Solid State and Solution Self-Assembly of Helical Polypeptides
Tethered to Polyhedral Oligomeric Silsesquioxanes
Shiao-Wei Kuo,*,†Hsin-Fang Lee,‡Wu-Jang Huang,§Kwang-Un Jeong,| and Feng-Chih Chang*,‡
Department of Materials and Optoelectronic Science, Center for Nanoscience and Nanotechnology, National Sun Yat-Sen UniVersity, Kaohsiung, Taiwan, Institute of Applied Chemistry, National Chiao Tung UniVersity, Hsin Chu, Taiwan, Department of EnVironmental Science and Engineering, National Ping-Tun UniVersity of Science and Technology, Ping-Tun, Taiwan, and Department of Polymer-Nano Science and Technology, Chonbuk National UniVersity, Jeonju 561-756, Korea
ReceiVed October 22, 2008; ReVised Manuscript ReceiVed January 8, 2009
ABSTRACT: New macromolecular self-assembling building blocks, polyhedral oligo- silsesquioxane (POSS)-helical polypeptide copolymers, have been synthesized through the ring-opening polymerization (ROP) of γ-benzyl-L-glutamate N-carboxyanhydride (γ-Bn-Glu NCA) using aminopropyl isobutyl-POSS as a macroinitiator. The incorporation of POSS units at the chain ends of PBLG moieties has two important effects: allowing intramolecular hydrogen bonding between the POSS and PBLG units to enhance the latter’s R-helical conformations in the solid state and preventing the aggregation of nanoribbons through the POSS blocks’ protrusion from the ribbons to allow the formation of clear gels in solution.
Introduction
The secondary structure of peptide chains plays a crucial role in the formation of the well-defined tertiary structure of proteins. Unlike proteins, most synthetic polymers lack the ability to retain specific 3-D structures in solution or in bulk phase due to their random coil conformations. Such irregular conformations hamper the potential of synthetic polymers as useful building blocks for the creation of self-assembled structures with a well-defined size and 3-D shape.1 As a result, rod-coil block copolymers containing a well-defined rigid segment in their architectures have received considerable attention due to their unique self-assembly behaviors which differ from that of coil-coil diblock copolymer.2Poly(γ-benzyl-L-glutamate) (PBLG) is a synthetic polypeptide, which can form both R-helix and β-sheet secondary structures stabilized by intramolecular and intermolecular hydrogen bonding, respectively.3The R-helical structure of PBLG serves as a rigid-rod like structure in solid and solution state,4which provides its unique bulk and solution behavior such as thermotropic liquid crystalline ordering5,6and thermo-reversible gelation, respectively.7,8
Rod-coil block copolymers based on a rigid PBLG helix have been studied extensively for several decades.9Recently, Manners et al. have found that random coil-PBLG block copolymers with flexible polyferrocenylsilane blocks10 form thermo-reversible gels in toluene solution.11 The underlying mechanism for the PBLG diblock copolymer gelation was suggested to be different from that of the gelation of pure PBLG because the random coil-PBLG diblock copolymers could not form a liquid crystalline phase in dilute solutions, which is essential for pure PBLG gelation.12The proposed mechanism for the self-assembly behavior of the random coil-PBLG block copolymers implies that a wide variety of PBLG block
copolymers with novel architectures other than random coil-helix could generate well-defined supramolecular structures in solu-tion.
Polyhedral oligomeric silsesquioxane (POSS) derivatives comprise a family of molecularly precise, near-isotropic mol-ecules that have diameters ranging from 1 to 3 nm, depending on the number of silicon atoms in the central cage and the nature of its peripheral substituent groups.13-19A cubic T
8 silsesqui-oxane unit possesses a cubic inorganic Si8O12core surrounded by eight tunable substituent groups. POSS has been recognized as a well-defined building block for nanostructured materi-als.20-25The cubic silsesquioxane unit can not only be viewed as a nanoparticle for both its size and filler function, but also a well-defined macromonomer for its ability to undergo polym-erization. It appears logically that two well-defined macromo-lecular architectures, POSS and PBLG, could be combined to generate polymer building blocks with well-defined 3-D shapes, which dictate the self-assembly process and resulting in su-pramolecular structures. Because of their unique structures, POSS derivatives are useful building blocks for the preparation of nanostructured materials.26,27
We were interested in combining the well-defined macro-molecular architectures of POSS and PBLG to generate polymeric building blocks having distinct 3-D shapes for the self-assembly of supramolecular structures. Herein, we report the synthesis of chain end-tethered PBLG-POSS copolymers and their self-assembly in solid state and in solution. We synthesized the POSS-PBLG copolymers through the ring-opening polymerization (ROP) of γ-benzyl-L-glutamate N-carboxyanhydride (γ-Bn-Glu NCA) using aminopropyl isobutyl-POSS as a macroinitiator (Scheme 1).
Experimental Section
Materials. Aminopropyl isobutyl-POSS (Hybrid Plastics),
tet-rahydrofuran (anhydrous; Aldrich), and N,N-dimethylformamide (anhydrous; Aldrich) were used as received.γ-Benzyl-L-glutamate N-carboxyanhydride (Bn-Glu NCA) was prepared according to a literature procedure28and stored at -30 °C. Toluene was used
without further purification.
Synthesis of POSS-PBLG Copolymers. A Schlenk flask fitted
with a stirrer bar and drying tube was charged with the appropriate
* To whom correspondence should be addressed. E-mail: changfc@ mail.nctu.edu.tw, telephone/fax: 886-3-5131512 (F.-C.C.). E-mail: kuosw@ faculty.nsysu.edu.tw, fax: 886-7-5254099 (S.-W.K.).
†Department of Materials and Optoelectronic Science, Center for Nanoscience and Nanotechnology, National Sun Yat-Sen University.
‡Institute of Applied Chemistry, National Chiao Tung University. §Department of Environmental Science and Engineering, National Ping-Tun University of Science and Technology.
|
Department of Polymer-Nano Science and Technology, Chonbuk National University.
10.1021/ma802370y CCC: $40.75 2009 American Chemical Society Published on Web 02/12/2009
amount of Bn-Glu NCA in a THF/DMF mixture (0.1 g/mL). The required amount of macroinitiator-aminopropyl isobutyl-POSS was added, and the reaction mixture was stirred at room temperature as shown in scheme 1. After 72 h, the block copolymers were precipitated in diethyl ether, filtered, and vacuum-dried.
Preparation of Gels. For organogels, POSS-PBLG copolymers
and toluene were mixed in a sealed vial and heated until the mixture became a homogeneous solution. The homogeneous solution was stored at 22°C for 24 h. After 24 h, the organogel was subjected
to gravity for 24 h. When no flow was observed, we defined the state as a gel. The lowest gelation concentration in this condition was determined as Cgel. Tgelwas measured by the procedure reported
by Hirst et al.29All C
geland Tgelvalues were measured three times.
Characterization. 1H NMR spectra were recorded at room
temperature on a Bruker AM 500 (500 MHz) Spectrometer using the residual proton resonance of the deuterated solvent as the internal standard. High-resolution solid-state13C NMR experiments
were carried out at room temperature using a Bruker DSX-400
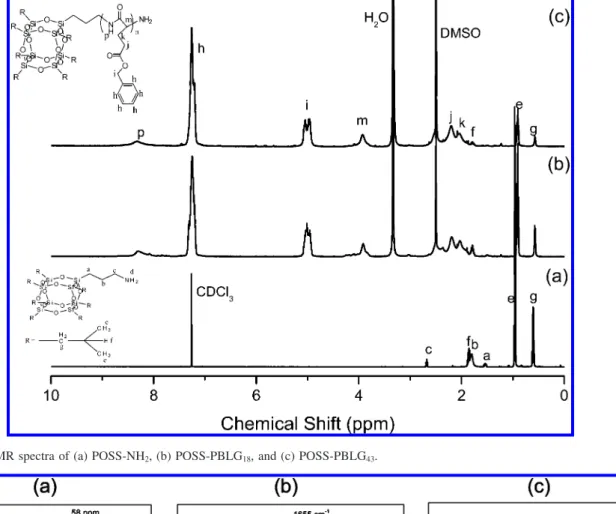
Figure 1.1H NMR spectra of (a) POSS-NH
2, (b) POSS-PBLG18, and (c) POSS-PBLG43.
Figure 2.(a) Solid state13C NMR spectra, (b) IR spectrum and (c) Raman spectrum of POSS-PBLG 18. Scheme 1. Synthesis of POSS-PBLG Copolymers
Spectrometer operating at resonance frequencies of 399.53 and 100.47 MHz for1H and13C, respectively.
Thermal analysis through differential scanning calorimetry (DSC) was performed using a DuPont 910 DSC-9000 controller at a scan rate of 10 °C/min over a temperature range from 25 to 120 °C under a nitrogen atmosphere.
IR spectra were obtained using a Nicolet Avatar 320 FT-IR Spectrometer; 32 scans were collected with a spectral resolution of 1 cm-1. The holder was placed in the sample chamber and the spectrum was recorded under a N2 purge to maintain the film’s
dryness. 2D IR correlation analysis was conducted using “Vector 3D” software (from Bruker Instrument Co.). All of the spectra were normalized before being subjected to the 2D correlation analyses. Raman spectroscopy was performed using a Jobin-Yvon LA-BRAM HR800 with 514.5-nm Ar laser excitation.
Transmission electron microscopy (TEM) images were obtained on a Hitachi H-7500 Electron Microscope operating at 100 kV. The TEM specimen was prepared by gently placing a carbon-coated copper grid on the fraction of organogel. The sample specimen was stained with the vapor from an aqueous solution of RuO4to enhance
phase contrast.
The oriented fiber 2D WAXD patterns were obtained using a Rigaku X-ray imaging system with an 18 kW rotating anode X-ray generator. The patterns were recorded on an image plate (Rigaku, R-AXIS-IV). A 30 min exposure time was required for a high-quality pattern.
Small angle X-ray scattering (SAXS) experiments were carried out at the BL17B3 beamline of the National Synchrotron Radiation Research Center (NSRRC), Taiwan. We used an X-ray beam of
0.5-mm diameter and a wavelength of 1.24 Å for the SAXS measurement and collected SAXS (with a Q range of 0.015-0.2 Å-1) with linear position-sensitive detectors (20 cm in length). The Q values of the SAXS and WAXS profiles were calibrated using standard samples of polyethylene, Ag-behenate, and tripalmitat. The block copolymer samples were sealed between two Kapton windows of 1 mm thickness and measured at room temperature. X-ray diffraction data were collected on the wiggler beamline BL17A1 of the NSRRC, Taiwan. A triangular bent Si(111) single crystal was employed to obtain a monochromated beam of wavelengthλ)1.3344Å.
The XRD patterns were collected using imaging plate (IP; Fuji BAS III, area ) 20× 40 cm2) curved with a radius equivalent to
the sample-to-detector distance of 280 mm. With 100 µm pixel resolution of the IP and a typical exposure time of 15 min, the XRD was measured cover a range of values of Q from 0.05 to 2.2 Å-1, where the X-ray vector transfer Q ) 4π sin(θ)/λ is defined by the scattering angle 2θ and λ. The two-dimensional X-ray diffraction patterns observed for the sample (typical diameter 10 mm; thickness 1 mm) were circularly averaged to one-dimensional diffraction profile I(Q), with Q value calibrated using standard samples of Ag-Behenate and Si powder (NBS 640b).
Results and Discussion
Self-Assembly Behavior of POSS-PBGL in Solid State. The
POSS-PBLG copolymers were synthesized by the ring-opening polymerization (ROP) of γ-benzyl-L-glutamate N-carboxyan-hydride (γ-Bn-Glu-NCA) with the aminopropyl isobutyl-POSS
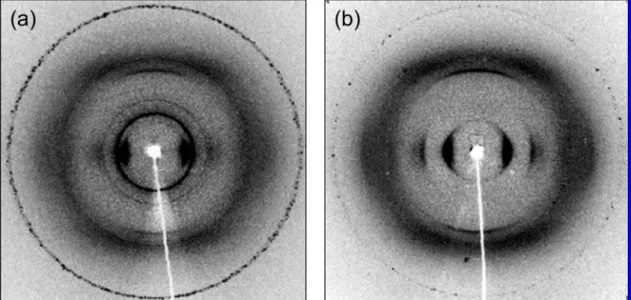
Figure 3. 2D WAXD fiber patterns of (a) POSS-PBLG18and (b) pure PBLG53.
as a macroinitiator. The polymerization was performed in a THF/DMF mixture at room temperature for 72 h. The precipita-tions of the reaction soluprecipita-tions into diethyl ether gave the POSS-PBLG copolymers in good yields. The POSS-POSS-PBLG copolymers were characterized by1H NMR as shown in Figure 1. In the1H NMR spectrum of the amino-terminated POSS (POSS-NH2; (Figure 1a), the appearance of a signal at 2.73 ppm represents the primary amino-terminated. Next, we used POSS-NH2 to initiate the polymerization ofγ-benzyl-L-glutamate N-carboxy-anhydride (Bn-Glu NCA), which was conducted in DMF solution at room temperature. The length of the γ-benzyl- L-glutamate segment was controlled by adjusting the molar ratio of the Bn-Glu NCA and the primary amino end-functionalized POSS initiator. After precipitation and drying, these POSS-b-PBLG copolymers were characterized using1H NMR spectros-copy (Figure 1, parts b and c). Table 1 summarizes the average number of amino acid units in the PBLG block calculated by 1H NMR integration.
Pure PBLG systems having degrees of polymerization (DP) below 18 exist as mixtures of columnar hexagonal arrangements of R-helices and lamellar assemblies ofβ-sheets.1
The incor-poration of a POSS unit at the chain end of PBLG (DP ) 18), however, resulted only in the columnar hexagonal arrangement of R-helices, as evidenced from the solid state13C NMR, FT-IR, FT-Raman spectra, and 2D WAXD analyses as shown in Figures 2 and 3. The secondary structures of PBLG can be identified on the basis of the distinctly different resonances observed in their NMR spectra (δ ) 58 ppm for the R-helix; δ ) 53 ppm for the β-sheet).1,3 Of these two signals, POSS-PBLG18exhibited (Figure 2a) only the resonance at 58 ppm, implying that only its R-helix form existed in the bulk. FTIR and FT-Raman spectra of these peptides can also be used to verify the types of secondary structures based on the amide I band as shown in Figure 2, parts b and c, respectively. For the R-helical conformation, this signal appears at 1655 cm-1. A band at 1630 cm-1is characteristic of theβ-sheet conformation.1Our POSS-PBLG18sample exhibited its amide I band at 1655 cm-1, with no signal present at 1630 cm-1, in its FTIR and FT-Raman spectra, again suggesting that only the R-helical conformation exists in the bulk.
2D WAXD patterns of oriented samples by shearing also can be used to identify secondary peptide structures.1,3Parts a and b of Figure 3 present the fiber patterns, measured at room temperature, of POSS-PBLG18and pure PBLG53samples that had been annealed at 373 K for 2 days. Pure PBLG53(Figure 3b) displays strong equatorial reflections with relative positions at 1:31/2:41/2, indicating hexagonal packing of cylinders com-posed of 18/5 (18 residues in five turns) R-helices; the lines at meridional reflections account for a pitch length of these R-helices of ca. 0.52 nm.1POSS-PBLG
18(Figure 3a) exhibits a similar pattern to that of pure PBLG53at its equatorial and meridional reflections, also suggesting that its R-helix form exists in the solid state. POSS-PBLG18exhibits, however, some less-oriented rings relative to those of POSS crystals, as has been reported previously.30 As a result, it seems that the incorporation of the POSS moiety at the PBLG chain end leads to intramolecular hydrogen bonding between the POSS and PBLG units that enhances the formation of the PBLG R-helix; recall that this conformation is stabilized through such interac-tions.
Scheme 2. Intramolecular Hydrogen Bonding Interaction of POSS-PBLG Diblock Copolymer
Table 1.Characterizations of POSS-PBLG Copolymers and Pure PBLG Homopolymer
sample yield (%) Mna Cgelb(wt %) Tgelb(°C) Wrc(nm)
POSS-PBLG18 63 4800 weak gel weak gel 3.3e
POSS-PBLG37 58 9000 0.8 41 5.5e
POSS-PBLG43 57 10300 0.3 44 8.1
POSS-PBLG56 62 13100 0.2 46 11.8
pure PBLG53 60 11600 1.7 33
aMolecular weight and degree of polymerization calculated by1H NMR integration.bCritical gelation concentrations in wt % and gel-solution
transition temperature.cThe width of the ribbon formed in toluene measured
by SAXS.dThe width of the PBLG helix calculated by1H NMR integration. (Lhelix(nm) ) NPBLG× 0.15 nm where NPBLGis the average number of residues in the PBLG helix determined by1H NMR).
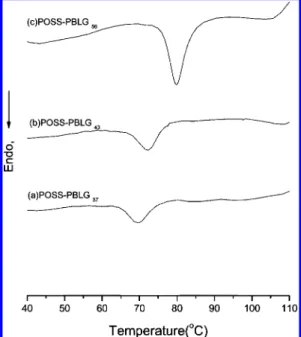
Figure 5.DSC profiles of dried toluene gels: (a) POSS-PBLG37, (b) POSS-PBLG43, and (c) POSS-PBLG56.
We used generalized two-dimensional (2D) IR correlation spectroscopy to confirm the presence of intramolecular hydrogen
bonds between the POSS and PBLG units. A 2D IR spectrum is obtained as a function of two independent wavenumber axes; the peaks located on the spectral plane can be used to study intra- and intermolecular interactions between functional groups.31In general, two types of spectra, 2D synchronous and asynchronous, are obtained; the correlation intensities in the 2D synchronous and asynchronous maps reflect the relative degrees of in-phase and out-of-phase responses, respectively. The 2D synchronous spectra are symmetric with respect to the diagonal line in the correlation map. Auto peaks, which represent the degree of autocorrelation of perturbation-induced molecular vibrations, are located at the diagonal positions of a synchronous 2D spectrum; their values are always positive. When an auto peak appears, the signal at that wavenumber would change greatly under environmental perturbation. Cross-peaks located at off-diagonal positions of a synchronous 2D spectrum (they may be positive or negative) represent the simultaneous or coincidental changes of the spectral intensity variations mea-sured at V1and V2. Positive cross-peaks result when the intensity variations of the two peaks at V1 and V2 occur in the same direction (i.e., both increase or both decrease) under the environmental perturbation; negative cross-peaks reveal that the intensities of the two peaks at V1and V2change in opposite directions (i.e., one increases while the other decreases) under perturbation.32-35
Figure 4a presents the synchronous 2D correlation map (from 800 to 2000 cm-1) for the POSS-PBLG18sample. The bands at 1730, 1655, and 1550 cm-1correspond to CdO, amide I, and amide II bands of the PBLG, respectively. The band at 1087 cm-1represents Si-O-Si stretching of the POSS cage. These four absorptions exhibit many positive auto and cross peaks; in addition, a new autopeak appeared at 1150 cm-1, which was absent in the 1D FTIR spectra, indicating that hydrogen bonding occurs between the amide and carbonyl groups of the PBLG unit and the siloxane groups of the POSS moiety. In contrast, the corresponding asynchronous 2D correlation map (Figure 4b) exhibits no such auto or cross peaks within the same wave-number range. These results demonstrate that the incorporation
Figure 6. TEM images of the (a) PBLG53homopolypeptide, and copolymers of (b) POSS-PBLG18, (c) POSS-PBLG43, and (d) POSS-PBLG56from a polymer concentration of 0.2 wt. % in toluene. Samples were observed after RuO4staining.
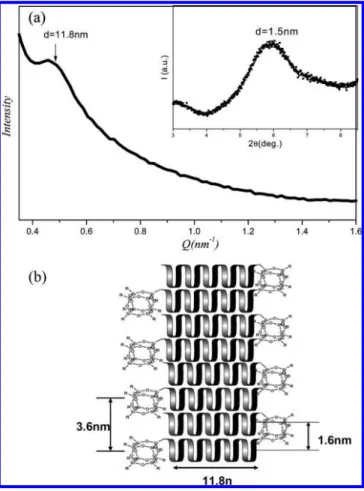
Figure 7. (a) SAXS profiles for dried toluene gels of POSS-PBLG56 showing the width of the ribbon (11.8 nm). The inset denotes the diffraction at 1.5 nm indicating the distance between two PBLG helices obtained by WAXD. (b) Schematic presentation of the nanoribbon formed in the network structure of the toluene gel of POSS-PBLG56.
of a POSS moiety at the PBLG chain end leads to intramolecular hydrogen bonding between the POSS and PBLG units such that the latter units form R-helices in the solid state as shown in Scheme 2.
Self-Assembly Behavior of POSS-PBGL in Solution State. In toluene solution, we observed thermoreversible gelation
of the POSS-PBLG copolymers. The POSS-PBLG copolymers having PBLG helices with 18, 37, 43, and 56 amino acid residues formed transparent gels at relatively low critical gelation concentrations (<1 wt %) in toluene (inset of Figure 6c). In contrary, the PBLG53homopolypeptide formed a turbid gel at higher critical gelation concentrations (>1.5 wt %) in toluene (inset of Figure 6a). The different gelation behavior between POSS-PBLG and PBLG homopolymer implies different gelation mechanism between these two systems. The length of the PBLG helix has an important effect on the gelation of the POSS-PBLG copolymers because theπ-π interaction between the PBLG helices is believed to be the main driving force of the self-assembly and accounts for the stability of the assembled structures.11As shown in Table 1, the critical gelation concen-tration decreases with increasing PBLG length; but the critical gelation temperature increases with increasing PBLG length. Furthermore, the gels retained their shape and thermorevers-ibility on the addition of methanol (5 vol % to toluene). This suggests that there is no end-to-end intermolecular hydrogen bonding between the PBLG blocks, as the methanol can readily disrupt the hydrogen bonding between this PBLG chain.36,37 This result indicates that hydrogen bonding is not a key factor for the gelation.
The thermal behavior of the dry gels was studied by differential scanning calorimetry (DSC). Upon heating at a rate of 10 °C/min from room temperature, the dry gels of POSS-PBLG37, POSS-PBLG43, and POSS-PBLG56 exhibited
endo-thermic peaks at 69, 72, and 80°C, respectively (Figure 5a-c). Optical microscopies of these gels also showed complete melting of the bundle fibers at these particular temperatures. These broad endothermic peaks correspond to the melting temperature (Tm)
of the self-assembled structure, as observed by transmission electron microscopy (TEM) shown below. The elevation of melting temperatures of these dry gels with increasing PBLG length can be attributed to theπ-π interaction between the PBLG helices enhancing with increasing PBLG length and thus reinforces the rigidity of the self-assembled structures.
The self-assembled structures formed by POSS-PBLGn
copolymers in toluene were studied by transmission electron microscopy (TEM), small-angle X-ray scattering (SAXS), and wide-angle X-ray diffraction (WAXD). Parts a-d of Figure 6 are TEM images of the fibrous structures formed from PBLG53 homopolypeptide and copolymers of POSS-PBLG18, POSS-PBLG43, and POSS-PBLG56. Pure PBLG samples form thermo-reversible gels in solvents such as toluene and benzyl alcohol.38 The turbid gel that we prepared from the pure PBLG53exhibits disorder and irregular fiber aggregation with widths ranging from 500 to 1000 nm. One aspect of the current understanding of the gelation of PBLG homopolypeptide in solution is based on phase separation.39 The PBLG homopolypeptide undergoes microscopic phase separation when the warm homogeneous solution is cooled. Phase separation increases the concentration of PBLG locally, and the increased concentration forces the PBLG rods into ordered liquid crystalline phases where the rods align parallel to the long axis of the phase-separated domains. Strong dipolarπ-π interactions between the phenyl groups of the PBLG rods stabilize the microfibers that are intertwined into an irregular 3-D network structure.
In contrast, the clear gels of the POSS-PBLG copolymer exist as bundles of ordered fibers in which the fiber units have a width
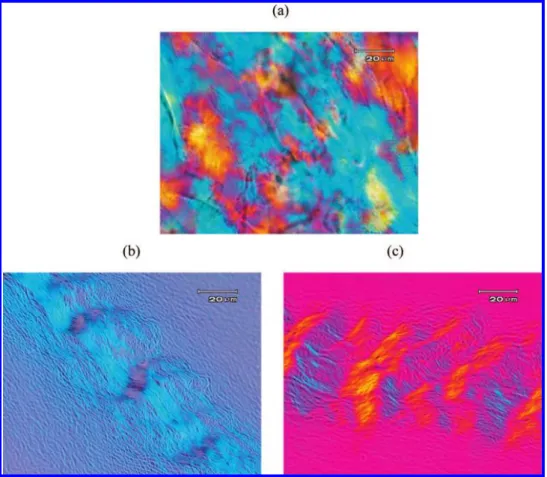
Figure 8. Optical birefringent texture of dried toluene gels (2 wt %) of (a) PBLG53homopolypeptide, (b) POSS-PBLG43, and (c) POSS-PBLG56 copolymers.
of ca. 10 nm. Comparing parts b-d of Figure 5 of POSS-PBLG toluene gels with different PBLG length, the width of fibers increased significantly with increasing PBLG length and sum-marized in Table 1. The width variation suggests that the fibrous structures may consist of the stacking of these POSS-PBLG copolymers and the long axis of the PBLG helix is parallel to the direction of fiber growth.
We performed SAXS measurements on the dried gels because each gel displayed a strong birefringence when examined by using polarized optical microscopy, indicating the presence of regular structures. The SAXS data for the dried POSS-PBLG56 toluene gel (Figure 7a) is consistent with the TEM (Figure 6d). The widths of the individual fibers are indicated by a diffraction peak at 11.8 nm. We did not observe such a lateral dimension in the SAXS spectrum of the fibers in the pure PBLG53 gel because of lack of order. The WAXD profile (inset of Figure 7a) of the dried POSS-PBLG56 gel displays diffraction peaks at 3.6, 1.5, and 0.52 nm, corresponding to the distance between two neighboring POSS blocks, the distance between the R-helices of PBLG, and the pitch length of the R-helix, respectively. Our experimental results strongly suggest that the nanoribbon mechanism for the self-assembly of random coil-PBLG block copolymers11also applies to the POSS-PBLG copolymers. We believe that the POSS-PBLG copolymer self-assembles in an antiparallel manner to form nanoribbon structures (Figure 7b) in which steric interactions between POSS blocks and unfavorable orientations of PBLG helical dipole moments40 are minimized. The long axis of the PBLG helix exists parallel to the plane of the ribbon. In an antiparallel stacking mode, we expect that the distance between two neighboring POSS blocks would be ca. 3.6 nm; i.e., the bulky POSS moieties could be accommodated in the planar nanoribbon structures without a change in the ribbon’s morphology. The POSS blocks protrude from the ribbon because of their high solubility in toluene; this arrangement prevents aggregation of the nanoribbons and results in the formation of clear gels. On the basis of the TEM, SAXS, and WAXD results, a mechanism
for the gelation of POSS-PBLG was observed. The experimental results strongly suggest that the nanoribbon mechanism for the self-assembly of random coil-helical polypeptide block co-polymers11can also apply to the POSS-PBLG copolymers.
The POSS block can prevent the POSS-PBLG copolymers from aligning into a nematic phase during self-assembly in dilute solution, which was found in the homo PBLG gelation. In addition, the strong dipolarπ-π interactions involving phenyl groups between the PBLG helices stabilize the nanoribbon structure and provide the driving force for the self-assembly. The nanoribbon mechanism proposed here explains the depen-dence of Cgelof the block copolymers on the length of the PBLG helix and the independence of gelation on the presence of hydrogen bonding disrupting solvents such as methanol.
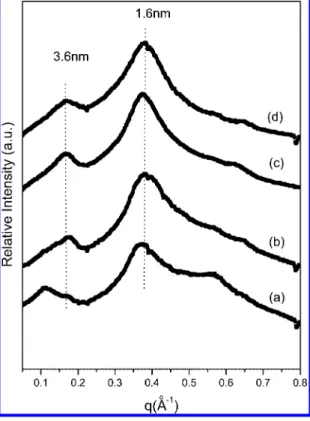
For PBLG itself, a smectic liquid crystalline phase in solution has rarely been observed, due to the polydispersed nature of PBLG made by the conventional ROP of NCA polymerization. Tirrell and co-workers showed that the monodispersed PBLG prepared by a genetic engineering method undergoes smectic ordering in concentrated solution.6 Gallot and co-workers demonstrated that PBLG block copolymers form lamellar structures in concentrated solution, in spite of the PBLG polydispersity, via hexagonal packing of the PBLG helices.41 We anticipate that the POSS-PBLG copolymers form lyotropic liquid crystalline phases in concentrated solution because of the unique structural characteristics of the POSS stabilize the liquid crystalline phase of copolymer molecules and, at the same time, does not prevent the alignment of the helices into the liquid crystalline phase due to its compact chemical structure. Also, the POSS copolymers differ from conventional PBLG block copolymers because of the presence of the POSS which possesses a well-defined structure with monodispersity in molecular weight. When we examined solutions of the POSS-PBLG43and POSS-PBLG56copolymers in toluene, we found that these solutions show very different birefringent textures (Figure 8, parts b and c) from PBLG53homopolypeptide (Figure 8a) under the polarized optical microscope. X-ray diffraction experiments on the dried gel from the concentrated toluene solutions of POSS-PBLG copolymers show two diffraction peaks at 1.6 and 3.6 nm (Figure 9), which suggests that POSS-PBLG copolymers exhibit smectic-like order in concentrated solutions. We propose that a layered structure of POSS-PBLG copolymers in the liquid crystalline phase resulting from an antiparallel stacking of POSS-PBLG copolymers into a 2-D layer in a manner similar to the nanoribbon formation. This result strongly suggests that smectic ordering of the POSS-PBLG copolymers in solutions is feasible with a POSS molecule as a nonrandom coil block even though the PBLG block is poly-disperse in chain length.
Conclusions
We have synthesized and investigated the solid state and solution self-assembly behaviors of a series of POSS-PBLG copolymers. The POSS moiety incorporated at the chain end of the PBLG unit plays two important roles: (a) allowing intramolecular hydrogen bonding to occur between the POSS and PBLG units to enhance the latter’s R-helical conformations in the solid state and (b) preventing the aggregation of nanoribbons although the POSS blocks’ protrusion from the ribbons results in the formation of clear gels in solution. Such a hierarchical self-organization is interesting from a biomimetic point of view, and also provides a new strategy for the design of micrometer-scale superstructured materials with nanometric precision.
Acknowledgment.This work was supported financially by the National Science Council, Taiwan, Republic of China, under Contract Nos. NSC 97-2221-E-110-013-MY3 and NSC
97-2120-Figure 9.X-ray diffraction patterns of dried toluene gels (2 wt %) of (a) POSS-PBLG18, (b) POSS-PBLG37, (c) POSS-PBLG43, and (d) POSS-PBLG56copolymers.
M-009-003. The SAXS experiments were conducted at beamline BL17B3 at the National Synchrotron Radiation Research Center (NSRRC), Taiwan.
References and Notes
(1) Klok, H. A.; Lecommandoux, S. AdV. Mater. 2001, 13, 1217. (2) Lee, M.; Cho, B. K.; Zin, W. C. Chem. ReV. 2001, 101, 3869. (3) Papadopoulous, P.; Floudas, G.; Klok, H. A.; Schnell, I.; Pakula, T.
Biomacromolecules 2004, 5, 81.
(4) Flory, P. J. Proc. R. Soc. London Ser. A 1956, 234, 73. (5) Robinson, C.; Ward, J. C. Nature 1957, 180, 1183.
(6) Yu, S. M.; Conticello, V. P.; Zhang, G.; Kayser, C.; Fournier, M. J.; Mason, T. L.; Tirrell, D. A. Nature 1997, 389, 167.
(7) Tohyama, K.; Miller, W. G. Nature 1981, 289, 813.
(8) Kuo, S. W.; Lee, H. F.; Chang, F. C. J. Polym. Sci., Polym. Chem.
Ed. 2008, 46, 3108.
(9) Lecommandoux, S.; Achard, M. F.; Langenwaiter, J. F.; Klok, H. A.
Macromolecules 2001, 34, 9100.
(10) Kim, K. T.; Vandermeulen, G. W. M.; Winnik, M. A.; Manners, I.
Macromolecules, 2005, 38, 4958.
(11) Kim, K. T.; Park, C.; Vandermeulen, G. W. M.; Rider, D. A.; Kim, C.; Winnik, M. A; Manners, I. Angew. Chem., Int. Ed. 2005, 44, 7964. (12) Tadmor, R.; Khalfin, R. L; Cohen, Y Langmuir 2002, 18, 7146. (13) Xu, H.; Kuo, S. W.; Lee, J. S.; Chang, F. C. Macromolecules 2002,
35, 8788.
(14) Li, G. Z.; Wang, L. C.; Ni, H. L.; Pittman, C. U. J. Inorg. Organomet.
Polym. 2001, 11, 123.
(15) Phillips, S. H.; Haddad, T. S.; Tomczak, S. J. Curr. Opin. Solid State
Mater. Sci. , 204 (8), 21.
(16) Joshi, M.; Butola, B. S. J. Macromol. Sci. Polym. ReV. 2004, C44, 389.
(17) Mark, J. E. Acc. Chem. Res. 2004, 37, 946.
(18) Pielichowaski, K.; Niuguna, J.; Janowski, B.; Pielichowski, J. AdV.
Polym. Sci. 2006, 201, 225.
(19) Lickiss, P. D.; Rataboul, F. AdV. Organmetal. Chem. 2008, 57, 1. (20) Choi, J.; Harcup, J.; Yee, A. F.; Zhu, Q.; Laine, R. M. J. Am. Chem.
Soc. 2001, 123, 11420.
(21) Tamaki, R.; Tanaka, Y.; Asuncion, M. Z.; Choi, J.; Laine, R. M. J. Am.
Chem. Soc. 2001, 123, 12416.
(22) Huang, C. F.; Kuo, S. W.; Lin, F. J.; Huang, W. J.; Wang, C. F.; Chen, W. Y.; Chang, F. C. Macromolecules 2006, 39, 300. (23) Lin, H. C.; Kuo, S. W.; Huang, C. F.; Chang, F. C. Macromol. Rapid
Commun. 2006, 27, 537.
(24) Chan, S. C.; Kuo, S. W.; She, H. S.; Lee, H. F.; Chang, F. C. J. Polym.
Sci.: Polym. Chem. Ed. 2007, 45, 125.
(25) Chan, S. C.; Kuo, S. W.; Chang, F. C. Macromolecules 2005, 38, 3099.
(26) Lee, Y. J.; Huang, J. M.; Kuo, S. W.; Lu, J. S.; Chang, F. C. Polymer 2005, 46, 173.
(27) Lee, Y. J.; Huang, J. M.; Kuo, S. W.; Chen, J. K.; Chang, F. C. Polymer 2005, 46, 2320.
(28) Daly, W. H.; Poche, D. Tetrahedron Lett. 1988, 29, 5859. (29) Hirst, A. R; Smith, D. K; eiters, M. C.F; Geurts, H. P. M.; Wright,
A. C. J. Am. Chem. Soc. 2003, 125, 9010. (30) Cui, L.; Zhu, L. Langmuir 2006, 22, 5982. (31) Noda, I. J. Am. Chem. Soc. 1989, 111, 8116.
(32) Noda, I.; Ozaki, Y. Two-Dimensional Correlation Spectroscopy; John Wiley & Sons: New York, 2004.
(33) Kuo, S. W. Polymer 2008, 49, 4420.
(34) Kuo, S. W.; Huang, W. J.; Huang, C. F.; Chan, S. C.; Chang, F. C.
Macromolecules 2004, 37, 4164.
(35) Kuo, S. W.; Huang, C. F.; Tung, P. H.; Huang, W. J.; Huang, J. M.; Chang, F. C. Polymer 2005, 46, 9348.
(36) Rohrer, P.; Elias, H.-G. Makromol. Chem. 1972, 151, 281. (37) Kim, C.; Kim, K. T.; Chang, Y.; Song, H. H.; Cho, T.-Y.; Jeon, H.-J.
J. Am. Chem. Soc. 2001, 123, 5586.
(38) Doty, P.; Bradbury, J. H.; Holtzer, A. M. J. Am. Chem. Soc. 1956,
78, 947.
(39) Miller, W. G.; Lee, K.; Tohyama, K.; Voltaggio, V. J. Polym. Sci.
Polym. Symp. 1978, 65, 91.
(40) Ludwigs, S.; Krausch, G.; Reiter, G.; Losik, Antonietti M.; Schlaad, H. Macromolecules 2005, 38, 7532.
(41) Perly, B.; Douy, A.; Gallot, B. Makromol. Chem. 1976, 177, 2569. MA802370Y