Intensely Luminescent Alkynyl-Phosphine Gold(I)-Copper(I)
Complexes: Synthesis, Characterization, Photophysical, and
Computational Studies
Igor O. Koshevoy,*,†Yi-Chih Lin,‡Antti J. Karttunen,†Pi-Tai Chou,*,‡Pirjo Vainiotalo,† Sergey P. Tunik,§Matti Haukka,†and Tapani A. Pakkanen*,†
Department of Chemistry, UniVersity of Joensuu, FI-80101, Joensuu, Finland, Department of Chemistry, National Taiwan UniVersity, Taipei 106, Taiwan, and Department of Chemistry, St.-Petersburg State UniVersity, UniVersitetskii pr. 26, 198504, St.-Petersburg, Russia
Received October 17, 2008
The reactions between the diphosphino-alkynyl gold complexes (XC6H4C2Au)PR2-C6H4-PR2(AuC2C6H4X) with
Cu+ lead to the formation of a family of heterometallic clusters of the general formula [{Au3Cu2(C2C6H4X)6
}-Au3(PR2C6H4PR2)3][PF6]2(X ) NO2, H, OMe, NMe2; R ) C6H5, NC4H4). These complexes adopt the same structural
pattern and consist of a heterometallic alkynyl cluster [Au3Cu2(C2C6H4X)6]- “wrapped” by the cationic
[Au3(PR2C6H4PR2)3]3+ “belt”. The novel compounds were characterized by NMR spectroscopy and ESI-MS
measurements. A systematic study of their luminescence properties revealed efficient room-temperature phosphorescence in solution with remarkably weak quenching by molecular oxygen. The photophysical experiments demonstrate that the increase in the electron donor ability of the alkynyl ligands and the electron-withdrawing character of the diphosphines results in the bathochromic shift of emission maxima (in the 576-686 nm range) and a decrease in the luminescence quantum yield. The electronic structure calculations showed that variations of X or R substituents have very little effect on the structural parameters but display a significant influence on the electronic properties of the clusters and characteristics of luminescence. The metal-centered triplet emission within the heterometallic alkynyl cluster is suggested to play a key role in the observed phosphorescence.
Introduction
Luminescent d10 transition metal complexes attract
in-creasing attention because of their rich photophysical and photochemical properties and possible practical applications, for example, in OLED display technology or as dopant emitters.1In addition, the chemistry of the gold(I) compounds is of considerable interest due to their unique featuresformation of the secondary bonds through inter- or intramolecular
aurophilic interactions.2The ability of gold(I) ions to form heterometallophilic Au · · · M bonding is of particular impor-tance because of the influence this interaction exerts onto the molecular structure and physical properties (e.g., onto luminescence) of the heterometallic aggregates.3
Among the Au(I) complexes, phosphino-alkynyls repre-sent a specific class of organogold compounds, which are able to build up supramolecular architectures based on the
* To whom correspondence should be addressed. E-mail: igor.koshevoy@ joensuu.fi (I.O.K.); [email protected] (T.A.P.); [email protected] (P.-TC.).
†University of Joensuu. ‡National Taiwan University. §St.-Petersburg State University.
(1) (a) Ma, Y.; Che, C.-M.; Chao, H.-Y.; Zhou, X.; Chan, W.-H.; Shen, J. AdV. Mater. 1999, 11, 852–857. (b) Zhang, Q.; Zhou, Q.; Cheng, Y.; Wang, L.; Ma, D.; Jing, X.; Wang, F. AdV. Mater. 2004, 16, 432– 436. (c) Fave, C.; Cho, T.-Y.; Hissler, M.; Chen, C.-W.; Luh, T.-Y.; Wu, C.-C.; Reau, R. J. Am. Chem. Soc. 2003, 125, 9254–9255. (d) Baldo, M. A.; Thompson, M. E.; Forrest, S. R. Pure Appl. Chem.
1999, 71, 2095–2106. (e) Jia, W. L.; McCormick, T.; Tao, Y.; Lu,
J.-P.; Wang, S. Inorg. Chem. 2005, 44, 5706–5712.
(2) (a) Forward, J. M.; Fackler, J. P. J.; Assefa, Z. In Optoelectronic
Properties of Inorganic Compounds; Roundhill, D. M., Fackler, J. P. J.,
Eds.; Plenum: New York, 1999; pp 195-231. (b) Laguna, A. In Gold:
Chemistry, Biochemistry and Technology; Schmidbauer, H.; Eds.;
Wiley: Chichester, U.K., 1999; pp 349-429. (c) Dyson, P. J.; Mingos, D. M. P. In Gold: Chemistry, Biochemistry and Technology; Schmid-bauer, H., Eds.; Wiley: Chichester, U. K. 1999; pp 511-557. (d) Gimeno, M. C.; Laguna, A. In ComprehensiVe Coordination Chemistry
II; McCleverty, J. A., Meyer, T. J., Eds.; Elsevier: New York, 2003;
Vol. 6, pp 911-1145. (e) Schmidbaur, H.; Schier, A. Chem. Soc. ReV.
2008, 37, 1931–1951. (f) Yam, V. W.-W.; Cheng, E. C.-C. Chem.
Soc. ReV. 2008, 37, 1806–1813.
(3) Fernandez, E. J.; Laguna, A.; Lopez-de-Luzuriaga, J. M. Dalton Trans.
2007, 1969–1981.
Inorg. Chem. 2009, 48, 2094-2102
2094 Inorganic Chemistry, Vol. 48, No. 5, 2009 10.1021/ic801987t CCC: $40.75 2009 American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
metallophilic interactions and the ability of the alkynyl units to function as linear rigid-rod bridging building blocks. The supramolecular aggregates are stabilized by ancillary phos-phine ligands that allow for the preparation of a number of the luminescent heterometallic polynuclear complexes.4-6 In our recent studies,5,6 we reported an investigation of the Cu(I)-promoted assembly of the Au-Cu alkynyl clusters supported by the [Au3(diphospine)3]3+ triangular “belts”
(diphosphine ) PPh2(C6H4)nPPh2; n ) 1, 2, 3). Variations
in the length of the diphosphine ligand spacers resulted in the successful synthesis of a series of heterometallic com-plexes of the general formula [{AuxCuy(C2Ph)2x
}-Au3{PPh2-(C6H4)n-PPh2}3]3+(y-x)(n ) 1, 2, 3; x ) (n +
1)(n + 2)/2; y ) n(n + 1)). These compounds display very similar structural patterns and contain the central heterome-tallic clusters {AuxCuy(C2Ph)2x} wrapped about by the
tricationic [Au3(PP)3]3+“belts”, anchored to the central parts
by the Au-Au bonds. The complexes obtained display very intense room-temperature long-wavelength luminescence both in solution and in the solid state with a maximum quantum yield of 92% (n ) 1). However, it was experimen-tally demonstrated that the increase in the size of the aggregates leads to the decrease in photostability and luminescence efficiency that was supported by the compu-tational results, which also indicated that the phosphorescence observed originates from the metal-centered transitions within the heterometallic Au-Cu cores.6 This prompted us to experimental and theoretical studies related to modification of the electronic structure of this type of compounds via variation of the coordination environment. The most stable and effective luminophore [{Au3Cu2(C2Ph)6}Au3{PPh2
-C6H4-PPh2}3]2+ was chosen as a parent compound to
prepare its analogues containing substituted phenylalkynes and diphosphines with different electron-donor properties and to study the effect of these variations on the luminescence properties of these supramolecular aggregates.
Experimental Section
General Comments. 1,4-Bis(diphenylphosphino)benzene7and
1,4-bis(dichlorophosphino)benzene8were synthesized according to
published procedures. Complexes {Au(1-C2-4-X-C6H4)}n (X )
NO2, OMe, NMe2) were prepared analogously to (AuC2Ph)n9using
commercially available alkynes 1-HC2-4-X-C6H4. Tetrahydrofuran
was distilled over Na-benzophenoneketyl under a nitrogen atmo-sphere prior to use. Other reagents and solvents were used as
received. The solution1H,13C, and31P NMR spectra were recorded
on Bruker Avance 400 and Bruker DPX 300 spectrometers. Mass spectra were measured on a Bruker APEX-Qe ESI FT-ICR instrument, in the ESI+mode. Microanalyses were carried out in the analytical laboratories of St.-Petersburg State University and the University of Joensuu.
P(NC4H4)2C6H4P(NC4H4)2(1). Synthesis was carried under a
nitrogen atmosphere. Pyrrole (1.6 g, 23.9 mmol) was added dropwise to a cooled (-40°C) solution of n-BuLi in hexanes (16 cm3, 1.6 M). A pale suspension was stirred at -40°C for 30 min,
warmed to room temperature, and stirred for an additional 30 min. Then, the reaction mixture was cooled to -78 °C and treated dropwise with a solution of 1,4-bis(dichlorophosphino)benzene (1.64 g, 5.85 mmol) in THF (25 cm3). When the addition was
completed, the reaction mixture was warmed to room temperature and stirred for 1 h. The resulting yellow-brownish solution was filtered to remove a small amount of white precipitate. The solvents were evaporated. An oily residue was extracted with CH2Cl2(3×
4 cm3), and extracts were diluted with hexanes (10 cm3) and passed
through a layer of silica (2 × 5 cm). The solvents again were removed; the resulting yellow solid was washed with methanol (3 × 5 cm3) to give white material of sufficient (ca. 97%) purity (1.05
g, 45%). An analytically pure sample was obtained by additional chromatographic purification on silica (2:3 eluent CH2Cl2/hexane
v/v). 31P{1H} NMR (CDCl
3; δ): 63.2 (s). 1H NMR (CDCl3; δ):
7.08 (m, C6H4, 4H), 6.95 (m, Pyr, 8H), 6.35 (m, Pyr, 8H). Anal.
calcd for C22H20N4P2: C, 65.67; H, 5.01; N, 13.92. Found: C, 65.54;
H, 5.11; N, 13.77.
(AuC2C6H4NO2)2PPh2C6H4PPh2(2). (AuC2C6H4NO2)n(150 mg,
0.437 mmol) was suspended in CH2Cl2(10 cm3), and a slight excess
of 1,4-bis(diphenylphosphino)benzene (102 mg, 0.229 mmol) was added. The reaction mixture was stirred for 30 min. The suspension obtained was evaporated, dissolved in CHCl3(ca. 20 cm3), diluted
with toluene (5 cm3), and passed through a layer of neutral Al 2O3
(1× 4 cm). The solution was concentrated to a volume of ca. 7 cm3. A pale solid was separated by centrifugation, suspended in
CH2Cl2 (2 cm3), diluted with toluene (4 cm3), and centrifugated
again. Subsequent washing with toluene (5 cm3) and diethyl ether
(3× 5 cm3) and vacuum drying gave a pale-yellow powder (235
mg, 95%). An analytically pure sample was obtained by recrys-tallization from CH2Cl2/diethyl ether at +5 °C. 31P{1H} NMR
(CDCl3; δ): 41.4 (s).1H NMR (CDCl3; δ): 8.14 (dm, C6H4-NO2,
J(H-H) 8.9 Hz, 4H), 7.62-7.49 (m, 28H). Anal. calcd for
C46H32Au2N2O4P2: C, 48.78; H, 2.85; N, 2.47. Found: C, 48.82; H,
2.80; N, 2.35.
(AuC2C6H4OMe)2PPh2C6H4PPh2(3). (AuC2C6H4OMe)n(150
mg, 0.457 mmol) was suspended in CH2Cl2 (10 cm3), and
1,4-bis(diphenylphosphino)benzene (106 mg, 0.238 mmol) was added. The reaction mixture was stirred for 30 min. The colorless transparent solution was diluted with toluene (5 cm3) and passed
through a layer of neutral Al2O3 (1 × 4 cm). The solution was
concentrated to a volume of ca. 5 cm3. A white solid was separated
by centrifugation, washed with toluene (5 cm3) and diethyl ether
(3× 5 cm3), and vaccuum-dried (232 mg, 92%). An analytically
pure sample was obtained by recrystallization from CH2Cl2/diethyl
ether at +5 °C. 31P{1H} NMR (CDCl
3; δ): 41.8 (s). 1H NMR
(CDCl3; δ): 7.62-7.47 (m, 24H), 7.45 (dm, C6H4-OCH3, J(H-H)
8.2 Hz, 4H), 6.80 (dm, C6H4-OCH3, J(H-H) 8.2 Hz, 4H), 3.80
(s, OCH3, 6H). Anal. calcd for C48H38Au2O2P2: C, 52.28; H, 3.47.
Found: C, 52.23; H, 3.46.
(AuC2C6H4NMe2)2PPh2C6H4PPh2(4). (AuC2C6H4NMe2)n(150
mg, 0.440 mmol) was suspended in CH2Cl2 (10 cm3), and
1,4-bis(diphenylphosphino)benzene (102 mg, 0.229 mmol) was added.
(4) (a) Wei, Q.-H.; Yin, G.-Q.; Zhang, L.-Y.; Shi, L.-X.; Mao, Z.-W.; Chen, Z.-N. Inorg. Chem. 2004, 43, 3484–3491. (b) Wei, Q.-H.; Zhang, L.-Y.; Yin, G.-Q.; Shi, L.-X.; Chen, Z.-N. J. Am. Chem. Soc. 2004,
126, 9940–9941. (c) Yin, G.-Q.; Wei, Q.-H.; Zhang, L.-Y.; Chen,
Z.-N. Organometallics 2006, 25, 580–587. (d) de la Riva, H.; Nieu-whuyzen, M.; Fierro, C. M.; Raithby, P. R.; Male, L.; Lagunas, M. C.
Inorg. Chem. 2006, 45, 1418–1420.
(5) Koshevoy, I. O.; Koskinen, L.; Haukka, M.; Tunik, S. P.; Serdobintsev, P. Y.; Melnikov, A. S.; Pakkanen, T. A. Angew. Chem., Int. Ed. 2008,
47, 3942–3945.
(6) Koshevoy, I. O.; Karttunen, A. J.; Tunik, S. P.; Haukka, M.; Selivanov, S. I.; Melnikov, A. S.; Serdobintsev, P. Y.; Khodorkovskiy, M. A.; Pakkanen, T. A. Inorg. Chem. 2008, 47, 9478–9488.
(7) Baldwin, R. A.; Cheng, M. T. J. Org. Chem. 1967, 32, 1572–1577. (8) Evleth, E. M. J.; Freeman, L. D.; Wagner, R. I. J. Org. Chem. 1962,
27, 2192–2197.
(9) Coates, G. E.; Parkin, C. J. Chem. Soc. 1962, 3220–3226.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
The reaction mixture was stirred for 30 min. The yellow transparent solution was diluted with toluene (5 cm3), concentrated to a volume
of ca. 7 cm3. The yellow solid was separated by centrifugation,
washed with toluene (5 cm3) and diethyl ether (3× 5 cm3), and
vaccuum-dried (236 mg, 95%). An analytically pure sample was obtained by recrystallization from CH2Cl2/diethyl ether at +5°C. 31P{1H} NMR (CDCl
3; δ): 42.0 (s).1H NMR (CDCl3; δ): 7.63-7.46
(m, 24H), 7.41 (dm, C6H4-N(CH3)2, J(H-H) 9.0 Hz, 4H), 6.61
(dm, C6H4-N(CH3)2, J(H-H) 9.0 Hz, 4H), 2.96 (s, N(CH3)2, 12H).
Anal. calcd for C50H44Au2N2P2: C, 53.20; H, 3.93; N, 2.48. Found:
C, 53.01; H, 3.95; N, 2.40.
(AuC2C6H4NO2)2P(NC4H4)2C6H4P(NC4H4)2 (5). Complex 5
was generated analogously to 3, using (AuC2C6H4NO2)n(150 mg,
0.437 mmol) and 1,4-bis(dipyrrolylphosphino)benzene (92 mg, 0.229 mmol). Yield: 202 mg, 85%. An analytically pure sample was obtained by recrystallization from CH2Cl2/diethyl ether at +5
°C.31P{1H} NMR (CDCl
3; δ): 95.0 (s, br).1H NMR (CDCl3; δ):
8.15 (dm, C6H4-NO2, J(H-H) 8.9 Hz, 4H), 7.59 (dm, C6H4-NO2,
J(H-H) 8.9 Hz, 4H), 7.38 (m, P-C6H4-P, 4H), 7.10 (m, NC4H4,
8H), 6.52 (dm, NC4H4, J(H-H) 1.8 Hz, 8H). Anal. calcd for
C38H28Au2N6O4P2: C, 41.93; H, 2.59; N, 7.72. Found: C, 42.23; H,
2.59; N, 7.39.
(AuC2Ph)2P(NC4H4)2C6H4P(NC4H4)2 (6). (AuC2C6H5)n (150
mg, 0.503 mmol) was suspended in CH2Cl2 (10 cm3), and
1,4-bis(dipyrrolylphosphino)benzene (106 mg, 0.264 mmol) was added. The reaction mixture was stirred for 30 min. The resulting suspension was evaporated, dissolved in CHCl3(ca. 35 cm3), diluted
with toluene (5 cm3), and treated as 2. A pale yellowish solid was
formed, 230 mg, 92%. An analytically pure sample was obtained by recrystallization from CH2Cl2/diethyl ether at +5°C.31P{1H}
NMR (CDCl3; δ): 95.5 (s, br).1H NMR (CDCl3; δ): 7.50 (m, 4H),
7.37-7.23 (m, 10H), 7.11 (m, NC4H4, 8H), 6.50 (dm, NC4H4,
J(H-H) 2.1 Hz, 8H). Anal. calcd for C38H30Au2N4P2: C, 45.71; H,
3.03; N, 5.61. Found: C, 45.94; H, 3.08; N, 5.50.
(AuC2C6H4OMe)2P(NC4H4)2C6H4P(NC4H4)2(7). (AuC2C6H4
-OMe)n(150 mg, 0.457 mmol) was suspended in CH2Cl2(10 cm3),
and 1,4-bis(dipyrrolylphosphino)benzene (95 mg, 0.236 mmol) was added. The reaction mixture was stirred for 30 min. The white suspension was diluted with toluene (10 cm3), and the solid was
separated by centrifugation, washed with toluene (5 cm3) and diethyl
ether, and dried under a vacuum. The product (220 mg, 91%) was obtained as a white powder. An analytically pure sample was obtained by extraction with chloroform passing through a layer of neutral Al2O3(1× 4 cm), evaporation, and washing of the residue
with diethyl ether.31P{1H} NMR (CDCl
3; δ): 95.7 (s, br).1H NMR
(CDCl3; δ): 7.45 (dm, C6H4-OCH3, J(H-H) 8.8 Hz, 4H), 7.32
(m, P-C6H4-P, 4H), 7.10 (m, NC4H4, 8H), 6.83 (dm, C6H4-OCH3,
J(H-H) 8.8 Hz, 4H), 6.49 (m, NC4H4, 8H), 3.81 (s, OCH3, 6H).
Anal. calcd for C40H34Au2N4O2P2: C, 45.38; H, 3.24; N, 5.29.
Found: C, 45.53; H, 3.26; N, 5.24.
General Procedure for the Preparation of Heterometallic Au-Cu Complexes. Complexes 2-7 (0.100 mmol) were sus-pended in CH2Cl2(8 cm3) and diluted with diethyl ether (4 cm3),
and a solution of Cu(NCMe)4PF6 (0.067 mmol) in CH2Cl2 (3
cm3) was added dropwise to afford a transparent orange to red
solution (except 12, orange suspension formed). The solvent was removed on a rotovap (12 was separated from the pale solution by centrifugation). The solid residue was dissolved in acetone (3 cm3), filtered, and precipitated by the addition of hexane or
diethyl ether to give a microcrystalline solid. If necessary, the samples were recrystallized by the gas-phase diffusion of pentane
or diethyl ether into their acetone solutions at +5°C. Complex [{Au3Cu2(C2Ph)6}Au3(PPh2C6H4PPh2)3][PF6]2 (9) was reported
earlier.6
[{Au3Cu2(C2C6H4NO2)6}Au3(PPh2C6H4PPh2)3][PF6]2 (8). An
orange compound was formed. Yield: 82%. ES MS (m/z): [Au6Cu2(C2C6H4NO2)6(PPh2C6H4PPh2)3]2+, 1762 (calcd, 1762). 31P{1H} NMR (acetone-d 6; δ): 44.7 (s, 3P), -144.8 (sept, 1P, PF6). 1H NMR (acetone-d 6; δ), {Au(C2C6H4NO2)2}: 7.65 (d, H-meta, 12H, J(H-H) 8.8 Hz), 7.11 (dd, H-ortho, 12H, J(H-H) 8.8 Hz). 1H NMR (acetone-d
6; δ), diphosphine: 8.02 (dm(AXX′), H-ortho,
24H, J(H-H) 6.7, J(P-H) 13.6 Hz), 7.91 (m(A2X2), {P-C6H4-P}
12H, <J(P-H)> 3.4 Hz), 7.68 (t, H-para, 12H, J(H-H) 7.5 Hz), 7.51 (dd, H-meta, 24H, J(H-H) 6.7, 7.5 Hz).13C NMR
(acetone-d6; δ), {Au(C2C6H4NO2)2}: 148.33 (s, C-para-NO2), 134.47 and
124.78 (s, C-meta and C-ortho), 123.52 (s, C-ipso), 112.86 (s, CtC-Au), 117.39 (s, CtC-Au). 13C NMR (acetone-d
6; δ),
diphosphine: 137.12 (m (AXX′), C (-C6H4-), J(P-C) 59 Hz),
136.92 (m (AXX′), C-ortho, J(P-C) 16.5 Hz), 135.73 (m (AXX′), C(H) (-C6H4-), J(P-C) ca. 12 Hz), 134.63 (s, C-para), 131.61
(m (AXX′), C-meta, J(P-C) 11.9 Hz), 129.50 (m (AXX′), C-ipso,
J(P-C) 56.7 Hz). Anal. calcd for C138H96Au6Cu2F12N6O12P8: C,
43.45; H, 2.54; N, 2.20. Found: C, 43.34; H, 2.87; N, 2.16. [{Au3Cu2(C2C6H4OMe)6}Au3(PPh2C6H4PPh2)3][PF6]2(10). An
orange-red compound was formed. Yield: 86%. ES MS (m/z): [Au6Cu2(C2C6H4OMe)6(PPh2C6H4PPh2)3]2+, 1717 (calcd, 1717). 31P{1H} NMR (acetone-d 6; δ): 43.4 (s, 3P), -144.8 (sept, 1P, PF6). 1H NMR (acetone-d 6; δ), {Au(C2C6H4NO2)2}: 6.69 (d, H-ortho, 12H, J(H-H) 8.7 Hz), 6.39 (dd, H-meta, 12H, J(H-H) 8.7 Hz), 3.72 (s, 18H, OMe).1H NMR (acetone-d 6; δ), diphosphine: 7.97 (dm(AXX′), H-ortho, 24H, J(H-H) 6.7, J(P-H) 7.3 Hz), 7.75 (m(A2X2), {P-C6H4-P} 12H, <J(P-H)> 3.4 Hz), 7.66 (t, H-para, 12H, J(H-H) 7.5 Hz), 7.48 (dd, H-meta, 24H, J(H-H) 6.7, 7.5 Hz). 13C NMR (acetone-d 6; δ), {Au(C2C6H4NO2)2}: 161.24 (s,
C-para-OMe), 135.07 and 115.07 (s, C-meta and C-ortho), 117.67 (s, C-ipso), 114.43 (s, CtC-Au), 110.94 (s, CtC-Au), 56.35 (s, OMe).13C NMR (acetone-d
6; δ), diphosphine: 137.43 (m (AXX′),
C (-C6H4-), J(P-C) 59 Hz), 137.03 (m (AXX′), C-ortho, J(P-C)
16.4 Hz), 135.61 (m (AXX′), C(H) (-C6H4-), J(P-C) ca. 11 Hz),
134.55 (s, C-para), 131.61 (m (AXX′), C-meta, J(P-C) 11.8 Hz), 130.01 (m (AXX′), C-ipso, J(P-C) 55.2 Hz). Anal. calcd for C144H114Au6Cu2F12O6P8: C, 46.43; H, 3.08. Found: C, 46.51; H,
3.15.
[{Au3Cu2(C2C6H4NMe2)6}Au3(PPh2C6H4PPh2)3][PF6]2(11). A
very dark red compound was formed. Yield: 71%. ES MS (m/z): [Au6Cu2(C2C6H4NMe2)6(PPh2C6H4PPh2)3]2+, 1756 (calcd, 1756). 31P{1H} NMR (acetone-d
6; δ): 42.9 (s, 3P), -144.8 (sept, 1P, PF6). 1H NMR (acetone-d
6; δ), {Au(C2C6H4NMe2)2}: 6.58 (d, H-ortho,
12H, J(H-H) 8.8 Hz), 6.39 (dd, H-meta, 12H, J(H-H) 8.8 Hz), 2.90 (s, 36H, NMe2).1H NMR (acetone-d6; δ), diphosphine: 7.98
(dm(AXX′), H-ortho, 24H, J(H-H) 6.7, J(P-H) 6.7 Hz), 7.73 (m(A2X2), {P-C6H4-P} 12H, <J(P-H)> 3.3 Hz), 7.64 (t, H-para,
12H, J(H-H) 7.5 Hz), 7.45 (dd, H-meta, 24H, J(H-H) 6.7, 7.5 Hz).13C NMR (acetone-d
6; δ), {Au(C2C6H4NMe2)2}: 151.50 (s,
C-para-NMe2), 135.10 and 113.29 (s, C-meta and C-ortho), 112.91
(s, C-ipso), 115.70 (s, CtC-Au), 109.87 (s, CtC-Au), 41.24 (s, NMe2).13C NMR (acetone-d6; δ), diphosphine: 137.50 (m (AXX′),
C (-C6H4-), J(P-C) 59 Hz), 136.99 (m (AXX′), C-ortho, J(P-C)
16.4 Hz), 135.51 (m (AXX′), C(H) (-C6H4-), J(P-C) ca. 11 Hz),
134.36 (s, C-para), 131.62 (m (AXX′), C-meta, J(P-C) 11.7 Hz), 130.21 (m (AXX′), C-ipso, J(P-C) 54.5 Hz). Anal. calcd for C150H132Au6Cu2F12N6P8: C, 47.37; H, 3.50; N, 2.21. Found: C,
47.53; H, 3.46; N, 2.13.
Koshevoy et al.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
[{Au3Cu2(C2C6H4NO2)6}Au3(P(NC4H4)2C6H4P(NC4H4)2)3
]-[PF6]2 (12). An orange compound was formed. Yield: 84%. ES
MS (m/z): [Au6Cu2(C2C6H4NO2)6(P(NC4H4)2C6H4P(NC4H4)2)3]2+,
1696 (calcd, 1696).31P{1H} NMR (acetone-d
6; δ): 89.6 (s, 3P),
-144.8 (sept, 1P, PF6). 1H NMR (acetone-d6; δ),
{Au(C2C6H4NO2)2}: H-ortho 7.68 (d, 12H, J(H-H) 8.8 Hz), H-meta
7.22 (d, 12H, J(H-H) 8.8 Hz).1H NMR (acetone-d
6; δ),
diphos-phine: 8.05 (m(A2X2), {P-C6H4-P} 12H, <J(P-H)> 3.9 Hz), 7.53
(dm(AXX′), Pyr 2-CH, 24H, J(H-H) 2.1, J(P-H) 4.2 Hz), 6.49 (dm(AXX′), Pyr 3-CH, 24H, J(H-H) 2.1, J(P-H) 2.0 Hz).13C
NMR (acetone-d6; δ), {Au(C2C6H4NO2)2}: 149.97 (s, C-para-NO2),
135.09 and 124.95 (s, C-ortho and C-meta), 130.31 (s, C-ipso), 113.90 (s, CtC-Au), 116.48 (s, CtC-Au).13C NMR
(acetone-d6; δ), diphosphine: 137.69 (m (AXX′), C (-C6H4-), J(P-C) 82.6
Hz), 135.74 (m (AXX′), C(H) (-C6H4-), J(P-C) ca. 14 Hz),
127.78 (m (AXX′), Pyr 2-C, J(P-C) ca. 10 Hz), 117.63 (s, Pyr 3-C). Anal. calcd for C114H84Au6Cu2F12N18O12P8: C, 37.18; H, 2.30;
N, 6.85. Found: C, 37.16; H, 2.35; N, 6.57.
[{Au3Cu2(C2Ph)6}Au3(P(NC4H4)2C6H4P(NC4H4)2)3][PF6]2(13).
An orange-red compound was formed. Yield: 91%. ES MS (m/z): [Au6Cu2(C2Ph)6(P(NC4H4)2C6H4P(NC4H4)2)3]2+, 1561 (calcd, 1561). 31P{1H} NMR (acetone-d
6; δ): 89.8 (s, 3P), -144.8 (sept, 1P, PF6). 1H NMR (acetone-d
6; δ), {Au(C2Ph)2}: 7.13 (tt, H-para, 6H,
J(H-H) 7.3, 1.5 Hz). ABX system, H-ortho-H-meta: 6.89 (dd, 12H, J(H-H) 6.8, 1.5 Hz), 6.85 (dd, 12H, J(H-H) 6.8, 7.3 Hz).1H NMR
(acetone-d6; δ), diphosphine: 7.51 (dm(AXX′), Pyr 2-CH, 24H,
J(H-H) 2.1, J(P-H) 4.2 Hz), 7.82 (m(A2X2), {P-C6H4-P} 12H,
<J(P-H)> 3.9 Hz), 6.48 (dm(AXX′), Pyr 3-CH, 24H, J(H-H) 2.1, J(P-H) 2.0 Hz). 13C NMR (acetone-d
6; δ), {Au(C2Ph)2}:
134.00 and 129.85 (s, C-ortho and C-meta), 130.19 (s, C-para), 124.41 (s, C-ipso), 115.51 (s, CtC-Au), 111.65 (s, CtC-Au).
13C NMR (acetone-d
6; δ), diphosphine: 137.87 (m (AXX′), C
(-C6H4-), J(P-C) 82.5 Hz), 135.21 (m (AXX′), C(H) (-C6H4-),
J(P-C) ca. 13 Hz), 127.72 (m (AXX′), Pyr 2-C, J(P-C) ca. 9 Hz),
117.38 (s, Pyr 3-C). Anal. calcd for C114H90Au6Cu2F12N12P8: C,
40.12; H, 2.66; N, 4.93. Found: C, 39.73; H, 2.61; N, 4.77. [{Au3Cu2(C2C6H4OMe)6}Au3(P(NC4H4)2C6H4P(NC4H4)2)3
]-[PF6]2(14). A deep red complex was formed. Yield: 78%. ES MS
(m/z): [Au6Cu2(C2C6H4OMe)6(P(NC4H4)2C6H4P(NC4H4)2)3]2+, 1651
(calcd, 1651).31P{1H} NMR (acetone-d
6; δ): 90.0 (s, 3P), -144.8
(sept, 1P, PF6). 1H NMR (acetone-d6; δ), {Au(C2C6H4OMe)2}:
H-ortho 6.74 (d, 12H, J(H-H) 8.8 Hz), H-meta 6.41 (d, 12H, J(H-H) 8.8 Hz). 1H NMR (acetone-d 6; δ), diphosphine: 7.79 (m(A2X2), {P-C6H4-P} 12H, <J(P-H)> 3.6 Hz), 7.49 (dm(AXX′), Pyr 2-CH, 24H, J(H-H) 2.1, J(P-H) 4.2 Hz), 6.50 (dm(AXX′), Pyr 3-CH, 24H, J(H-H) 2.1, J(P-H) 2.0 Hz).13C NMR
(acetone-d6; δ), {Au(C2C6H4OMe)2}: 161.66 (s, C-para-OMe), 135.58 and
115.35(s, C-ortho and C-meta), 116.58 (s, C-ipso), 115.56 (s, CtC-Au), 109.85 (s, CtC-Au). 13C NMR (acetone-d
6; δ),
diphosphine: 137.97 (m (AXX′), C (-C6H4-), J(P-C) 83.2 Hz),
135.10 (m (AXX′), C(H) (-C6H4-), J(P-C) ca. 12 Hz), 127.78
(m (AXX′), Pyr 2-C, J(P-C) ca. 8 Hz), 117.37 (s, Pyr 3-C). Anal. calcd for C120H102Au6Cu2F12N12O6P8: C, 40.12; H, 2.86; N, 4.68.
Found: C, 39.66; H, 2.85; N, 4.50.
Photophysical Measurements. The steady-state absorption and emission spectra were recorded on a Hitachi (U-3310) spectropho-tometer and an Edinburgh (FS920) fluorometer, respectively. Both the wavelength-dependent excitation and emission response of the fluorometer have been calibrated. To determine the photolumines-cence quantum yield in solution, the samples were degassed by three freeze-pump-thaw cycles. 4-(Dicyanomethylene)-2-methyl-6-(paradimethylaminostyryl)-4H-pyran (DCM, λmax ) 615 nm,
Exciton) in methanol, with a quantum yield of∼0.43, served as
the standard for measuring the quantum yield.10Lifetime studies
were performed with an Edinburgh FL 900 photon-counting system using a hydrogen-filled lamp as the excitation source. The data were analyzed using the nonlinear least-squares procedure in combination with an iterative convolution method. The emission decays were analyzed by the sum of the exponential functions, which allows partial removal of the instrument time broadening and consequently renders a temporal resolution of∼300 ps.
Computational Details. The studied supramolecular Au(I)-Cu(I) complexes were fully optimized without any symmetry constraints using the BP86 density functional method. The copper and gold atoms were described with a triple-valence ζ-quality basis set with the polarization functions (def2-TZVP),11employing a 60-electron
relativistic effective core potential for gold.12A split-valence basis
set with the polarization functions on non-hydrogen atoms was used for all of the other atoms (def2-SV(P)).13The multipole-accelerated
resolution-of-the-identity technique was used to speed up the calculations.14All of the geometry optimizations were carried out
with TURBOMOLE versions 5.9.1 and 5.10.15The triplet states
were studied using spin-unrestricted formalism.
Results and Discussion
Synthesis and Characterization. Completely analogous
to the preparation of the previously described complex [Au2(CtCPh)2(µ-1,4-PPh2C6H4PPh2)],16 treatment of the
polymers [AuCtCC6H4X]n(X ) NO2, H, OMe, NMe2) with
the diphosphine ligands PR2C6H4PR2(R ) Ph, NC4H4) leads
tocleanformationofthedigoldcomplexes[Au2(CtCC6H4X)2
(µ-1,4-PR2C6H4PR2)] (2-7) in greater than 85% yield (Scheme
1). These compounds were characterized by1H and31P NMR
spectroscopy and elemental analysis.
The assembly of the heterometallic Au(I)-Cu(I) com-plexes of the general formula [{Au3Cu2(C2C6H4X)6}Au3
-(PR2C6H4PR2)3][PF6]2(X ) NO2, H, OMe, NMe2; R ) Ph,
NC4H4) was achieved by reacting the digold complexes 2-7
with stoichiometric amounts of Cu(NCMe)4PF6 in
dichlo-romethane at room temperature, similarly to the procedure reported earlier (Scheme 2).6Bright-orange to deep-red air-stable solids 8-14 were isolated in 71-91% yield after recrystallization. Formation of the heterometallic complex with R ) NC4H4 and X ) NMe2 (15) has been observed
using1H and 31P NMR spectroscopy, but it appeared to be
unstable, which prevented its isolation in a pure form and subsequent structural and photophysical studies. It is also worth noting that we attempted to prepare analogous heterometallic complexes (X ) NO2, H, OMe, NMe2) containing diphosphine
(10) Drake, J. M.; Lesiecki, M. L.; Camaioni, D. M. Chem. Phys. Lett.
1985, 113, 530–534.
(11) Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297– 3305.
(12) Andrae, D.; Ha¨uβermann, U.; Dolg, M.; Stoll, H.; Preuβ, H. Theor.
Chem. Acc. 1990, 77, 123–141.
(13) Scha¨fer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571– 2577.
(14) (a) Eichkorn, K.; Treutler, O.; O¨ hm, H.; Ha¨ser, M.; Ahlrichs, R. Chem.
Phys. Lett. 1995, 240, 283–290. (b) Sierka, M.; Hogekamp, A.;
Ahlrichs, R. J. Chem. Phys. 2003, 118, 9136–9148. (c) Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Theor. Chem. Acc. 1997, 97, 119–124. (d) Weigend, F. Phys. Chem. Chem. Phys. 2006, 8, 1057– 1065.
(15) Ahlrichs, R.; Ba¨r, M.; Ha¨ser, M.; Horn, H.; Ko¨lmel, C. Chem. Phys.
Lett. 1989, 62, 165–169.
(16) Yam, V. W.-W.; Choi, S. W.-K.; Cheung, K.-K. Organometallics 1996,
15, 1734–1739.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
with R ) i-Pr using the same synthetic approach. However, no aggregates of the structural type shown in Scheme 2 were formed judging by the NMR analysis of the crude products. The spectroscopic data showed that in these cases only simple coordination of Cu+ions to the π orbitals of the alkynyl ligands most probably occurred.
Compounds 8-14 were completely characterized by 1H, 13C, and 31P NMR and ESI-MS spectrometry. The crystal
structure of complex 9 was elucidated previously in an X-ray diffraction study.6
The choice of the alkynyl and phosphine ligands was dictated by their different electron-donor properties. Variation of the substituents in the alkynylbenzenes used in this study from the electron-withdrawing NO2to the electron-donating NMe2was
aimed to influence electronic characteristics of the central heterometallic fragment. Replacement of the P-bonded phenyls for pyrrolyls in the diphosphine ligand, which are known to be the strong electron-withdrawing groups, results in a dramatic increase of the phosphorus ligand’s π-acidity.17This obviously influences the properties of the tricationic [AuPR2(C6H4)2PR2]3
“belt”, thus making possible variations in the energy of the orbitals, which take part in the formation of the excited states. Therefore, it is sensible to divide the compounds studied into two groups according to the nature of the diphosphine ligand used to wrap the central gold-copper alkynyl fragment. The first group of com-plexes is based on the PPh2C6H4PPh2ligand, whereas in the second,
the diphosphine contains pyrrolyl substituents at the phosphorus atoms, P(NC4H4)2C6H4P(NC4H4)2.
The ESI mass spectra of compounds 8, 10, and 11 (Figure S1, S denotes Supporting Information) display signals of the
doubly charged [{Au3Cu2(C2C6H4X)6}Au3(PPh2C6H4
-PPh2)3]2+molecular ions, the m/z values and isotopic patterns
of which completely match the calculated ones. Comparison of the1H and13C NMR data obtained in the previous study
for the completely characterized complex 9,6with the relative intensities of the signals, their chemical shifts, and coupling constants observed in the spectra of the congeners 8, 10, and 11 containing NO2, OMe, and NMe2substituents in the
phenyl ring of the alkynyl moieties, clearly showed that these complexes retain the parent structure (Figure 1) in solution. The NMR parameters of the triangular belts of the substituted congeners are nearly identical (see the Experi-mental Section) to those of the parent compound, which is also indicative of a weak influence of the alkynyl “rod” substituents on the shielding of the nuclei in the tri(gold-diphosphine) belt. The only variations observed in the NMR spectra of these complexes are related to the chemical shifts of the substituted phenyl moieties. The order of the ortho-and meta-protons’ chemical shifts is clearly related to the donor ability of the corresponding substituents (NMe2 >
OMe > H > NO2). It is worth noting that the substituents
display a long-range electronic effect, which can be observed in the shielding of the remote alkynyl carbon atoms; for example, high-field shifts of the carbon bound to Au (109.87 ppm {NMe2}, 110.94 ppm{OMe}, 112.60 ppm{H}, 117.39
ppm{NO2}) very well follow the order of the substituents’
donor properties.
According to the elemental analysis and mass spectro-scopic data (Figure S1), the other group of compounds,
12-14, based on 1,4-bis(dipyrrolylphosphino)benzene (1) fits
(17) Moloy, K. G.; Petersen, J. L. J. Am. Chem. Soc. 1995, 117, 7696– 7710.
Scheme 1
Scheme 2
Figure 1. Schematic structure of the dications [{Au3Cu2(C2C6H4X)6}Au3 -(PR2C6H4PR2)3]2+(8-14), (X ) NO2, H, OMe, NMe2; R ) Ph, NC4H4). Koshevoy et al.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
the stoichiometry of the [{Au3Cu2(C2C6H4X)6}Au3(P(NC4H4)2
-C6H4P(NC4H4)2)3][PF6]2species. Analysis of the1H,13C, and 31P NMR data for this series of complexes (see the
Experimental Section and Figure S2,1H COSY spectrum of 13) indicates that the components of these polynuclear
compounds form structural patterns completely analogous to those of complexes 8-10 and contain the central [Au3Cu2(C2C6H4X)6] fragments, which are wrapped about
by the [AuP(NC4H4)2(C6H4)2P(NC4H4)2]3“belts” anchored
to the central parts by Au-Au bonds (Figure 1). The presence of only one signal in the 31P spectra of these
compounds is indicative of the D3h symmetry group the
Au3(PP)3belts belong to. The number of signals in the1H
and13C spectra is also completely compatible with the D 3h
structure of the belt and the [Au3Cu2(C2C6H4X)6] core, where
three equivalent rods form a right triangle with two copper ions in its center above and below the belt plane. Comparison of the corresponding chemical shifts observed in the spectra of these two groups of compounds again indicates a very weak shielding interaction between the “rods” and “belt”. The change of phenyl for pyrrolyl substituents in the diphosphine ligands has a nearly negligible and nonsystem-atic effect on the shielding of the alkynyl carbons disposed most closely (three to four bonds separation) to the phos-phorus atoms.
Photophysical Properties. Figures 2 and 3 show the UV/
vis absorption and emission spectra of complexes 8-14 in
dichloromethane. The broad, higher-energy absorptions below 270 nm for all of the titled compounds are ascribed to the intraligand π f π* (CtCAr) transitions of the alkynyl moieties. This assignment is consistent with the previous reports on the relevant alkynyl analogues, for which the dominant absorption band in the spectral region of 230-300 nm, in general, is ascribed to the characteristic band of the alkynyl-phosphine moieties in combination with their smaller congeners.5,16,18As for the lower-lying transitions, the bands centered at∼300 nm can be reasonably assigned to an electronic transition from the σ (Au-P) orbital to an empty π*(CtCPh or phosphine) antibonding orbital located at the bridging phenyl group.16,18The 300-400 nm absorp-tion bands for these complexes are mainly due to the Cu-π-alkynyl fragment metal- and cluster-centered transi-tions.5,16 The lowest energy transition featured with a shoulder and extended to∼500 nm is likely to possess some metal (d of Au and Cu) f alkynes (π*) charge transfer character, but due to the very mixed nature of the HOMO and the LUMO (see computational section), it cannot be described as a pure metal-to-ligand charge-transfer (MLCT) transition.
Remarkably strong luminescence was observed for all of the compounds in dichloromethane. Complexes 8-11 pos-sess the same bridging ligand, PPh2C6H4PPh2, while the
difference lies in the variation of the X-substituting group in the para position, as marked in Figure 1. As a result, the emission peak wavelength reveals a bathochromic shift from
8 (576 nm) to 11 (686 nm), as shown in Figure 2. Because
the electron-donating strength of the X-substituent gradually increases from 8 to 11, the result can be rationalized by the lifting of the HOMO energy level upon increasing the electron density at the alkyne moiety, giving rise to the reduction of the HOMO-LUMO energy gap. Similar results were obtained for the other class of complexes, 12-14, that possess another bridging ligand, PR2C6H4PR2(R ) NC4H4).
Anchoring the substituent in the X position with increasing electron donating strength from -NO2to -OMe causes the
emission red shift from 607 nm (12) to 671 nm (14) (see Figure 3). These results are also consistent with those deduced from the theoretical approach (vide infra). As for the bridging-ligand effect, for example, complexes 8 and 12, having different bridging ligands but identical X substituents, exhibit appreciably different emission peak wavelengths (576 nm for 8 versus 607 nm for 12). As elaborated in the computational section, the LUMOs of 8 and 12 are partially contributed by the bridging-ligand. Thus, in comparison to the PPh2C6H4PPh2 ligand in 8, the stronger
electron-withdrawing ligand PR2C6H4PR2(R ) NC4H4) in 12 causes
greater reduction of the LUMO energy, hence, the reduction of the emission energy gap. A similar argument holds for complexes 9 and 13 as well as for complexes 10 and 14.
The quantum yields (Q.Y.), observed lifetimes, and hence the deduced radiative decay rate constants (kr) for the titled
Au-Cu complexes are summarized in Table 1. The
mag-(18) Mu¨ller, T. E.; Choi, S. W.-K.; Mingos, D. M. P.; Murphy, D.; Williams, D. J.; Yam, V. W.-W. J. Organomet. Chem. 1994, 484, 209–224.
Figure 2. UV/vis absorption and normalized emission spectra of 8-11 in
deaerated CH2Cl2at room temperature, λexcit) 450 nm.
Figure 3. UV/vis absorption and normalized emission spectra of 12-14
in deaerated CH2Cl2at room temperature, λexcit) 450 nm.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
nitudes of the observed lifetimes of microseconds and the radiative decay rate constant of∼105s-1lead us to conclude
that the emission originates from the triple state manifold, that is, the phosphorescence. A striking feature of these complexes lies in that the phosphorescence intensity is subject to minor O2 quenching in aerated solutions. For
example, the Q.Y. of 8 (in CH2Cl2) was measured to be 0.93
(4.80 µs) in a degassed solution, while to our surprise, Q.Y. only decreased to 0.73 with an observed lifetime as long as 3.15 µs. On the basis of the collisional type of energy transfer, the rate of O2 quenching to the triplet state is
approximately 1/9 of the (solvent) diffusion-controlled rate. Accordingly, in the aerated solution, the observed lifetime of phosphorescence is commonly an order of magnitude lower, dropping the value to a few hundred nanoseconds.19
The high quantum yield and hence rather small O2quenching
rate manifest the uniqueness of the framework built by these titled supramolecules, in which the emission chromophores, that is, metal and alkynyl groups, are largely protected by the bulky ancillary and bridging ligands (see Figure 1). This unique scaffold greatly prevents O2collision and hence the
reduction of the O2 quenching rate. The nearly O2
-independent phosphorescence is remarkable and should render ample applications in, for example, phosphorescence dyes in time-resolved imaging.20
Computational Results. Density functional calculations
were performed to provide additional insight into the
structural and electronic properties of the supramolecular Au(I)-Cu(I) complexes (for computational details, see the Experimental Section). The computational results are sum-marized in Table 2 and Figure 4. In the case of complex 9, the availability of the X-ray crystal structure enabled the comparison between the theoretical and experimental ge-ometries. The DFT-optimized and experimental structural parameters were found to be in good agreement. All of the studied complexes 8-15 retain the “rods-in-belt” structural motif during the geometry optimization, and the changes in structural parameters are fairly small. Some structural trends can be observed, however. As X is changed from electron-withdrawing to electron-donating, the Au-Au bond contacts Table 1.Photophysical Properties of the Au-Cu Complexes in CH2Cl2.
complex λab/nm (10 -3 ε/cm-1M-1) λem/nma Q.Y.b τobs/µsa kr/s-1,c 8 aerated 265 (77.2), 351 (44.8), 408sh (28.3) 576 0.73 3.15 1.94× 105 degassed 0.93 4.80 9 aerated 263 (80.7), 336sh (21.1), 403 (26.0) 594 0.67 3.69 1.55× 105 degassed 0.96d 5.92 10 aerated 264 (79.6), 287sh (54.0), 402 (20.8) 620 0.33 2.60 1.33× 105 degassed 0.77 5.78 11 aerated 264 (70.8), 294 (58.8), 395 (32.5) 686 0.04 3.10 1.04× 105 degassed 0.10 9.20 12 aerated 333 (54.4), 607 0.55 4.81 9.14× 104 degassed 0.62 6.82 13 aerated 253 (72.8), 401 (18.3), 650 0.26 3.72 7.82× 104 degassed 0.61 7.80 14 aerated 260 (74.1), 398 (20.7) 671 0.12 1.61 7.22× 104 degassed 0.19 2.69 aλ
excit) 450 nm.bMeasured in dichloromethane solutions; DCM in methanol was used as a standard for the quantum yield measurements.ckris deduced from data obtained in the degassed solution.dThis value is slightly higher than that given in ref 6, but both are within the experimental error (8-10%). Table 2.Selected Structural Parameters and Frontier Orbital Characteristics of the Substituted Au(I)-Cu(I) Complexes 8-15
8 9 10 11 12 13 14 15
X NO2 H OMe NMe2 NO2 H OMe NMe2
R Ph Ph Ph Ph NC4H4 NC4H4 NC4H4 NC4H4
Structural Parameters Shown in Figure 4 (Å)a
R(Au-Au(c-b)) 2.91 2.89 (2.87)b 2.88 2.86 2.89 2.86 2.84 2.82 R(Au-Au(c-c)) 3.40 3.39 (3.33-3.36) 3.39 3.39 3.39 3.37 3.37 3.35 R(Au-Cu) 2.81 2.82 (2.75-2.91) 2.81 2.80 2.83 2.83 2.81 2.80 R(Au-C) 2.03 2.03 (1.97-2.01) 2.03 2.03 2.02 2.02 2.03 2.03 R(Cu-C) 2.07 2.07 (2.06-2.08) 2.07 2.07 2.07 2.07 2.07 2.07 R(Au-P) 2.39 2.38 (2.30-2.32) 2.38 2.38 2.37 2.36 2.36 2.36
Frontier Orbital Characteristics (eV)
HOMO-LUMO gap 1.82 1.80 1.63 1.38 1.66 1.45 1.17 0.87
HOMO -8.70 -7.80 -7.35 -6.76 -9.17 -8.32 -7.77 -7.12
LUMO -6.88 -6.00 -5.72 -5.38 -7.51 -6.87 -6.60 -6.24
aAverage bond lengths.bFor complex 9, the experimental structural parameters are listed in parentheses.
Figure 4. Optimized geometry of complex 9, showing the structural
parameters listed in Table 2 for all of the studied complexes. The complex is shown from a top view, omitting phenyl rings and hydrogen atoms for clarity.
Koshevoy et al.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
between the central and “belt” fragments become shorter. The Au-Au and Au-Cu distances in the central fragment show a similar decreasing trend. Because the electron-donating substituents in the X position contribute additional electron density to the central fragment, the Coulombic attraction between the anionic central fragment and the cationic “belt” fragment increases, shortening the supramo-lecular bond contacts. The structural effect of changing R from Ph to NC4H4 is less significant, although a slight
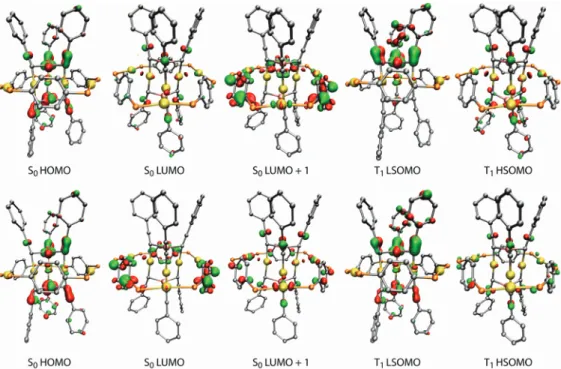
shortening of the Au-Au distances can be observed. The frontier molecular orbitals of the S0and T1electronic
states of complexes 9 and 13 (X ) H) are illustrated in Figure 5. In both complexes, HOMOs are located within the central fragment, the most significant contributions coming from the d(Cu), π(CtCPh), and d(Au) orbitals. In complex 9 (R ) Ph), the main contributions to the LUMO come from central fragment sp(Au), π*(CtCPh), and sp(Cu) orbitals. The LUMO+1 of 9 is delocalized over the “belt” fragment, with the largest contributions coming from the bridging Ph groups. Changing X from electron-withdrawing into electron-donat-ing affects the frontier orbitals mainly by increaselectron-donat-ing the contribution of the “belt” fragment on the LUMO. Changing R from Ph to NC4H4 further increases the contribution of
the “belt” fragment to the LUMOs, practically reversing their ordering in comparison to 9. For all studied complexes, geometry optimization of the lowest-lying triplet state results
in minor structural changes mainly within the Au-Au, Au-Cu, and Cu-C distances of the central fragment, but the “rods-in-belt” structural motif in general remains unper-turbed. For complexes with R ) Ph, the Au-Au bond contacts between the central and “belt” fragments shorten slightly, and the highest singly occupied molecular orbital (HSOMO) of the T1 state is closely related to the central
fragment-based S0LUMO. For complexes with R ) NC4H4,
the Au-Au bond contacts between the central and “belt” fragments are slightly elongated, and the T1 HSOMO is
related to the S0 LUMO+1. The lowest singly occupied
molecular orbital of the T1state is related to the S0HOMO
for both R ) Ph and R ) NC4H4.
Considering the strong contribution of the heterometallic core to the frontier orbitals of the S0and T1states, the
long-wavelength phosphorescence observed for the complexes is likely to involve a triplet state with a higher-energy SOMO of predominantly metal sp character and a lower-energy SOMO of mixed metal d and alkynyl π character. The initial excited states, such as MLCT [d(Cu, Au) f π*(CtPh)], end up in the emitting triplet state due to efficient intersystem crossing.6For complexes with R ) NC4H4, MLCT
excita-tions to “belt” ligands should be considered, as well. When R ) NC4H4, or X is an electron-donating substituent, a larger
contribution of the less protected “belt” fragment to the excited states might be one reason for the smaller quantum yields observed for such complexes.
The relative energies of the frontier orbitals are signifi-cantly altered by the modification of X and R (Table 2). As X changes from electron-withdrawing to electron-donating, the energy of the HOMO increases more than the energy of the LUMO, decreasing the magnitude of the HOMO-LUMO gap. The influence of changing the R is opposite of the effect of changing X, the more electron-withdrawing NC4H4 (19) (a) Tung, Y.-L.; Wu, P.-C.; Liu, C.-S.; Chi, Y.; Yu, J.-K.; Hu, Y.-H.;
Chou, P.-T.; Peng, S.-M.; Lee, G.-H.; Tao, Y.; Carty, A. J.; Shu, C.-F.; Wu, I. Organometallics 2004, 23, 3745–3748. (b) Hwang, F.-M.; Chen, H.-Y.; Chen, P.-S.; Liu, C.-S.; Chi, Y.; Shu, C.-F.; Wu, F.-I.; Chou, P.-T.; Peng, S.-M.; Lee, G.-H. Inorg. Chem. 2005, 44, 1344–1353. (c) Tung, Y.-L.; Lee, S.-W.; Chi, Y.; Chen, L.-S.; Shu, C.-F.; Wu, F.-I.; Carty, A. J.; Chou, P.-T.; Peng, S.-M.; Lee, G.-H.
AdV. Mater. 2005, 17, 1059–1064.
(20) Hanaoka, K.; Kikuchi, K.; Kobayashi, S.; Nagano, T. J. Am. Chem.
Soc. 2007, 129, 13502–13509.
Figure 5. Selected frontier molecular orbital isodensity plots for the Au(I)-Cu(I) complexes 9 (top) and 13 (bottom). Phenyl (9) and pyrrolyl rings (13) of
the “belt” fragment, and all hydrogen atoms omitted for clarity. Isodensity value of 0.04 au was used for all plots. SOMO ) singly occupied molecular orbital.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
decreasing the HOMO-LUMO gap in comparison to Ph. In this case, the decrease of the HOMO-LUMO gap is due to the energy of the LUMO decreasing more than the energy of the central fragment-based HOMO. The magnitude of the calculated HOMO-LUMO gaps decreases in the order 8 >
9 > 12 > 10 > 13 > 11 > 14. The decreasing trend of the
HOMO-LUMO gaps correlates well with the experimentally observed red-shift of the emission, except for complex 11, which was found to emit at slightly lower energy than complex 14. The HOMO-LUMO gaps calculated for the complexes also provide a qualitative estimate of their kinetic stability. For instance, experimental isolation of the complex where R ) NC4H4 and X ) NMe2 (15), which has a
considerably smaller HOMO-LUMO gap in comparison to other studied complexes, proved to be unsuccessful due to its instability toward decomposition.
Overall, the computational results suggest that, while modifying X or R has little effect on the structural properties of the supramolecular Au(I)-Cu(I) complexes, their elec-tronic properties are significantly altered by the modification of the substituents. Investigation of the electronic charac-teristics suggests that a metal-centered triplet emission within the central Au(I)-Cu(I)-alkynyl fragment plays a key role in the observed long-wavelength phosphorescence.
Conclusion
We have presented a systematic synthetic, photophysical, and theoretical study of a family of intensely luminescent heterometallic clusters of the general formula [{Au3Cu2
-(C2C6H4X)6}Au3(PR2C6H4PR2)3][PF6]2(X ) NO2, H, OMe,
NMe2; R ) C6H5, NC4H4). These complexes adopt the same
structural pattern and consist of heterometallic alkynyl clusters [Au3Cu2(C2C6H4X)6]
-“wrapped” by cationic [Au3(PR2C6H4PR2)3]3+“belts”. All of the compounds under
study exhibit strong room-temperature phosphorescence in solution with a maximum quantum yield of 0.96. The emission maxima wavelength reveals a bathochromic shift upon (a) the increase of the electron-donating strength of the X substituents of the alkynyl ligands and (b) the increase of the electron-withdrawing strength of the R groups of the diphosphines. The remarkable feature of these complexes consists of a very low degree of phosphorescence quenching by O2that is evidently determined by the uniqueness of their
framework, in which the emission chromophores, that is, metal and alkynyl groups, are protected by the bulky ancillary and bridging ligands. The theoretical calculations of the electronic structures showed that modifying X or R substit-uents has little effect on the structural features but signifi-cantly influences the electronic properties. The frontier orbital characteristics of the complexes suggest that the observed long-wavelength phosphorescence is associated with metal-centered triplet emission within the heterometallic alkynyl cluster.
Acknowledgment. Financial support from Academy of
Finland (I.O.K.) and Russian Foundation for Basic Research (grant 07-03-00908-a) is acknowledged.
Supporting Information Available: ESI-mass spectra of 8, 10-14; 1H-1H COSY spectrum of 13; and optimized Cartesian
coordinates of the studied systems in atomic units. This material is available free of charge via the Internet at http://pubs.acs.org.
IC801987T
Koshevoy et al.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
![Figure 1. Schematic structure of the dications [{Au 3 Cu 2 (C 2 C 6 H 4 X) 6 }Au 3 - -(PR 2 C 6 H 4 PR 2 ) 3 ] 2+ (8-14), (X ) NO 2 , H, OMe, NMe 2 ; R ) Ph, NC 4 H 4 ).](https://thumb-ap.123doks.com/thumbv2/9libinfo/8685520.197693/5.883.67.662.89.524/figure-schematic-structure-dications-au-cu-ome-nme.webp)