1
行政院國家科學委員會補助專題研究計畫
; 成 果 報 告
□期中進度報告
內前額皮質區長期神經塑性表現之研究
計畫類別:; 個別型計畫 □ 整合型計畫
計畫編號:NSC 96-2321 - B -006 - 001-
執行期間: 94 年 8 月 1 日至 97 年 7 月 31 日
計畫主持人:黃瓊君
共同主持人:許桂森
計畫參與人員: 梁嫈卿、楊秉鈞
成果報告類型(依經費核定清單規定繳交):□精簡報告 ;完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年;二年後可公開查詢
執行單位:國立成功大學醫學院藥理學科
中 華 民 國 97 年 9 月 22 日
2 中文摘要 前額皮質區一般被認為是哺乳動物大腦皮質區域最高導的聯結區域,其在大腦接受訊 息後的動作執行中扮演著相當重要的角色。在靈長類,背外側區的前額皮質被認為在抑制 性調控、工作記憶、注意力的選擇、注意力的轉移、規則性的學習及策略的替換中扮演著 相當重要的角色。然而,在囓齒性動物則為內前額皮質來執行此等功能;其在組織結構中 主要包含緣前區、下邊緣區及迴帶次區等。近年來對於大腦長期神經塑性之表現與學習記 憶之相關性已有相當多佐證,但截至目前為止對於內前額皮質區長期神經塑性之表現及其 分子機制的探討仍相當的匱乏。此外,在活體動物行為研究中也証實內前額皮質區可能參 與興奮性藥物濫用所產生之致敏感性及成癮作用中扮演著相當重要的角色。由於興奮性藥 物致敏感性及成癮作用發生的分子機制可能與長期神經塑性表現之分子機制相當,因此探 討內前額皮質區長期神經塑性之表現分子機制將有助於了解其與學習記憶之表現外,也將 有助於科學家了解藥物成癮發生之原因及開發有效的緩解藥物及策略。在本研究計畫的第 一年我們設計一系列的研究在離體內前額皮質區腦薄片標本上誘發長期增益現象的表現並 探討其生理及分子特徵。我們主要的發現包括在離體內前額皮質區腦薄片標本上直接投與 forskolin 會在 II-III 及 V 錐狀細胞層誘發 cAMP-dependent 型式的長期增益現象,並且此長期 增 益 現 象 的 誘 發 需 要 同 時 活 化 cAMP-dependent protein kinase(PKA) 及 p42/p44 mitogen-activated protein kinase (MAPK)等迅息傳遞逕路。直接活化乙型腎上腺素受體也可 以模擬 forskolin 的作用產生長期增益現象。此外,此長期增益現象的誘發可能透過促進突 觸前 glutamate 的釋放來產生。
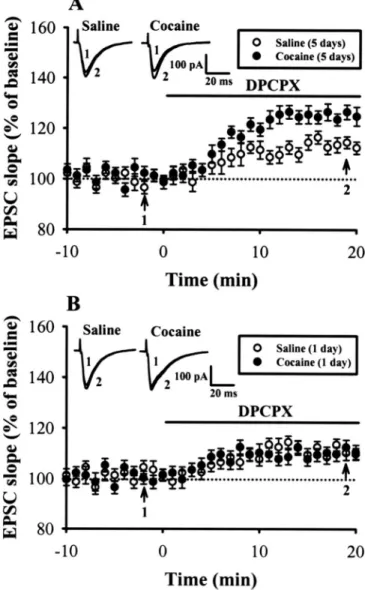
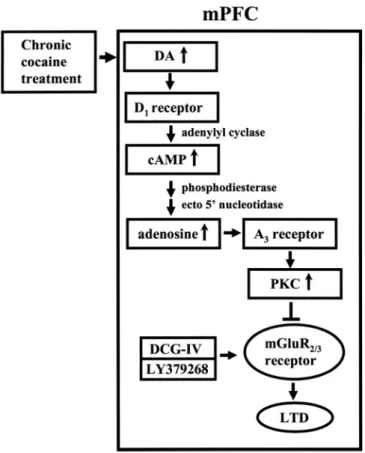
在研究計畫的第二年的研究中我們主要發現: 連續投予古柯鹼後對於在前額皮質第五 層錐狀神經元經由活化代謝型麩胺酸受體(metabotropic glutamate receptor)所誘發之長期抑 制作用(long-term depression)表現的影響。我們發現連續投予古柯鹼後會抑制經由投予(2S, 2’R, 3’R)-2-(2’, 3’-dicarboxycyclopropyl)glycine (DCG-IV) 或 LY379268 所誘發的長期抑制 作用。此作用的發生與古柯鹼所造成的多巴胺 D1 受體的活化有關。在正常大鼠的前額皮 質 腦 片 上 , 直 接 投 予 protein kinase C 的 活 化 劑 或 adenosine A3 受 體 的 作 用 劑 2-chloro-N6-(3-iodobenzyl)-adenosine-5-N-methyluronamide 也可以模擬連續投予古柯鹼的作 用 抑 制 長 期 抑 制 作 用 的 產 生 。 此 外 ﹐ 在 大 鼠 投 予 古 柯 鹼 前 , 先 處 理 PKC 阻 斷 劑 bisindolylmaleimide I 或 adenosine A3 受體抑制劑 MRS1220 則可以阻斷連續投予古柯鹼的 作用。另外抑制 cAMP 的代謝作用也可以抑制連續投予古柯鹼的作用。因此,我們主要的 模式為連續投予古柯鹼會經由增加活化多巴胺 D1 受體﹐所產生的 cAMP 代謝成 adenosine 作 用 在 adenosine A3 受 體 , 經 活 化 PKC , 抑 制 經 由 投 予 (2S, 2’R, 3’R)-2-(2’,
3’-dicarboxycyclopropyl)glycine (DCG-IV) 或 LY379268 所誘發的長期抑制作用。
在研究計畫的第三年的研究中我們主要發現連續投予古柯鹼後會抑制經由投予 5-HT 在前額皮質第五層錐狀神經元經由活化 5-HT2A 受體所產生之增加自發性 glutamate 釋放的 作用,並且此作用與經由連續投予古柯鹼後所導致 5-HT2A 受體的去敏感化作用有關,並 且與古柯鹼抑制 5-HT 再回收的藥理特性有關。 此等研究結果對於了解藥物成癮發生之原因及開發有效的緩解藥物提供了重要的策略 及方向。 關鍵字: 古柯鹼; 前額皮質區;長期抑制作用; 藥物成癮
3
英文摘要
The prefrontal cortex (PFC) is believed to be the highest association area in the mammalian cortex and is required for proper executive control. In primates, the dorsolateral PFC has been implicated in a wide variety of cognitive processes including inhibitory control, working memory, selective attention, attentional-set shifting, rule learning, and strategy switching. In rodents, where the architecture of the cortex is simpler than that of primates, the medial PFC (mPFC), the cortex that encompasses the prelimbic, infralimbic, and the anterior cingulated subregions, is considered to be homologous to the primate dorsolateral PFC. Although long-term changes in synaptic strength are thought to provide a cellular basis for information encoding and storage in the nervous system, little is known about whether such changes will occur in the PFC. Recently, the drugs of abuse and learning mechanisms were found to activate common neuronal circuits and signal transduction pathways and the PFC was shown to play an important role in mediating aspects of additive behavior such as impulsivity and reward-related learning. However, the influence of psychstimulants on the synaptic plasticity in PFC neurons remains to be elucidated. Because a full understanding of the molecular mechanisms underlying the long-term synaptic plasticity in PFC region and their influence by psychostimulant administration will ultimately open new avenues for research into the neurobiological basis of PFC-related memory formation and execution and may contribute to develop a new strategy or pharmacological management in the treatment of drug additive behavior, the primary goal of this study is to generate a more comprehensive picture of the synaptic plasticity occurring in PFC neurons from physiological and pathological bases. Toward these ends, in the first year of this project, we used forskolin, an adenylyl cyclase activator, to examine the effects of cAMP on excitatory synaptic transmission in the mPFC using whole-cell patch-clamp recordings from visually identified layer II-III or V pyramidal cells in vitro. We found that bath application of forskolin significantly increased the amplitude of excitatory postsynaptic currents (EPSCs) in a concentration- and age-dependent manner. This enhancement was completely abolished by coapplication of cAMP-dependent protein kinase (PKA) inhibitor and p42/p44 mitogen-activated protein kinase (MAPK) kinase inhibitor, but not application of either drug alone. The membrane-permeable cAMP analog, Sp-adenosine 3c',5c'-cyclic monophosphorothioate triethylammonium, or activation of β-adrenergic receptor by isoproterenol mimicked the effect of forskolin to potentiate EPSCs. However, neither exchange protein activated by cAMP (Epac) inhibitor, brefeldin A, nor hyperpolarization and cyclic nucleotide-activated channel blocker, ZD7288, affected forskolin response. The augmentation of EPSCs by forskolin was accompanied by a reduction of the
4
synaptic failure rate, coefficient of variation and paired-pulse ratio of EPSCs and an increase in release probability and number of releasable synaptic vesicles. Forskolin also significantly increased the frequency of miniature EPSCs without altering their amplitude distribution. These results indicate that cAMP acts presynaptically to elicit a synaptic potentiation on the layer V pyramidal neurons of mPFC through converging activation of PKA and p42/p44 MAPK signaling pathways.
In the second year of this project, we have found that repeated cocaine administration in vivo impaired the long-term depression (LTD) induced by bath application of group II metabotropic glutamate receptor (mGluR) agonists, (2S, 2’R, 3’R)-2-(2’, 3’-dicarboxycyclopropyl)glycine (DCG-IV) or LY379268, at excitatory synapses onto layer V pyramidal neurons of rat mPFC. In contrast, this impairment was not found in slices from rats treated with saline or a single dose of cocaine. Such effect of cocaine was selectively prevented when cocaine was co-administered with the selective D1-like receptor antagonist SCH23390. In slices from control rats, a brief
application of either protein kinase C (PKC) activator phorbol-12,13-dibutyrute or adenosine A3
receptor agonist 2-chloro-N6-(3-iodobenzyl)-adenosine-5-N-methyluronamide mimicked the effect of repeated cocaine treatment to impair the induction of LTD. Bilateral intra-mPFC infusion of PKC inhibitor bisindolylmaleimide I or adenosine A3 receptor antagonist MRS1220
before cocaine injection prevented cocaine-induced impairment of LTD induction. Furthermore, endogenous adenosine tone is greater in slices from cocaine-treated rats than from the saline-treated controls. When the metabolism of cAMP to adenosine was blocked, the extent of LTD in slices from saline and cocaine-treated rats was similar. These results suggest that cocaine-induced impairment of group II mGluR-mediated LTD is caused, at least in part, by an increase in adenosine subsequent to the rise in cAMP following D1-like receptor activation,
which leads to an adenosine A3 receptor-mediated upregulation of PKC activity and thereby
triggers an inhibition of group II metabotropic glutamate receptor function.
In the third year, we examine whether repeated cocaine exposure in vivo is capable of altering 5-HT-induced regulation of glutamatergic transmission in the mPFC. In mPFC layer V pyramidal neurons, application of 5-HT, through activation of 5-HT2A receptors, induced a
massive enhancement of spontaneous excitatory postsynaptic currents (sEPSCs). Repeated cocaine administration resulted in an attenuation in the ability of 5-HT to enhance sEPSCs. This effect was prevented when cocaine was coadministered with the selective 5-HT2A receptor
antagonist ketanserin and was mimicked by repeated 5-HT2A receptor agonist
(-)4-iodo-2,5-dimethoxyphenylisopropylamine administration. Repeated cocaine administration is not associated with any changes in the levels of 5-HT2A receptors or Gαq/11 proteins.
However, cocaine-treated rats exhibited a significant increase in phosphorylation of Gαq/11
proteins in the mPFC. These results suggest that cocaine-induced inhibition of 5-HT2A receptor
function is caused, at least in part, by increased phosphorylation of Gαq/11 proteins leading to a
reduction of the coupling of 5-HT2A receptor with Gαq/11 proteins.
Together these data, we provide a new insight into the molecular mechanisms underlying the development of drug addiction and offer a new opportunity to develop new strategies to treatment
5
Keywords: cocaine; medial prefrontal cortex; long-term depression; addiction
成果已部份發表於
1. Huang, C. C. and Hsu, K. S. (2006) Presynaptic mechanism underlying cAMP-induced
synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol. Pharmacol. 69: 846-856.
2. Huang, C. C., Yang, B. J., Lin, H. J. and Hsu, K. S. (2007) Repeated cocaine
administration blocks group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J. Neurosci. 27(11): 2958-2968.
3. Huang, C. C., Lin, H. J. and Hsu, K. S. (2007) Repeated cocaine administration facilitates
long-term potentiation induction in medial prefrontal cortex pyramidal neurons. Cere.
Cortex 17(8):1877-1888.
4. Huang, C. C. and Hsu, K. S. (2008) The role of NMDA receptors in regulating group II
metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. Neuropharmacology 54:1071-1078.
5. Huang, C. C., Liang, Y. C., Lee, C. C., Wu, M. Y. and Hsu, K. S. (2008) Repeated cocaine
administration decreases 5-hydroxytryptamine 2A receptor-mediated facilitation of synaptic activity in rat medial prefrontal cortex. (Submitted).
成果自評:
研究進度與原計畫內容相符,部分結果已發表。此等發現對於了解內前額皮質區的 神經突觸塑性表現有相當之助益。具基礎醫學發展之意義。
Presynaptic Mechanism Underlying cAMP-Induced Synaptic
Potentiation in Medial Prefrontal Cortex Pyramidal Neurons
□SChiung-Chun Huang and Kuei-Sen Hsu
Department of Pharmacology, College of Medicine, and Center for Gene Regulation and Signal Transduction Research, National Cheng Kung University, Tainan, Taiwan
Received August 16, 2005; accepted November 23, 2005
ABSTRACT
cAMP, a classic second messenger, has been proposed re-cently to participate in regulating prefrontal cortical cognitive functions, yet little is known about how it does so. In this study, we used forskolin, an adenylyl cyclase activator, to examine the effects of cAMP on excitatory synaptic transmission in the medial prefrontal cortex (mPFC) using whole-cell patch-clamp recordings from visually identified layer II–III or V pyramidal cells in vitro. We found that bath application of forskolin significantly increased the amplitude of excitatory postsynaptic currents (EPSCs) in a concentration- and age-dependent manner. This enhancement was completely abolished by coapplication of cAMP-dependent protein kinase (PKA) inhibitor and p42/p44 mitogen-activated protein kinase (MAPK) kinase inhibitor, but not application of either drug alone. The membrane-permeable cAMP analog adenosine 3⬘,5⬘-cyclic monophosphorothioate,
Sp-isomer, triethylammonium salt, or activation of-adrenergic
receptor by isoproterenol mimicked the effect of forskolin to potentiate EPSCs. However, neither exchange protein acti-vated by cAMP (Epac) inhibitor brefeldin A nor hyperpolariza-tion and cyclic nucleotide-activated channel blocker 4-ethyl-phenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288) affected forskolin response. The augmentation of EPSCs by forskolin was accompanied by a reduction of the synaptic failure rate, coefficient of variation and paired-pulse ratio of EPSCs, and an increase in release probability and number of releasable synaptic vesicles. Forskolin also signifi-cantly increased the frequency of miniature EPSCs without altering their amplitude distribution. These results indicate that cAMP acts presynaptically to elicit a synaptic potentiation on the layer V pyramidal neurons of mPFC through converging activation of PKA and p42/p44 MAPK signaling pathways.
The prefrontal cortex (PFC) is believed to subserve a wide variety of cognitive processes, including working memory (Goldman-Rakic, 1995), selection and remembering of rele-vant stimuli (Rainer et al., 1998), and remembering of con-textually relevant, cross-modal stimulus association (Fuster et al., 2000). In rodents, the medial aspect of PFC (mPFC),
encompassing the infralimbic and prelimbic areas, is consid-ered to be anatomically homologous to the human/primate dorsolateral PFC to execute these functions (Zahrt et al., 1997; Birrell and Brown, 2000). Prefrontal dysfunction is commonly found in many psychiatric disorders, such as attention-deficient/hyperactivity disorder, post-traumatic stress disorder, the affective disorders, schizophrenia, and drug abuse (Heidbreder and Groenewegen, 2003).
cAMP is one of the best-studied second messengers. A substantial body of evidence implicates that cAMP activates cAMP-dependent protein kinase (PKA), thereby eliciting a long-lasting increase in transmitter release at many central
This work was supported by research grants NSC94-2321-B-006-008 and NSC94-2752-B-006-002-PAE from the National Science Council, Taiwan.
□S The online version of this article (available at http://molpharm. aspetjournals.org) contains supplemental material.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.105.018093.
ABBREVIATIONS: PFC, prefrontal cortex; mPFC, medial prefrontal cortex; PKA, cAMP-dependent protein kinase; Epac, exchange protein activated by cAMP; MAPK, mitogen-activated protein kinase; HCN, hyperpolarization and cyclic nucleotide-activated; EPSC, excitatory postsyn-aptic current; mEPSC, miniature excitatory postsynpostsyn-aptic current; ACSF, artificial cerebrospinal fluid; AMPA, ␣-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; PPR, paired pulse ratio; Nq, number of release vesicle; PD98059, 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyrane-4-one; U0126, 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene; SB203580, 4-[5-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine; SP600125, anthrax[1–9-cd]pyrazol-6(2H)-one; 8CPT-2Me-cAMP, 8-(4-chlorophrnylthio)-2⬘-O-methyl-cAMP; H-89, N-[2-((p-bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide; KT5720, (9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3⬘,2⬘,1⬘-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid; Sp-cAMPS, adenosine 3⬘,5⬘-cyclic monophos-phorothioate, Sp-isomer; CNQX, 6-cyano-7-notroquinoxaline-2, 3-dione; ZD7288, 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride; D-APV, D-(⫺)-2-amino-5-phosponopentanoic acid; QX-314, lidocaine N-ethyl bromide; Dd-forskolin, 1,9-dideoxy-forskolin; NMDA,
N-methyl-D-aspartate; N, number of readily releasable quanta; P, release probability; MK-801, 5H-dibenzo[a,d]cyclohepten-5,10-imine.
0026-895X/06/6903-846 –856$20.00
MOLECULARPHARMACOLOGY Vol. 69, No. 3
Copyright © 2006 The American Society for Pharmacology and Experimental Therapeutics 18093/3084530
Mol Pharmacol 69:846–856, 2006 Printed in U.S.A.
synapses (Chavez-Noriega and Stevens, 1994; Weisskopf et al., 1994; Colwell and Levine, 1995; Salin et al., 1996). This effect of cAMP, together with that on cAMP response ele-ment-binding protein, is also believed to underlie long-term potentiation of synaptic efficacy and memory consolidation (Bailey et al., 1996). However, PKA-independent actions of cAMP, which enhance the release of transmitters and hor-mones, have been reported recently. For example, at the crayfish neuromuscular junction, cAMP activates presynap-tic exchange protein activated by cAMP (Epac) and hyperpo-larization and cyclic nucleotide-activated (HCN) channels, thereby increasing transmitter release (Beaumont and Zucker, 2000; Zhong and Zucker, 2005). At the calyx of Held, cAMP facilitates transmitter release via activating the Epac pathway in the nerve terminal (Kaneko and Takahashi, 2004). In pancreatic cells, the cAMP-sensitive guanine nucle-otide-exchanging factor may interact with Rim2, thereby en-hancing insulin secretion (Ozaki et al., 2000; Kashima et al., 2001). In addition, another study has revealed that elevated cAMP levels can potentiate the neuroprotective activity of the noradrenaline on dopamine neurons through the activa-tion of the MAPK signaling pathway (Troadec et al., 2002). These findings indicate that cAMP may operate a wide vari-ety of targets to exert its cellular functions.
Recent studies support a role for cAMP-regulated signaling in the cognitive function of the PFC. For instance, Aujla and Beninger (2001) have shown that cAMP/PKA inhibition in the PFC immediately before testing impaired working mem-ory performance when long delays were used. However, an-other study found a dose-dependent impairment of working memory performance on the delayed-alternation task when the cAMP analog Sp-cAMPS was infused into the PFC of young rats (Arnsten et al., 2005). A recent study has also described an inhibitory effect of cAMP/PKA signaling on inwardly retifying K⫹conductance of mPFC neurons (Dong and White, 2004). At present, however, very little is known about the molecular mechanisms by which cAMP regulates the glutamatergic synaptic transmission in the mPFC. In this study, we have demonstrated the first evidence that cAMP presynaptically facilitates synaptic transmission on the layer V pyramidal neurons of mPFC through activating two parallel signaling events, one involving PKA, and the other involving p42/p44 MAPK.
Materials and Methods
Slice Preparation. All experiments were performed according to the guidelines laid down by the Institutional Animal Care and Use Committee of National Cheng Kung University (Tainan, Taiwan). The coronal brain slices containing the prelimbic area PFC (1.5–2.5 mm anterior to bregma) were prepared from 8- to 23-day-old male Sprague-Dawley rats for whole-cell, patch-clamp recordings using procedures described previously (Hirsch and Crepel, 1990). Most experiments (unless otherwise noted) were performed on 14- to 16-day-old rats. In brief, rats were killed by decapitation under halo-thane anesthesia, and coronal slices (200m) were prepared using a vibrating microtome (Leica VT1000S; Leica, Nussloch, Germany). The anterior cingulated cortex and the shoulder or frontal area 2 region of the frontal cortex (Paxinos and Watson, 1998) were used for recording. The slices were placed in a storage chamber of artificial CSF (ACSF) oxygenated with 95% O2/5% CO2 and kept at room temperature for at least 1 h before recording. The composition of the ACSF solution was 117 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2
mM MgCl2, 25 mM NaHCO3, 1.2 mM NaH2PO4, and 11 mM glucose at pH 7.3 to 7.4 and equilibrated with 95% O2/5% CO2.
Electrophysiological Recordings. For whole-cell, patch-clamp recording, one slice was transferred to a recording chamber of stan-dard design and fixed at the glass bottom of the chamber with a nylon grid on a platinum frame. The chamber consisted of a circular well of low volume (1–2 ml) and was perfused constantly at 32.0⫾ 0.5°C with a speed of 2 to 3 ml/min. Visualized whole-cell patch-clamp recording of synaptically evoked EPSCs and miniature EPSCs (mEPSCs) was conducted using standard methods as described pre-viously (Huang et al., 2002). The layer II–III or V pyramidal neurons were identified by their pyramidal shape, presence of a prominent apical dendrites, and distance from the pial surface with an upright microscope (Olympus BX50WI; Olympus, Tokyo, Japan) equipped with a water-immersion ⫻40 objective lens and a Nomarski con-denser combined with infrared videomicroscopy. Patch pipettes were pulled from borosilicate capillary tubing and heat-polished. The elec-trode resistance was typically 3 to 6 M⍀. The composition of intra-cellular solution was 115 mM potassium gluconate, 20 mM KCl, 10 mM HEPES, 2 mM MgCl2, 0.5 mM EGTA, 3 mM Na2ATP, 0.3 mM Na3GTP, 5 mM QX-314, and sucrose to bring osmolarity to 290 to 295 mOsM and pH to 7.3.
After a high-resistance seal (⬎2 G⍀ before breaking into whole-cell mode) was obtained, suction was applied lightly through the pipette to break through the membrane. The cell was then main-tained at ⫺70 mV for several minutes to allow diffusion of the internal solution into the cell body and dendrites. Recordings were made using an Axopatch 200B (Molecular Devices, Sunnyvale, CA) amplifier. Electrical signals were low-pass-filtered at 3 kHz, digi-tized at 10 kHz using a 12-bit analog-to-digital converter (Digidata 1200; Molecular Devices). An Intel Pentium-based computer with pCLAMP software (version 8.0; Molecular Devices) was used for online acquisition and offline analysis of the data. For measurement of synaptically evoked EPSCs, a bipolar stainless steel stimulating electrode was placed on layer II–III approximately 150 to 200m away from the apical dendrites of the recorded neurons (Fig. 1A) and the superfusate routinely contained bicuculline methiodide (10M) to block inhibitory synaptic responses. The strength of synaptic transmission was mostly quantified by measuring the initial rising slope of EPSC (2-ms period from its onset; picoamperes per millisec-ond), which contains only a monosynaptic component. In some ex-periments, synaptic currents were recorded in the presence of NMDA receptor antagonistD-APV (50M) or AMPA/kainate receptor an-tagonist CNQX (20M), and the amplitude was measured over a 0.5-to 2-ms window concentrated around the peak. Series resistance (Rs) was calculated according to the equation Rs⫽ 10 mV/I, where I was the peak of transient current (filtered with 10 kHz) evoked by the 10-mV testing pulse when the pipette capacitance was compensated fully. Only cells demonstrating⬍25 M⍀ series resistance (usually 10–20 M⍀) were used in these experiments.
Miniature EPSCs were recorded from layer V pyramidal neurons held in voltage clamp at a potential of⫺70 mV in the presence of bicuculline methiodide (10M), tetrodotoxin (1 M), and CdCl2(100 M) and analyzed offline using a commercially available software (Mini Analysis 4.3; Synaptosoft, Leonia, NJ). mEPSCs were abol-ished by CNQX (20M) plusD-APV (50M), indicating that these are glutamatergic events. The software detects events based on amplitudes exceeding a threshold set just above the baseline noise of the recording (3 pA). All detected events were re-examined and accepted or rejected based on subjective visual examination. The program then measured amplitudes and intervals between succes-sive detected events. Background current noise was estimated from the baseline with no clear event and was subtracted from signals for analysis. The mEPSC frequencies were calculated by dividing the total number of detected events by the total time sampled. Periods of 5 to 10 min were analyzed for forskolin treatment. Events were ranked by amplitude and interevent interval for preparation of
cu-mulative probability distribution. Amplitude histograms were binned in 1-pA intervals.
Drug Application. All drugs were applied by manually switching the superfusate. Drugs were diluted from stock solutions just before application. Forskolin, 1,9-dideoxy-forskolin (Dd-forskolin), KT5720, H-89, PD98059, U0126, SB203580, SP600125, and brefeldin A were dissolved in dimethyl sulfoxide stock solutions and stored at⫺20°C until the day of experiment. Other drugs used in this study were dissolved in distilled water. The concentration of dimethyl sulfoxide in the perfusion medium was 0.1%, which alone had no effect on basal synaptic transmission. Forskolin, isoproterenol, propranolol, 8-(4-chlorophrnylthio)-2⬘-O-methyl-cAMP (8CPT-2Me-cAMP), aden-osine 3⬘,5⬘-cyclic monophosphorothioate, Sp-isomer (Sp-cAMPS) triethylammonium salt, ZD7288, PD98059, U0126, SB203580, SP600125, CNQX, bicuculline methiodide, and D-APV were
pur-chased from Tocris Cookson (Bristol, UK); Dd-forskolin, brefeldin A, strychnine hydrochloride, and tetrodotoxin were obtained from Sigma (St. Louis, MO); H-89 was purchased from Calbiochem (La Jolla, CA).
Statistical Analysis. The data for each experiment were normal-ized relative to baseline and are presented as means⫾ S.E.M. The numbers of experiments are indicated by n. The significance of the difference between the mean was calculated by paired or unpaired Student’s t test. Probability values (p) of less than 0.05 were consid-ered to represent significant differences. Comparisons between con-trol and experimental distributions of mEPSCs amplitude and inter-event intervals were made by performing a Kolmogorov-Smirnov
test. Distributions were considered different using a conservative critical probability level of p⬍ 0.01.
Results
A photograph of a slice used for recording is shown in Fig. 1A. The area used for recording is outlined by a circle. Figure 1B shows an infrared microscope image of a representative layer V pyramidal neuron. The results of the present study were obtained from 176 pyramidal neurons. These neurons had a mean resting membrane potential, spike height, and input resistance of ⫺68.7 ⫾ 0.8 mV, 93.7 ⫾ 2.3 mV, and 286⫾ 29 M⍀ (n ⫽ 62), respectively, which are comparable with the values reported previously (Wang and O’Donnell, 2001). In all experiments, layer V pyramidal neurons were held at a holding potential of ⫺70 mV, and EPSCs were evoked by the stimulation of layer II–III fibers with bipolar-stimulating electrodes every 20 s in the presence of GABAA
receptor antagonist bicuculline methiodide (10M). Because EPSCs were completely blocked by CNQX (20 M) plus D -APV (50M) (Fig. 1C), they were predominantly mediated by ionotropic glutamate receptors.
Potentiation of EPSCs by cAMP. We initially examined
the effect of the adenylyl cyclase activator, forskolin, on the evoked EPSCs. Typical responses are shown in Fig. 2A. Bath application of forskolin (25M) produced a rapid and sus-tained enhancement of evoked EPSCs on layer V pyramidal neurons. The mean EPSC slope measured 20 min after fors-kolin application was increased by 53.6⫾ 5.8% of the control baseline (n⫽ 8; p ⬍ 0.05, paired Student’s t test). No signif-icant recovery was visible after drug washout of at least 20-min intervals. The magnitude of EPSC potentiation by forskolin was concentration-dependent (Fig. 2B), with an estimated EC50value of 21M. Because forskolin has been
reported to possess many cAMP-independent actions, includ-ing the blockade of several types of K⫹ conductance, it is possible that the effect of forskolin on EPSCs is caused by its nonspecificity (Laurenza et al., 1989). To exclude this possi-bility, an analog of forskolin, Dd-forskolin, which has no effect on adenylyl cyclase but does mimic many of the cAMP-independent actions of forskolin, was used. As shown in Fig. 2A, Dd-forskolin (25M) had no significant effect on EPSCs (4.2⫾ 2.3%; n ⫽ 4; p ⬎ 0.05, paired Student’s t test). As on layer V pyramidal neurons, forskolin (25M) also increased the slope of EPSCs of layer II–III pyramidal neurons. The mean EPSC slope 20 min after forskolin application was increased by 48.7⫾ 6.9% of the control baseline (n ⫽ 5; p ⬍ 0.05, paired Student’s t test) (Fig. 2C). Moreover, the facili-tatory effect of forskolin on the layer V pyramidal neurons was robust at P8-P10 but became weaker as animal matu-rated (Fig. 2D), with the magnitude of potentiation by fors-kolin (25M) being 82.5 ⫾ 7.8% at P8–P10 (n ⫽ 5), 53.6 ⫾ 5.8% at P14–P16 (n⫽ 10), and 32.8 ⫾ 4.2% at P21–P23 (n ⫽ 5), indicating that the enhancement of synaptic transmission by forskolin is age-dependent. In the present study, forskolin was not observed to significantly change the holding current under voltage-clamp conditions (16.2⫾ 2.2 pA for before and 19.3⫾ 3.1 pA for 20 min after forskolin application; n ⫽ 35;
p⬎ 0.05, paired Student’s t test).
Additional evidence that elevation of cAMP levels can in-duce a synaptic potentiation of glutamatergic transmission on the layer V pyramidal neurons of mPFC came from
exper-Fig. 1. Pharmacological properties of EPSCs. A, photomicrograph of a
coronal slice used for recording. The circle indicates the area typical used for recording. The location of the stimulation (S) and recording electrode (R) is also shown. B, an infrared microscope image of a representative layer V mPFC pyramidal neuron (N) in a slice from a 14-day-old rat. The patch recording pipette (P) can be seen on the right. C, averaged EPSCs (five consecutive sweeps) evoked in the layer V pyramidal neurons of mPFC by stimulating the layer II–III at 0.05 Hz in the presence of bicuculline methiodide (10M). Perfusing the slices with NMDA receptor antagonist D-APV (50M) significantly attenuated the duration and amplitude of EPSCs and further addition of the AMPA/kainate receptor antagonist CNQX (20M) completely abolished the synaptic currents.
iments using the nonhydrolyzable cAMP analog Sp-cAMPS. Similar to forskolin, bath application of Sp-cAMPS (100M) for 20 min produced a significant enhancement of EPSCs by 42.5⫾ 5.7% of the control baseline (n ⫽ 5; p ⬍ 0.05, paired Student’s t test) (Fig. 3A).
Forskolin Enhances Synaptic Transmission through Converging Activation of PKA and p42/p44 MAPK Sig-naling Pathways. An established signal transduction
path-way for forskolin action is the activation of adenylyl cyclase, leading to an increase in intracellular cAMP levels and acti-vating PKA, which in turn modulates the function of a series of cellular substrates by increasing their phosphorylation state. We first examined whether the cAMP-dependent syn-aptic potentiation is mediated by PKA activation. To test this possibility, two structurally unrelated PKA inhibitors, KT5720 and H-89, were used. In the presence of either of KT5720 (1 M) or H-89 (1 M), forskolin was still able to potentiate EPSCs but the magnitude of potentiation was significantly smaller than that induced in the interleaved control condition (p⬍ 0.05; unpaired Student’s t test). On average, EPSC slope measured 20 min after forskolin appli-cation was increased by 38.4⫾ 4.2% (n ⫽ 8) in the presence of KT5720 and 36.6⫾ 4.1% (n ⫽ 5) in the presence of H-89 (Fig. 3B). However, neither KT5720 nor H-89 alone had effects on EPSCs (Supplemental Fig. S1A). These results suggest that in addition to the PKA signaling cascade, other signal mechanisms are necessary for the induction of synap-tic potentiation by forskolin.
At the calyx of Held, an increase in cAMP levels in the nerve terminal can facilitate transmitter release via the ac-tivation of Epac pathway (Kaneko and Takahashi, 2004). We next examined whether the cAMP-dependent synaptic poten-tiation seen in this study is mediated by Epac activation. To test this possibility, we used a selective Epac agonist, 8CPT-2Me-cAMP, which has been shown to have little effect on PKA (Enserink et al., 2002). To identify the saturating concentration of 8CPT-2Me-cAMP for activating Epac by ex-ternal bath application, we treated slices with varying con-centrations of 2Me-cAMP. As shown in Fig. 3C, 8CPT-2Me-cAMP induced a concentration-dependent potentiation
of EPSCs, and the magnitudes of potentiation caused by 50 and 100M 8CPT-2Me-cAMP were not significantly differ-ent (Fig. 3C). Thus, 50M 8CPT-2Me-cAMP was chosen to examine whether forskolin- and 8CPT-2Me-cAMP-induced synaptic potentiation use a similar induction mechanism. The approach used to address this question is to determine whether the induction of one form of potentiation reduces or occludes the induction of the other form of potentiation. As shown in Fig. 3D, after 8CPT-2Me-cAMP-induced potentia-tion was fully established, applicapotentia-tion of forskolin still caused synaptic potentiation. On average, EPSC slope measured 20 min after forskolin application was increased by 51.3⫾ 4.6% (n⫽ 5), which was not significantly different from that found in slices without receiving 8CPT-2Me-cAMP. Because the small G-protein antagonist brefeldin A has recently been shown to effectively antagonize 8CPT-2Me-cAMP’s action on synaptic transmission at the crayfish neuromuscular junc-tions (Zhong and Zucker, 2005), we then used brefeldin A to characterize the role of Epac activation in forskolin-induced synaptic potentiation. Brefeldin A alone (100 M) had no effect on basal synaptic transmission and did not affect for-skolin-induced EPSC potentiation (Fig. 3E). On average, EPSC slope measured 20 min after forskolin application was increased by 49.7⫾ 6.5% (n ⫽ 5), which was not significantly different from that of potentiation elicited under control con-dition. These results suggest that activation of Epac is not necessary for forskolin-induced synaptic potentiation in the layer V pyramidal neurons of mPFC.
To clarify the involvement of HCN channels in the induc-tion of forskolin-induced synaptic potentiainduc-tion, we compared the magnitude of synaptic potentiation in control slices with that obtained in slices preincubated in HCN channel blocker ZD7288. As shown in Fig. 3E, ZD7288 (30M) did not affect forskolin-induced EPSC potentiation (Fig. 3E). On average, EPSC slope measured 20 min after forskolin application was increased by 48.4⫾ 5.7% (n ⫽ 5), which was not significantly different from that of potentiation elicited under control con-ditions. Thus, these results rule out an involvement of HCN channels in the forskolin response.
In a number of systems, cAMP-induced MAPK activation
Fig. 2. Potentiation of EPSCs by forskolin. A, a
represen-tative experiment showing the time course of the action of forskolin and Dd-forskolin on the slope of evoked EPSCs on layer V mPFC pyramidal neuron. EPSCs were evoked ev-ery 20 s by a single pulse and were recorded from a holding potential of⫺70 mV. Each data point represents a single response evoked before, during, and after the application of forskolin or Dd-forskolin. Bath application of forskolin (25 M, E) potentiated EPSCs, whereas the inactive analog Dd-forskolin (25M, F) had no such effect. Sample traces are averages of five consecutive EPSCs recorded at times indicated by the numbers on the graph. Horizontal bars denote the period of delivery of forskolin or Dd-forskolin. B, the concentration-dependence of forskolin effect. Ordinate indicates the percentage of EPSC potentiation measured 20 min after forskolin application. Data are derived from 4 to 10 cells. C, a representative experiment showing the time course of the action of forskolin and Dd-forskolin on the slope of evoked EPSCs on layer II–III mPFC pyramidal neuron at a holding potential of⫺70 mV. Bath application of forskolin (25M, E) potentiated EPSCs, whereas the inactive analog Dd-forskolin (25M, F) had no such effect. D, EPSC potentiation induced by forskolin (25 M) at different postnatal ages. Data are derived from 5 to 10 cells. The potentiation at P21–P23 was significantly smaller than that at P8 –P10.
independently of PKA has been reported (Iacovelli et al., 2001; Troadec et al., 2002). To determine whether the cAMP-dependent synaptic potentiation is mediated by MAPK activation, forskolin-induced synaptic potentiation was attempted in the presence of multiple types of MAPK inhib-itors. As shown in Fig. 3F, forskolin response was strongly reduced by p42/p44 MAPK signaling pathway inhibitors, PD98059 and U0126. On average, EPSC slope measured 20 min after forskolin application was increased by 18.6⫾ 3.5% in the presence of PD98059 (50M) (n ⫽ 8; p ⬍ 0.05 com-pared with forskolin alone, unpaired Student’s t test) and 15.3⫾ 3.4% in the presence of U0126 (10M) (n ⫽ 6; p ⬍ 0.05 compared with forskolin alone, unpaired Student’s t test). Neither PD98059 nor U0126 alone had effects on EPSCs (Supplemental Fig. S1B). In contrast, inhibition of p38 MAPK signaling pathway with SB203580 (1M) or c-Jun N-terminal kinase inhibitor SP600125 (20M) failed to af-fect the forskolin-induced synaptic potentiation (SB203580, 56.3⫾ 4.6%, n ⫽ 4; SP600125, 51.6 ⫾ 3.9%, n ⫽ 4) (Fig. 3H). In another experiment, we found that forskolin failed to potentiate EPSCs when H-89 and PD98059 or H-89 and U0126 were applied together (H-89⫹ PD98059, 2.8 ⫾ 1.5%,
n⫽ 5; H-89 ⫹ U0126, 2.4 ⫾ 1.3%, n ⫽ 4) (Fig. 3, G and H),
suggesting that cAMP elevated by forskolin activates both PKA and p42/p44 MAPK signaling pathways to induced syn-aptic potentiation.
Presynaptic Expression of Forskolin-Induced Syn-aptic Potentiation. To dissect the synSyn-aptic site of action for
forskolin, the effects of forskolin on the AMPA and NMDA receptor-mediated component of synaptic currents were amined. If forskolin-induced synaptic potentiation were ex-pressed presynaptically, changes in both of the magnitude of AMPA receptor-mediated EPSC (EPSCAMPA) and NMDA
re-ceptor-mediated EPSC (EPSCNMDA) by forskolin would be
expected. The EPSCAMPA was recorded from the layer V
pyramidal neurons in the presence of NMDA receptor antag-onistD-APV (50M) at a holding potential of ⫺70 mV, and
the EPSCNMDAwas recorded in the presence of
AMPA/kai-nate receptor antagonist CNQX (20M) at a holding poten-tial of ⫹40 mV to remove the voltage-dependent block of Mg2⫹. As illustrated in Fig. 4, A and B, forskolin (25 M)
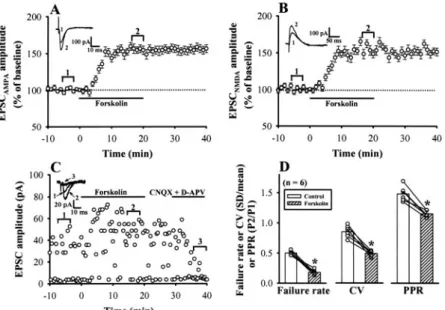
increased the amplitude of EPSCAMPAby 51.4⫾ 5.6% (n ⫽ 5)
of control baseline. Comparable results were obtained with EPSCNMDA(48.9⫾ 5.2% of control baseline, n ⫽ 5).
Fig. 3. Coapplication of PKA and p42/p44 MAPK inhibitors
blocks the forskolin-induced EPSC potentiation. A, sum-mary of experiments (n⫽ 5) showing that bath application of nonhydrolyzable cAMP analog Sp-cAMPS increased EPSCs. B, summary of experiments showing that KT5720 (1 M, n ⫽ 8; E) or H-89 (1 M, n ⫽ 5; F) treatment partially blocked the forskolin-induced synaptic potentia-tion. C, summary of experiments showing that bath appli-cation of Epac agonist 8CPT-2Me-cAMP (10 –100M) en-hanced EPSCs in a concentration-dependent manner. Data are derived from four to six cells. D, summary of experi-ments (n⫽ 5) showing that treatment of 8CPT-2Me-cAMP did not affect the forskolin-induced synaptic potentiation. E, summary of experiments showing that forskolin-induced synaptic potentiation was not significantly affected by prior treatment with either brefeldin A (BFA, 100M; n ⫽ 5; E) or ZD7288 (30M; n ⫽ 5; F). F, summary of experi-ments showing that PD98059 (50M, n ⫽ 8; E) or U0126 (10M, n ⫽ 6; F) treatment significant reduced the fors-kolin-induced synaptic potentiation. G, summary of exper-iments showing that forskolin-induced EPSC potentiation was completely abolished by prior coapplication of H-89 (1 M) and PD98059 (50 M) (n ⫽ 5, E) or H-89 (1 M) and U0126 (10M) (n ⫽ 4, F). Sample traces are averages of five consecutive EPSCs recorded at time indicated by the numbers on the graph. H, histogram comparing the effects of different antagonists on the forskolin-induced synaptic potentiation. The magnitude of potentiation was measured 20 min after forskolin application. Data are taken from B, E, F, and G.ⴱ, significant difference from control group at p⬍ 0.05.
To further test the possibility that forskolin induces syn-aptic potentiation through a presynsyn-aptic mechanism, we ex-amined the effect of forskolin on the failure rate of single-fiber EPSCs evoked by minimal stimulation, which reflects changes in the presynaptic transmitter release (Stevens and Wang, 1994). As a typical example shown in Fig. 4C, the expression of forskolin-induced synaptic potentiation was ac-companied by a decrease in synaptic failure rate. On average, the failure rate was decreased from 0.51⫾ 0.03 to 0.18 ⫾ 0.03 after forskolin application (n⫽ 6; p ⬍ 0.05, paired Student’s
t test) (Fig. 4D). We also addressed the synaptic locus of
forskolin-induced synaptic potentiation by examining the trial-to-trial amplitude fluctuation in EPSCs with the vari-ance analysis. The CV value varies with quantal content but is independent of changes in the postsynaptic response to a fixed amount of transmitter and is a useful measure of changes in presynaptic function (Bekkers and Stevens, 1990). Because variance analysis is best done on unitary synaptic responses, our strategy was to carry out a variance analysis of unitary single-fiber EPSCs evoked by minimal stimulation before and after forskolin application. We found that after the induction of synaptic potentiation by forskolin, the value of CV for unitary EPSCs was decreased from 0.85⫾ 0.04 to 0.49⫾ 0.03 (n ⫽ 6; p ⬍ 0.05, paired Student’s t test) (Fig. 4D).
When the excitatory afferents to the central neurons are activated twice with a short interval between each stimulus, the response to the second stimulus is generally facilitated in relation to the initial stimulus. This phenomenon is called paired-pulse facilitation and is attributed to an increase in the amount of transmitter release to the second stimulus (Zucker, 1989). On the other hand, the manipulation of pre-synaptic transmitter release may result in the change in the magnitude of paired-pulse facilitation. If the forskolin-in-duced synaptic potentiation involves a presynaptic mecha-nism of action, it will be associated with a decrease in the ratio of paired-pulse (PPR). To test this hypothesis, PPR (using an interpulse interval of 50 ms) was determined before and during application of 25M forskolin for 20 min. Under control conditions, the ratio of the slope of the second EPSC divided by the first one was 1.48⫾ 0.04 (n ⫽ 6). We found
that forskolin significantly decreased the PPR to 1.15⫾ 0.05 (n⫽ 6; p ⬍ 0.05, paired Student’s t test) (Fig. 4D), suggesting an increase in glutamate release probability after forskolin application.
Forskolin Enhances Frequency of mEPSCs. To
fur-ther confirm the possibility that forskolin potentiates synap-tic transmission through a presynapsynap-tic mechanism, we ex-amined the effects of forskolin on mEPSCs in the presence of tetrodotoxin (1 M) and CdCl2 (100 M). mEPSCs in the
layer V pyramidal neurons were measured under voltage clamp at⫺70 mV and were pharmacologically isolated from spontaneous inhibitory currents by the inclusion of 10M bicuculline methiodide in the ACSF perfusing the slices. The mEPSCs were totally blocked by bath coapplication of CNQX (20 M) and D-APV (50 M), confirming them to be true
glutamate receptor-mediated events (data not shown). Under control conditions, mEPSCs had a mean amplitude of 5.98⫾ 0.23 pA and a variable frequency ranging from 1.9 to 2.7 Hz (mean, 2.13⫾ 0.19 Hz; n ⫽ 5). In five pyramidal neurons tested, forskolin (25M) markedly increased the mean fre-quency of the mEPSCs from 2.13⫾ 0.19 to 5.23 ⫾ 0.21 Hz (p⬍ 0.05, paired Student’s t test) (Fig. 5, A and F). Signifi-cant differences in cumulative interevent interval distribu-tions were observed in all five cells tested during forskolin application (i.e., forskolin shifted the interevent interval dis-tribution of mEPSCs to shorter intervals: p⬍ 0.01, Kolmog-orov-Smirnov test). A typical example of recorded cell is shown in Fig. 5D. However, there was no significant effect of forskolin (25 M) on the mEPSC amplitude. This can be observed by a lack of effect of forskolin on either the ampli-tude histogram (Fig. 5B) or the cumulative probability plots (Fig. 5C, p ⫽ 0.94; Kolmogorov-Smirnov test). The mean amplitude of mEPSCs recorded in the presence of forskolin (25M) was 6.21 ⫾ 0.19 pA, which was of comparable am-plitude with that of mEPSCs recorded under control condi-tions (5.98 ⫾ 0.23 pA; p ⫽ 0.78, paired Student’s t test). Therefore, these data further suggest that forskolin may act presynaptically to enhance the amount of glutamate release without changing the postsynaptic sensitivity to glutamate.
Forskolin Increases the Number of Releasable Vesi-cles and Release Probability. Although the above results Fig. 4. Forskolin increases both AMPA receptor- and
NMDA receptor-mediated EPSCs and decreases the failure rate, CV, and PPR. A, average time course of the effect of forskolin (25 M) on EPSCAMPA amplitude (n ⫽ 5). EPSCAMPAwas recorded every 20 s by a single pulse stim-ulation in the presence of NMDA receptor antagonist D -APV (50M) at a holding potential of ⫺70 mV. B, average time course of the effect of forskolin (25M) on EPSCNMDA amplitude (n⫽ 5). EPSCNMDAwas recorded in the presence of AMPA receptor antagonist CNQX (20M) at a holding potential of⫹40 mV. Sample traces are averages of five consecutive EPSCs recorded at the times indicated by the numbers on the graph. C, plot of EPSC amplitude recorded at⫺70 mV holding potential against time, before, during, and after bath application of forskolin for 20 min, evoked at 0.1 Hz by minimal stimulation. It is clear that forskolin produced a significant decrease in the number of failures. At the end of the experiment, CNQX (20M) andD-APV (50M) were applied to the bath to make sure that syn-aptic response was a glutamatergic EPSC. D, changes in failure rate, CV, and PPR associated with forskolin-in-duced synaptic potentiation (n⫽ 6). PPR was calculated from responses to paired-pulse stimulation with an inter-pulse interval of 50 ms.ⴱ, significant difference from con-trol group at p⬍ 0.05.
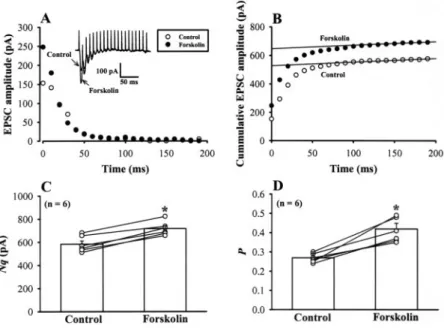
are consistent with a presynaptic site for the expression of forskolin-induced synaptic potentiation, they may be attrib-uted to a number of presynaptic mechanisms, including an increase in the number of readily releasable quanta (synaptic vesicles) (N) or an increase in release probability (P) (Trudeau et al., 1996; Kaneko and Takahashi, 2004). To determine which mechanism underlies the forskolin-induced synaptic potentiation, we first used the approach of high-frequency repetitive stimulation (20 stimuli at 100 Hz) (Schneggenburger et al., 1999), which induced a strong de-pression of EPSCs (Fig. 6A). In this approach, assuming that depression is largely caused by depletion of readily releasable quanta, N multiplied by mean quantal size (q) can be esti-mated from zero time intercept of a line fitted to a cumulative amplitude plot of EPSCs (Fig. 6B), and P can be estimated from the first EPSC amplitude divided by Nq. During stim-ulation at 100 Hz, EPSCs underwent a marked depression and reached a steady low level. Forskolin (25M) potenti-ated the first few EPSCs during a train of tetanic stimulation (Fig. 6A). Cumulative amplitude plot of EPSCs before and after forskolin application indicated that forskolin increased
Nq by 23.2⫾ 4.1% (n ⫽ 6; p ⬍ 0.05, paired Student’s t test)
(Fig. 6C) and P by 55⫾ 11% (n ⫽ 6; p ⬍ 0.05, paired Student’s
t test) (Fig. 6D). Given that forskolin had no significant effect
on q (Fig. 5E), these results suggest that forskolin increases both the number of readily releasable quanta and release probability.
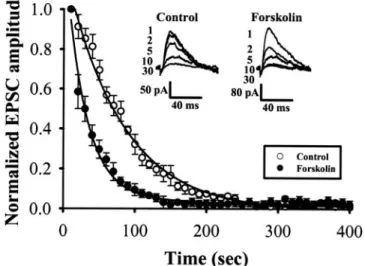
Another way of assessing the release probability is to eval-uate the speed of block of NMDA receptors by irreversible
open channel blocker MK-801 (Rosenmund et al., 1993). Re-peated activation of synapses in the presence of MK-801 results in a progressive decline in the amplitude of the NMDA receptor-mediated synaptic current, and the rate of decay depends on the probability of transmitter release at synapses. If the probability of transmitter release is higher, a larger proportion of postsynaptic NMDA receptors is blocked at any one time, resulting in a faster decline of EPSCNMDA
(Rosenmund et al., 1993). The EPSCNMDA was recorded in
the presence of CNQX (20M) and at a holding potential of ⫹40 mV. After confirming a stable baseline at a basal stim-ulus frequency of 0.1 Hz, the stimulation was stopped, and MK-801 (40M) was applied. Stimulation was restarted 10 min later, and the EPSCNMDAwas recorded in the
continu-ous presence of MK-801. From five such experiments, the time course of decline could be fitted by a double exponential function, with a fast time constant of 82 s and a slow time constant of 186 s. In the presence of forskolin (25M), the fast and slow time constants were 31 and 285 s, respectively (Fig. 7). Forskolin also apparently increased the proportion of the fast component. Because P is inversely proportional to the decay time constant in the MK-801 experiments (Rosen-mund et al., 1993), these results further support the proposal that forskolin increases P.
-Adrenergic Receptor Agonist Isoproterenol
Poten-tiates EPSCs. The final test was to determine whether
activation of receptors that are positively coupled to elevate cAMP can mimic forskolin to potentiate synaptic transmis-sion on the layer V pyramidal neurons of mPFC. The
-ad-Fig. 5. Effects of forskolin on mEPSCs. A, sample traces
(three traces superimposed) of mEPSCs before (baseline, in the presence of 1M tetrodotoxin to block Na⫹channels and 100M CdCl2to block voltage-dependent Ca
2⫹ chan-nels) and after application of forskolin (25 M). Lower traces are the averaged mEPSCs of 25 events each before and after forskolin application with increasing time reso-lution, demonstrating the lack of effect on the amplitude and kinetics of mEPSCs. B1and B2, amplitude histograms of mEPSCs. The threshold for peak detection was set at⫺3 pA. Data were binned in 1 pA intervals. C, cumulative probability plots of mEPSCs before (solid line) and during (broken line) application of forskolin (p⫽ 0.94; Kolmog-orov-Smirnov test). D, cumulative interevent interval dis-tribution illustrating a significant decrease in the inter-event interval (i.e., increased frequency; p ⬍ 0.01, Kolmogorov-Smirnov test) during forskolin application. E and F, summary of the effect of forskolin (25M) on the average amplitude and frequency of mEPSCs (n⫽ 5). Data are presented as means⫾ S.E.M. ⴱ, significant difference from baseline at p⬍ 0.05. The data shown in A to D were taken from the same cell. Holding potential,⫺70 mV.
renergic receptor agonist isoproterenol has been shown to mimic the enhancement effect of cAMP elevation on synaptic transmission of many brain regions (Herrero and Sa´nchez-Prieto, 1996; Huang et al., 1996). Although the action of -adrenergic receptors on the glutamatergic transmission of the mPFC region has not been established,-adrenergic re-ceptors have been shown to mediate many noradrenaline functions in the mPFC (Bing et al., 1992), and noradrenaline has been reported to facilitate the release of glutamate from presynaptic terminals that synapse onto layer V pyramidal neurons of mPFC (Marek and Aghajanian, 1999). Thus, we conducted a series of experiments to test the hypothesis that activation of -adrenergic receptors would induce a cAMP-mediated synaptic potentiation. As shown in Fig. 8A, bath application of isoproterenol (15M) for 20 min induced EPSC potentiation by 43.5⫾ 4.3% of the control baseline (n ⫽ 5; p ⬍ 0.05, paired Student’s t test). The response to isoproterenol was completely blocked by propranolol (20M), a selective -adrenergic receptor antagonist, suggesting that this effect is indeed mediated by the activation of -adrenergic recep-tors (Fig. 8B). In addition, isoproterenol was still able to potentiate EPSCs in the presence of H-89 (1 M), but the magnitude of potentiation was significantly smaller than that induced in the interleaved control condition (p⬍ 0.05, unpaired Student’s t test) (Fig. 8C). On average, EPSC slope measured 20 min after isoproterenol application was in-creased by 28.5 ⫾ 3.5% (n ⫽ 5) in the presence of H-89. Coapplication of H-89 (1 M) and PD98059 (50 M) com-pletely blocked isoproterenol-induced EPSC potentiation (n⫽ 5; 2.5 ⫾ 1.2% of preisoproterenol baseline) (Fig. 8D). These data are consistent with the hypothesis that the en-hancement action of-adrenergic receptor activation on glu-tamatergic transmission in the mPFC is mediated by both PKA and p42/p44 MAPK signaling cascades.
Discussion
We have, for the first time, systematically examined the role of cAMP elevation in the regulation of synaptic trans-mission on layer V pyramidal neurons of mPFC. Our results indicate that forskolin and membrane-permeable cAMP
an-alogs induce synaptic potentiation through presynaptic mechanisms. Most importantly, we provide pharmacological evidence that cAMP acts on both PKA and p42/p44 MAPK signaling pathways to enhance transmitter release from pre-synaptic nerve terminal. Furthermore, activation of -adren-ergic receptor with isoproterenol mimics forskolin and elicits a cAMP-dependent synaptic potentiation.
Presynaptic Locus of Expression of cAMP-Depen-dent Synaptic Potentiation. Various approaches were
taken to determine the site of action of forskolin in enhancing transmission on layer V pyramidal neurons of mPFC. Based on these experiments, it is likely that forskolin-induced syn-aptic potentiation is primarily of presynsyn-aptic origin. Three lines of evidence support this conclusion. First, forskolin increases equally the AMPA and NMDA receptor-mediated component of EPSCs (Fig. 4, A and B). Second, the increase in synaptic transmission by forskolin was accompa-nied by a decrease in the synaptic failure rate, magnitude of CV, and PPR (Fig. 4D), which are generally considered to indicate a presynaptic mode of drug action (Zucker, 1989; Bekkers and Stevens, 1990; Stevens and Wang, 1994). Third, forskolin significantly increased the frequency of mEPSCs but did not affect the amplitude of mEPSCs (Fig. 5). A change in the amplitude of mEPSCs has traditionally been inter-preted as a postsynaptic modification, whereas a change in their frequency is typically associated with mechanisms that increase the probability of transmitter release. Thus, the lack of effect of forskolin on the amplitude of mEPSCs also implies that forskolin-induced synaptic potentiation is not mediated by a change in postsynaptic sensitivity to glutamate.
Forskolin Increases the Number of Releasable Vesi-cles and Release Probability. Using three independent
approaches, the paired-pulse stimulation, high-frequency stimulation, and MK-801 protocols, we have shown that for-skolin increases the release probability, P. The high-fre-quency stimulation protocol also indicates that forskolin in-creases the number of releasable vesicles, N. In cerebellar parallel-Purkinje cell synapses, the effect of forskolin has been attributed primarily to an increase in P (Chen and Regehr, 1997). At the calyx of Held, forskolin has also been
Fig. 6. Forskolin increases both the number of releasable
vesicles and release probability. A, depressions of EPSCs during trains (20 stimuli) of 100-Hz stimulation before (control, E) and after application of forskolin (25M, F). Sample records of the EPSCs before and after forskolin application are shown (superimposed) in the inset. B, cu-mulative amplitudes of EPSCs during the 100-Hz train before (E) and after forskolin application (F). Amplitudes of EPSCs from 16 to 20th were fitted with a linear regression line and extrapolated to time 0 for estimating the readily releasable pool size. C, mean number of releasable vesicles (N) multiplied by q significantly increased after forskolin application (n ⫽ 6). D, the mean release probability (P), which was estimated from the ratio of the first EPSC amplitude divided by Nq, underwent a significant increase after forskolin application (n⫽ 6). ⴱ, significant difference from control group at p⬍ 0.05.
proposed to facilitate transmitter release by increasing both
P and N (Kaneko and Takahashi, 2004). cAMP also regulates
release from dentate granule cells in the hippocampus by changing both P and N (Weisskopf et al., 1994). The fit of a double exponential function to the data in MK-801 experi-ments predicts vesicle populations having different release probability (Rosenmund et al., 1993). The finding that fors-kolin selectively accelerated the fast decay time constant and increased the relative proportion of the fast-decaying compo-nent suggests that forskolin increases P and the proportion of vesicles with high P as reported previously (Kaneko and Takahashi, 2004).
Molecular Mechanism of cAMP-Mediated Synaptic Potentiation. Forskolin has been reported to possess many
cAMP-independent actions, including the blockade of several
types of potassium currents, which could result in prolonga-tion of presynaptic acprolonga-tion potentials and consequent increase in transmitter release (Hoshi et al., 1988). However, the cAMP-independent action of forskolin could be mimicked by its analog Dd-forskolin, which is unable to activate adenylyl cyclase. In our experiments, Dd-forskolin had no significant effect on EPSCs (Fig. 2, A and C). Thus, the effect of forskolin is not caused by its nonspecificity. This idea was also sup-ported by the finding that nonhydrolyzable cAMP analog Sp-cAMPS mimicked forskolin to potentiate EPSCs (Fig. 3A). Consistent with this idea, we have found that the activation of-adrenergic receptors that are coupled to Gsproteins and
activation of cAMP-dependent signaling pathways also elicit a cAMP-mediated synaptic potentiation (Fig. 8).
What is the molecular target of cAMP? Previous studies have shown that cAMP-dependent synaptic potentiation is mediated mainly by activating PKA in a variety of brain regions, including the hippocampus, amygdala, cerebellum, and striatum (Chavez-Noriega and Stevens, 1994; Huang et al., 1996, 2002; Salin et al., 1996; Chen and Regehr, 1997). In contrast, at the calyx of Held, presynaptic cAMP is proposed to facilitate synaptic transmission via activating Epac path-way (Kaneko and Takahashi, 2004). The cAMP-dependent potentiation of synaptic transmission at crayfish glutamater-gic neuromuscular junctions is mediated by acting on Epac and HCN (Zhong and Zucker, 2005). However, we found that forskolin still caused synaptic potentiation when forskolin was applied in the presence of PKA, Epac, or HCN inhibitors. Furthermore, although the application of a selective Epac agonist 8CPT-2Me-cAMP potentiated EPSCs, it did not oc-clude the subsequent forskolin-induced synaptic potentia-tion. However, antagonists of PKA and p42/p44 MAPK each reduced forskolin-induced synaptic potentiation, and to-gether they almost fully abolished the potentiation. Our find-ings that the effects of PKA and p42/p44 MAPK activation seem to be additive, suggesting the possibility that coincident activation of these two signaling pathway is required for the induction of cAMP-dependent synaptic potentiation on layer V pyramidal neurons of mPFC. The biological step down-stream of PKA and p42/p44 MAPK responsible for the cAMP-induced synaptic potentiation remains to be determined.
Fig. 7. Forskolin accelerates the time course of blocking NMDA
receptor-mediated EPSC (EPSCNMDA) by MK-801. EPSCNMDAwas evoked at 0.1 Hz at a holding potential of⫹40 mV in the presence of MK-801 (40M). Data are derived from a different group of cells, one group in the presence of forskolin (25M; F, n ⫽ 5) and the other in its absence (E, n ⫽ 5). The numbers of sample traces (superimposed) indicate the sequence of stim-ulation. Ordinate indicates the amplitude of EPSCNMDAnormalized to the initial amplitude. Abscissa indicates the time after starting stimula-tion in the presence of MK-801. Mean relative amplitudes derived each from five cells were fitted with double exponential functions.
Fig. 8.-Adrenergic receptor agonist isoproterenol enhances
synaptic transmission. A, summary of experiments (n⫽ 5) showing that bath application isoproterenol (15M) for 20 min increased EPSCs. B, summary of experiments (n⫽ 4) showing that isoproterenol-induced EPSC potentiation was completely abolished by prior treatment with-adrenergic receptor antagonist propanolol (20 M). C, summary of experiments showing that H-89 (1 M, n ⫽ 5) treatment partially blocked the isoproterenol-induced synaptic potenti-ation. D, summary of experiments showing that isoprotere-nol-induced synaptic potentiation was completely abolished by prior coapplication of H-89 (1M) and PD98059 (50 M, n⫽ 5). Sample traces are averages of five consecutive EPSCs recorded at the times indicated by the numbers on the graph.
Given that forskolin increases both N and P, the target of these kinases seems to be in both the vesicular trafficking mechanism and the exocytotic mechanism. Indeed, at many synapses, activation of PKA has been shown to phosphory-late one or more proteins, either associated with or part of the protein complex that is necessary for the exocytosis of syn-aptic vesicles and underlies synsyn-aptic facilitation (Nagy et al., 2004). Activation of p42/p44 MAPK was also reported to facilitate glutamate release from rat brain synaptosomes by phosphorylating the synaptic vesicle membrane protein syn-apsin I, thereby regulating its interaction with the actin cytoskeleton, leading to the recruitment of releasable synap-tic vesicles from a distal pool (Jovanovic et al., 2000).
We were surprised to find that that the facilitatory effect of forskolin decreased with postnatal development (Fig. 2D). This developmental decline of forskolin-induced synaptic po-tentiation is not unique to excitatory afferents to layer V pyramidal neurons of mPFC; it was also reported at the calyx of Held (Kaneko and Takahashi, 2004). Although the molec-ular mechanism underlying this phenomenon remains un-clear, a developmental decrease in the molecular target downstream of PKA and/or p42/p44 MAPK seems to contrib-ute to this phenomenon. Further work, involving the use of functional knockout of candidate proteins, is needed to assess this hypothesis.
Recent results suggest that p42/p44 MAPK inhibitors have nonspecific effects in modulating glutamate release. Pereira et al. (2002) showed that PD98059 inhibited glutamate re-lease from hippocampal synaptosomes stimulated with KCl when used at concentrations that inhibited p42/p44 MAPK activity. U0126, however, did not significantly affect KCl-induced glutamate release at concentrations shown to inhibit p42/p44 MAPK activity. Thus, the nonspecific effects of p42/ p44 MAPK inhibitors may mask the forskolin response. This possibility, however, was ruled out by our observations that both PD98059 and U0126 were effective to block forskolin-induced synaptic potentiation. In addition, U0126 was also reported to block forskolin-induced increase in p42/p44 MAPK activation in hippocampal area CA1 (Selcher et al., 2003).
Physiological Significance. What is the physiological
significance of cAMP-dependent synaptic potentiation in the mPFC? It was reported recently that PKA inhibitor can at-tenuate the induction of long-term potentiation in the mPFC (Huang et al., 2004). Thus, our findings might be implicated in the induction of long-term synaptic plasticity in the mPFC. The pyramidal neurons within the deep layers of mPFC integrate multiple excitatory and inhibitory inputs and send projections to many other brain regions (Heidbreder and Groenewegen, 2003). Through this network, the mPFC guides complex cognitive responses, such as working memory and the planning and execution of goal-directed behaviors (Goldman-Rakic, 1995; Fuster et al., 2000). Thus, potentia-tion of mPFC glutamatergic transmission by elevapotentia-tion of cAMP levels would expect to lead to alter these cognitive functions. Consistent with this prediction, infusion of the cAMP analog Sp-cAMPS into the PFC of young rats has been shown to produce a dose-dependent impairment in working memory performance on the delayed-alternation task (Arn-sten et al., 2005). However, Aujla and Beninger (2001) have shown that cAMP/PKA inhibition in the PFC impaired work-ing memory performance under condition that requires
hip-pocampal interactions with PFC, suggesting that cAMP/PKA activation might be beneficial for working memory perfor-mance. Together, these results suggest that the effects of PFC cAMP on working memory performance may follow an inverted U-shape dose-response relationship (Arnsten et al., 2005). Much work remains to be done to verify this hypoth-esis. In addition, other findings support a role for increased cAMP/PKA signaling in cocaine-induced long-lasting neuro-nal adaptation in PFC pyramidal neurons (Dong et al., 2005), which may be involved in the development of cocaine additive behaviors.
Conclusion
In summary, cAMP acts presynaptically, via activating the PKA and p42/p44 MAPK signaling pathways, to induce syn-aptic potentiation on layer V pyramidal neurons of mPFC. This enhancement is a result of increase in both release probability and number of releasable vesicles. The finding that -adrenergic receptor activation mimics the forskolin action provides a major advance in establishing a role for more physiologically relevant stimuli in eliciting such synap-tic modification. Given the importance of mPFC for cognitive functions, our findings may provide novel pharmacological strategies to treat human cognitive deficits in the future.
References
Arnsten AF, Ramos BP, Birnbaum SG, and Taylor JR (2005) Protein kinase A as a therapeutic target for memory disorders: rationale and challenges. Trends Mol
Med 11:121–128.
Aujla H and Beninger RJ (2001) Hippocampal-prefrontocortical circuits: PKA inhi-bition in the prefrontal cortex impairs delayed nonmatching in the radial maze in rats. Behav Neurosci 115:1204 –1211.
Bailey CH, Bartsch D, and Kandel ER (1996) Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA 93:13445–13452.
Beaumont V and Zucker RS (2000) Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ihchannels. Nat Neurosci 3:133–141.
Bekkers JM and Stevens CF (1990) Presynaptic mechanism for long-term potentia-tion in the hippocampus. Nature (Lond) 346:724 –729.
Bing G, Stone EA, Zhang Y, and Filer D (1992) Immunohistochemical studies of noradrenergic-induced expression of c-fos in the rat CNS. Brain Res 592:57– 62. Birrell JM and Brown VJ (2000) Medial frontal cortex mediates perceptual
atten-tional set shifting in the rat. J Neurosci 20:4320 – 4324.
Chavez-Noriega LE and Stevens CF (1994) Increased transmitter release at excita-tory synapses produced by direct activation of adenylate cyclase in rat hippocam-pal slices. J Neurosci 14:310 –317.
Chen C and Regehr WG (1997) The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci 17:8687– 8694.
Colwell CS and Levine MS (1995) Excitatory synaptic transmission in neostriatal neurons: regulation by cyclic AMP-dependent mechanisms. J Neurosci 15:1704 – 1713.
Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, and White FJ (2005) Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci 25:936 – 940.
Dong Y and White FJ (2004) Dopamine D1-class receptors selectively modulate a
slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci 23:2686 –2695.
Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, and Bos JL (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4:901–906. Fuster JM, Bodner M, and Kroger JK (2000) Cross-modal and cross-temporal
asso-ciation in neurons of frontal cortex. Nature (Lond) 405:347–351.
Goldman-Rakic PS (1995) Architecture of the prefrontal cortex and the central executive. Ann NY Acad Sci 769:71– 83.
Heidbreder CA and Groenewegen HJ (2003) The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev 27:555–579.
Herrero I and Sa´nchez-Prieto J (1996) cAMP-dependent facilitation of glutamate release by-adrenergic receptors in cerebrocortical nerve terminals. J Biol Chem
271:30554 –30560.
Hirsch JC and Crepel F (1990) Use-dependent changes in synaptic efficacy in rat prefrontal neurons in vitro. J Physiol 427:31– 49.
Hoshi T, Garber SS, and Aldrich RW (1988) Effect of forskolin on voltage-gated K⫹ channels is independent of adenylate cyclase activation. Science (Wash DC) 240: 1652–1655.
Huang CC, Chen YL, Liang YC, and Hsu KS (2002) Role for cAMP and protein phosphatase in the presynaptic expression of mouse hippocampal mossy fibre depotentiation. J Physiol 543:767–778.