects of DEPEs on H9c2 cells
2.1 DEPEs reduced H9c2 cell viability.
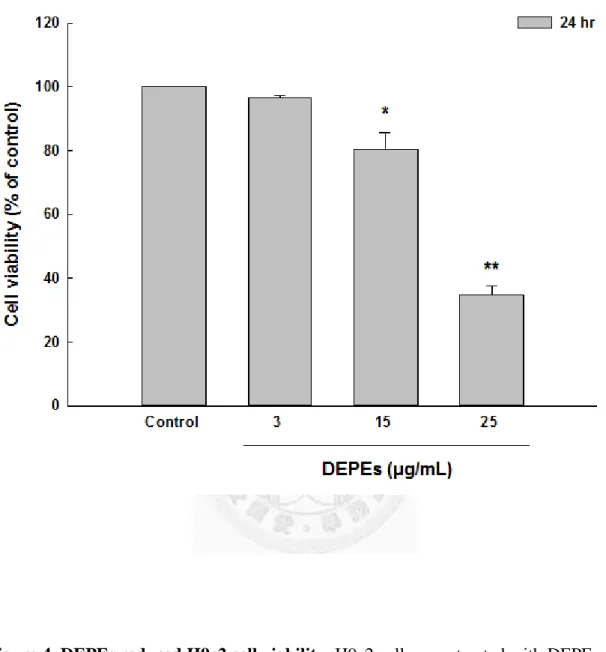
To examine the cytotoxic effects of high dose of DEPEs on H9c2 cell viability, cells were treated with various concentrations of DEPEs (3, 15, and 25 μg/mL) for 24 hours. The cell viability of H9c2 cells was significantly reduced at concentrations 15 and 25 μg/mL in a dose-dependent manner (Figure 4).
2.2 DEPEs induced apoptosis in H9c2 cells.
The externalization of phosphatidylserine (PS) is an early event in apoptosis.
Annexin V was shown to interact with PS and can be used to detect apoptosis by targeting for the loss of plasma membrane asymmetry (van Engeland et al, 1998). To examine whether apoptosis is involved in DEPEs-induced cytotoxicity in H9c2 cells, cells were incubated with 15 and 25 μg/mL DEPEs for 24 hours. Exposure to DEPEs (25 μg/mL) statistically increased the annexin V-PI positive cell population (Figure 5).
These results indicated that exposure to high dose of DEPEs could induce apoptosis in H9c2 cells.
2.3 Exposure to DEPEs increased activation of JNK, ER chaperone GRP78 and pro-apoptotic factor CHOP.
ER stress has shown to be associated with apoptosis. To examine whether ER stress plays an important role in DEPEs-induced apoptosis in H9c2 cells, cells were treated with 3, 15, and 25 μg/mL DEPEs for 24 hours. There was an increase in expression of GRP78 and CHOP after 25 μg/mL DEPEs treatment for 24 hours (Figure 6). H9c2 cells were then exposed to 25 μg/mL DEPEs for various time periods (3, 6, and 24 hours),
and the protein levels of GRP78 and CHOP were elevated after incubation for 24 hours (Figure 7A). Many studies have demonstrated that JNK is related to ER stress (Schröder
& Kaufman, 2005b; Szegezdi et al, 2006). And a short-term exposure to DEPEs found that JNK was phosphorylated within 1 hour (Figure 7B). These results showed that DEPEs activated JNK and induced the expression of ER stress markers GPR78 and CHOP.
2.4 P-JNK mediated DEPEs-induced upregulation of GRP78 and CHOP.
DEPEs activated of JNK prior to activation of ER stress markers, GRP78 and CHOP. In order to further identify the signal transduction pathway involved in the mechanism behind DEPEs-induced cytotoxicity, H9c2 cells were pretreated with SP600125 (JNK inhibitor) for 1 hour and followed by the administration of DEPEs (25 μg/mL) for 1 hour (Figure 8A) and 24 hours (Figure 8B). The cells were lysed and subsequently subjected to western blot assay to assess the effects of the JNK inhibitor on DEPEs-induced ER stress markers. The results showed that SP600125, JNK inhibitor, completely suppressed DEPEs-induced expression of GRP78 and CHOP, suggesting that DEPEs induced ER stress through JNK pathway.
2.5 Necrosis is also involved in DEPEs-induced cytotoxicity.
In figure 5, treatment of 25 μg/mL DEPEs also caused an increase in PI-positive H9c2 cell population. To confirm if necrosis is involved in DEPEs-induced cytotoxicity, H9c2 cells were treated with 3, 15, and 25 μg/mL DEPEs for 24 hours. LDH leakage, as a marker of cell membrane damage due to necrosis, was measured. And LDH released from H9c2 cells was significantly increased at 24 hours after DEPEs administration (Figure 9A). Loss of intracellular ATP level is another characteristic of necrotic cell
death, and it was decreased after DEPEs treatment (Figure 9B). These results indicated that DEPEs induced necrosis in H9c2 cells.
CHAPTER IV Discussion
Part I: Effects of DEPEs on neonatal rat cardiomyocytes
Cardiac hypertrophy is a common endpoint of many cardiovascular diseases and is characterized by an increase in cardiomyocyte size and protein content. Because of the small sizes of DEPs, they can penetrate the epithelium and vascular walls, and enter the bloodstream. Therefore, the toxicity of organic and inorganic constituents of the particles is important. The present study demonstrated that DEPEs induced cardiomyocyte hypertrophy, in which PKC-mediated MAPK pathway might be involved.
In this study, 1 to 15 μg/mL DEPEs had no significant effects on neonatal rat cardiomyocyte viability. Under this condition, the toxic effects of DEPEs on cardiomyocytes were investigated. DEPEs (5 μg/mL) increased cardiomyocyte size and the ratio of total protein/cell, which are characteristics of cardiac hypertrophy.
Meanwhile, the mRNA levels of hypertrophic markers BNP and β-MHC were enhanced.
These data suggested that DEPEs can induce cardiomyocyte hypertrophy. This finding is consistent with the observation that traffic exposure is associated with higher left ventricular mass in adults (Van Hee et al, 2009).
Hypertrophy can cause alterations in contractile protein compositions, including reactivation of β-MHC, skeletal α-actin, and myosin light chain-2 genes (Chien et al, 1991). It can also result in induction of non-contractile protein genes such as ANP and BNP (Cameron & Ellmers, 2003; Rohini et al, 2010). ANP and BNP defend against
increased hemodynamic load by decreasing blood pressure, regulating fluid homeostasis by increasing salt and water excretion, and regulating several hormones, such as angiotensin II, endothelin-1, and vasopressin. ANP and BNP are predominantly located in the cardiac atria and ventricle, respectively. BNP is also found in atria. Both ANP and BNP decrease plasma volume and blood pressure in response to an increased tension of the respective cardiac chamber (Krishnaswami, 2008). Activation of ERK and p38 MAPKs by hypertrophic agonist or by activated upstream kinase was reported to lead to phosphorylation of GATA-4, and analysis of the ANP and BNP promoter regions has also revealed binding sites for GATA-4 (Kerkela et al, 2002). Besides, full induction of β-MHC in pressure-overloaded rat hearts requires intact GATA binding elements in the
promoter regions (Pikkarainen et al, 2004). And Induction of ERK and JNK pathways could induce BNP promoter activity independently of GATA-4 binding (Kerkela et al, 2002). Thus, MAPKs are able to induce gene expression of hypertrophic markers, ANP, BNP, and β-MHC. In the present study, DEPEs induced the activation of MAPKs and
these hypertrophic markers in neonatal rat cardiomyocytes, which indicated that DEPEs might elevate gene expression levels of the hypertrophic markers through MAPKs pathway.
It has been hypothesized that ER stress is a pathogenic factor responsible for inducing cardiac hypertrophy. In most cell types, phosphorylation of eIF2α is involved in the initiation of the UPR, leading to a general inhibition of protein synthesis (Schröder & Kaufman, 2005a); however, dephosphorylation of eIF2α via ATF4 induced expression of CHOP and induction of GADD34 reverses the inhibition (Wek et al, 2006). ER stress may induce protein synthesis directly in cardiomyocytes, and thereby cause cell enlargement and cardiac hypertrophy (Dickhout et al, 2011), but the mechanisms by which ER stress induces protein synthesis in cardiomyocytes are still
unknown. On the other hand, angiotensin II, a known mediator of cardiac hypertrophy, has been shown to induce ER stress in rat cardiomyocytes by increased ER chaperone and CHOP expression (Okada et al, 2004). Therefore, ER stress can be involved in DEPEs-induced cardiomyocyte hypertrophy.
A variety of organic and inorganic constituents of DEPs have the potential to cause ROS generation. Inhaled particles that deposit within the distal lung have the potential to generate ROS, depending on the surface area of the particle and the reactive composition of the particle. Many transition metals present on particles serve as catalysts for ROS production. DEPs and lipopolysaccharide from gram negative bacteria have also been shown to stimulate generation of ROS in alveolar macrophages (Bonner, 2007). Besides, angiotensin II increases β-MHC gene expression in part via the generation of ROS (Shih et al, 2001). It was reported that DEPEs decreased neonatal rat cardiomyocytes viability and it might be due mainly to ROS formation (Okayama et al, 2006). And ROS has shown to participate in activation of p38 and JNK via apoptosis signal-regulating kinase 1 (Saitoh et al, 1998). Collectively, it suggested that ROS might be involved in DEPEs-induced cardiomyocyte hypertrophy.
Part II: Effects of DEPEs on H9c2 cells
Many studies have reported that DEPs induce oxidative stress, which played a key role for cascade activation during DEPEs-induced cellular injury (Bonner, 2007;
Okayama et al, 2006). In the present study, 15 and 25 μg/mL DEPEs significantly reduced H9c2 cell viability, where apoptosis and necrosis coexist.
After exposure to DEPEs extracted at different times, sometimes apoptotic and necrotic cell population both could be observed, and sometimes only necrotic cell population could be detected. DEPEs extracted at different times may vary in contents of organic compounds. Although the procedures of extraction were standardized as possible, there still may be some inaccuracies. Components of DEPEs should be standardized after every extraction, and this is the disadvantage of extraction.
Compared with DEPEs-induced cytotoxicity in neonatal rat cardiomyocytes at same dose, DEPEs seemed to cause more severe cytotoxicity in H9c2 cells. Although H9c2 cells and neonatal rat cardiomyocytes were reported to show similar hypertrophic responses in vitro (Watkins et al, 2010), they are still different in some ways. First, H9c2 cells are premature cardiac myoblasts, whereas neonatal rat cardiomyocytes are mature. Second, H9c2 cells have good proliferation ability but neonatal rat cardiomyocytes do not. Third, neonatal rat cardiomyocytes have contractile characteristics, and H9c2 cells are not contractile. The reason that DEPEs have more cytotoxic effects on H9c2 cells than that on neonatal rat cardiomyocytes may be that the latter have encountered some stress before being isolated from rats. But the exact differences of cytotoxicity between neonatal rat cardiomyocytes and H9c2 cells should be determined under the same dose treatment.
The present study showed that DEPEs induced not only ER stress but also apoptosis in H9c2 cells. Previous study has demonstrated that JNK inhibition prevented the
UPR-dependent human aortic smooth muscle cell death induced by 7-ketocholesterol (Pedruzzi et al, 2004). However, lead induces ER stress, but the induction of GRP78 and GRP94 expression via the JNK-AP-1 pathway functions as a defense mechanism against lead-induced cytotoxicity in vascular endothelial cells (Shinkai et al, 2010).
Therefore, ER stress induced by DEPEs in H9c2 cells may be the cause of apoptosis, or it may play a protective role. Besides, 7-ketocholesterol and lead both could induce ER stress marker GRP78 through the activation of JNK, which is consistent with the responses of H9c2 cells to DEPEs in this study.
In mouse macrophage cell line, DEPs induced apoptosis via p53 and Mdm2 (Yun et al, 2009). DEPs also produced DNA damage in the Chinese hamster lung fibroblast (V79) cell line (Gu et al, 2005). In neonatal rat cardiomyocytes, DEPEs extracted from PBS containing 0.05% Tween 80 induced cytotoxicity via ROS production. And in this study, DEPEs extracted from organic solvent induced neonatal rat cardiomyocyte hypertrophy and H9c2 cell death.
CHAPTER V
Conclusions and future applications
The present study demonstrated that low concentrations of DEPEs prompted neonatal rat cardiomyocyte hypertrophy. On the other hand, high concentrations of DEPEs induced apoptosis as well as necrosis in myocardial cells. These data provide a better understanding of potential mechanisms that link DEPs to toxicological regulation of cardiomyocytes. And the understanding of DEPEs-induced cardiomyocyte toxicity can help to find the ways to decrease the risks of DEPs-induced heart diseases.
Figures and Tables
Table 1. Certified Concentrations for Selected PAHs in SRM 2975
Table 2. Reference Concentrations for Selected PAHs in SRM 2975
Table 3. Concentrations for Selected Nitro-Substituted PAHs in SRM 2975
Table 4. Primers used in this study
Figure 1. Exposure to DEPEs had no effects on cardiomyocyte viability. Neonatal rat cardiomyocytes were treated with DEPEs (1, 5, 10, 12.5 and 15 μg/mL) and incubated for 24 hours. The cytotoxicity was analyzed by MTT assay as described under Materials and Methods. DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments.
A.
B.
C.
Figure 2. DEPEs induced cardiomyocytes hypertrophy. Cultured cardiomyocytes were exposed to vehicle control and various concentrations of DEPEs for 24 hours. (A) DEPEs-induced morphologic changes of cardiomyocytes. Cardiomyocytes were immunostained with an anti-desmin antibody (green), and the nucleus was stained with Hoechst 33258 (blue). The bar graph represents the relative cellular size to control. (B) Dose response effects of DEPEs with detection of the hypertrophy marker total protein/cell number. (C) Pathologic hypertrophy marker ANP, BNP, and β-MHC mRNA expression measured by real-time RT-PCR after 24 hours DEPEs treatment. The mRNA expression was standardized to expression of GAPDH. DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control; ** P<0.01 versus control
Figure 3. DEPEs induced phosphorylation of PKC, p38, ERK and JNK in cardiomyocytes. Cells were treated with 5 μg/mL DEPEs for the indicated times.
Whole cell lysates were processed for western blot analysis to detect changes in P-PKC, P-p38, P-ERK, and P-JNK. β-actin was used as an equal loading control. DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control; ** P<0.01 versus control
Figure 4. DEPEs reduced H9c2 cell viability. H9c2 cells were treated with DEPEs (3, 15 and 25 μg/mL) for 24 hours. To determine the cell viability, cells were subjected to the MTT assay as described under Materials and Methods. DMSO was used as a control.
Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control; ** P<0.01 versus control
Figure 5. DEPEs induced apoptosis in H9c2 cells determined by annexin V-FITC/PI staining. Cells were treated with DEPEs (15 and 25 μg/mL) for 24 hours, and cell apoptosis was measured by annexin V-FITC binding. Histograms show the percentages of early (annexin V-FITC positive and PI-negative) and late (annexin V-FITC positive and PI-positive) apoptotic cells in control and DEPEs-treated cells.
DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control; ** P<0.01 versus control
Figure 6. DEPEs induced expression of GRP78 and CHOP proteins. H9c2 cells were exposed to DEPEs (3, 15 and 25 μg/mL) for 24 hours, and total cell lysates were subjected to western blot analysis using the indicated antibodies. β-actin was used as internal standard. DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control; ** P<0.01 versus control
A.
B.
Figure 7. DEPEs induced phosphorylation of JNK and expression of GRP78 and CHOP proteins. H9c2 cells were exposed to DEPEs (25 μg/mL) for various time periods, and total cell lysates were subjected to western blot analysis using the indicated antibodies. β-actin was used as internal standard. DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control; ** P<0.01 versus control
A.
B
Figure 8. JNK inhibitor reversed DEPEs-induced expression of ER stress markers.
H9c2 cells were pre-incubated with 15 μM JNK inhibitor, SP600125, for 1 hour, and treated with 25 μg/mL DEPEs for 1 hour (A) and 24 hours (B). Lysates were then immunoblotted for P-JNK, GRP78, and CHOP. β-actin was used as internal standard.
DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control; ** P<0.01 versus control; ## P<0.01 versus DEPEs treatment alone without JNK inhibitors
A.
B.
Figure 9. Intracellular ATP levels and LDH release in H9c2 cells after DEPEs treatment. Cells were exposed to DEPEs for 24 hours, and intracellular ATP levels (A) and LDH release (B) were determined. DMSO was used as a control. Data are presented as mean values ± S.E.M. from three independent experiments. * P<0.05 versus control;
** P<0.01 versus control
.
References
Andersson H, Piras E, Demma J, Hellman B, Brittebo E (2009) Low levels of the air pollutant 1-nitropyrene induce DNA damage, increased levels of reactive oxygen species and endoplasmic reticulum stress in human endothelial cells. Toxicology 262:
57-64
Barry SP, Davidson SM, Townsend PA (2008) Molecular regulation of cardiac hypertrophy. The International Journal of Biochemistry & Cell Biology 40: 2023-2039
Belmont PJ, Chen WJ, San Pedro MN, Thuerauf DJ, Gellings Lowe N, Gude N, Hilton B, Wolkowicz R, Sussman MA, Glembotski CC (2009) Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circulation Research 106: 307-316
Bonner JC (2007) Lung fibrotic responses to particle exposure. Toxicologic Pathology 35: 148-153
Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, Luepker R, Mittleman M, Samet J, Smith SC, Tager I (2004) Air pollution and cardiovascular disease: a statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation 109: 2655-2671
Brook RD, Rajagopalan S, Pope CA, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC, Whitsel L,