國 立 交 通 大 學
奈 米 科 技 研 究 所
碩 士 論 文
功能性奈米金粒子對增強酵素催化活性之動力學探討
An Enzymatic kinetics Investigation into the Significantly Enhanced
Activity of Functionalized Gold Nanoparticles
研 究 生 : 吳中書
Chung-Shu Wu
指導教授 : 柯富祥 教授
Prof. Fu-Hsiang Ko
功能性奈米金粒子對增強酵素催化活性之動力學探討
An Enzymatic kinetics Investigation into the Significantly Enhanced
Activity of Functionalized Gold Nanoparticles
研 究 生:吳中書
Student:Chung-Shu Wu
指導教授:柯富祥 教授 Advisor:Prof. Fu-Hsiang Ko
國 立 交 通 大 學
奈 米 科 技 研 究 所
碩 士 論 文
A ThesisSubmitted to Institute of Nanotechnology College of Engineering
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in
Nanotechnology July 2008
Hsinchu, Taiwan, Republic of China
Acknowledgment
習慣了看reference,連寫致謝也要參考一下別人的。"謝謝來,謝謝去,不 如謝天吧!"這兩年來照顧我最多的學長所說的ㄧ句話。 我想,致謝是很隨性的吧! 難道還要像寫paper一樣要求起承轉合和注意 template中的字體,最後還不能忘記格式要正確…。這樣致謝的開頭,我還沒見 過,這樣算創新嗎? 還記得剛到新竹的我,對於整個環境還不熟悉的時候,學長的幫忙是我在這 兩年中最感謝的ㄧ件事;之後的生活,同學們的陪伴,應該是我接下來要感謝的 部份;至於教授給予的幫助以及指導,這份感謝在未來的幾年依舊會存在,我相 信我會用最大的努力來回應教授對我的肯定以及期待。 這段路,有高興有悲傷,值得慶幸的是ㄧ路相陪的夥伴們,因為擁有共同奮 鬥的目標,讓我們更加的靠在一起而密不可分。畢業了,或許會有些感傷,但記 憶會將我們這份感情刻印在腦中。還記得你嗆我的話語;還記得妳體貼的溫柔; 也還記得妳無理的任性…,不想把名字一一的列出,畢竟該感謝的人太多,而只 用一張A4 page寫下,是否會有種不被重視的感覺…。 寫到這邊,也該做個結尾! 兩年的時光,隨著這篇致謝也該落幕,相信未來 再次看到這篇致謝時,時光會倒流而歷歷在目。該感謝的人,ㄧ個都不能少,也 一個不能多,不然,就失去了這頁的意義!功能性奈米金粒子對增強酵素催化活性之動力學探討
研究生:吳中書 指導教授:柯富祥 教授
國立交通大學奈米科技研究所 碩士班
摘 要
在本篇研究中,我們利用奈米金粒子本身對生物分子的選擇性將
酵素固定於粒子的表面並探討其酵素行為的差異,在之前有關這方面
的研究,都會有修飾奈米粒子表面的步驟,然而這些動作不但費時且
複雜,對於酵素的影響也難以估計,更重要的是,這樣的方法無法確
定奈米粒子的表面覆蓋率而造成錯估其動力學參數,因此我們在保持
金奈米粒子的穩定性下,發展不需表面修飾的量測系統以減少對酵素
活性的影響並找出生物分子與奈米粒子之間的最佳比例。
我們發現酵素固定於奈米金上其活性的確有顯著性的增強,進ㄧ
步的實驗結果顯示其增強的行為是由於反應物與酵素之間親合力發
生變化所導致,這樣的結果支持奈米金粒子在這反應中為一項影響酵
素催化的因子,像這樣基礎性理論的探討對於日後在奈米生物領域上
的發展有所貢獻。
An Enzymatic kinetics Investigation into the Significantly
Enhanced Activity of Functionalized Gold Nanoparticles
Student : Chung-Shu Wu
Advisor : Dr. Fu-Hsiang Ko
Institute of Nanotechnology
National Chiao Tung University
Abstract
Enzymes immobilize onto gold nanoparticles (AuNPs) in the absence of a linker, using rapid and uncomplicated processes, generally possess higher activity bound to the surfaces through chemical modification. In previous reports, catalytic activity of enzyme-functionalized AuNPs has been investigated with the surface modification of linkers. Although the surface modification of linkers protects AuNPs from aggregation upon reactant’s entering, it will be perplexed at examination the coverage of the enzyme onto nanoparticles because less or much enzyme can not be identified by salt titration. Herein, linker-free immobilization is beneficial for developing precise activity assays. AuNP-bound lipase
exhibits significantly enhanced catalytic activity relative to that of the free enzyme. In this work, we have systematically investigated the interactions between the NP monolayer and the affected substrates by quantifying the kinetic parameters kcat
and KM to understand the enzymatic behavior of AuNP-bound lipase. Investigation of the kinetic parameters reveals that these two systems operate with the same value of
kcat; i.e., the AuNPs exert no influence on the process of product release in the
rate-limiting step. The Michaelis–Menten curves revealed that the AuNP-bound lipase provided the lower value of KM. Thus, the addition of AuNPs is an efficacious means of tuning enzyme–substrate association. We suspect that such fundamental research will aid in the development of new nanobiotechnological applications.
Contents
Acknowledgment...i Abstract in Chinese...ii Abstract in English...iii Contents...v List of Tables...vii List of Figures...viiiChapter 1: Introduction
...1 1.1 General Introduction...1 1.1.1 Why Nanomaterials? ...3 1.1.2 Nanobiotechnology...4Chapter 2: Literatures Review & Motivation
...92.1 The Application of Gold Nanoparticles on Nanobiotechnology...9
2.1.1 Oligonucleotides-Modified Gold Nanoparticles...9
2.1.2 Enzyme-Modified Gold Nanoparticles...13
2.2 Lipases...16
2.2.1 Interfacial Enzymes with Attractive Applications...16
2.2.2 Structure and Reaction Mechanism of Esterase...17
2.3 Motivation...19
Chapter 3: Experiments
...203.1 General Introduction...20
3.2 Experimental Methods...22
3.2.1 Preparation of Gold Nanoparticles...22
3.2.2 AuNPs-Bound Lipase...23
3.2.3 Assaying the Free and AuNPs-Bound Lipase...24
3.2.4 Michaelis-Menten Kinetics...24
3.2.5 Immobilization of Gold Nanoparticles with Lipase on Silicon wafer...25
Chapter 4: Results and Discussion
...274.1 Mechanism of the Catalytic Reaction...27
4.3 Determining the Immobilization of Lipase-functionalized on Gold
Nanoparticles...30
4.4 Determining the Coverage of the Gold Nanoparticles with the Lipase...34
4.5 Kinetic Assays...40
4.5.1 Activity Assays of the Free and Gold Nanoparticles -Bound Lipase...40
4.5.2 The Effect of the Concentration of Gold Nanoparticles on Catalysis...41
4.5.3 Michaelis–Menten and Arrhenius Plots...42
Chapter 5: Conclusions
...45List of Tables
Table 2.1...11
Melting temperatures and enthalpies for different sized nanoparticles in the solution aggregate system.
Table 2.2...11
Melting temperatures and enthalpies for different surface densities of probe oligonucleotides on the nanoparticles in the solution aggregate system.
List of Figures
Chapter 1: Introduction
Figure 1.1...2
Plot of the number of articles published on nanoparticles since 1990.
Figure 1.2...4
Sizes, shapes, and compositions of metal nanoparticles can be systematically varied to produce materials with distinct light-scattering properties.
Figure 1.3...5
Chemistry is the central science for the development of applied disciplines such as materials research and biotechnology. Materials science, which is based on classic chemical research fields and engineering technologies, has led to enormous advances in tailoring advanced modern materials.
Figure 1.4...5
A gap currently exists in the engineering of small-scale devices. Whereas conventional top-down processes hardly allow the production of structures smaller than about 100-200 nm, the limits of regular bottom-up processes are in the range of about 2-5 nm.
Figure 1.5...7
Integrated nanoparticle–biomolecule hybrid systems.
Chapter 2: Literatures Review
Figure 2.1...10
In the presence of complementary target DNA, oligonucleotide-functionalized gold nanoparticles will aggregate (A), resulting in a change of solution color from red to blue (B). The aggregation process can be monitored using UV-vis spectroscopy or simply by spotting the solution on a silica support (C).
Figure 2.2...12
The diagram of using of gold nanoparticle-oligonucleotide complexes as intracellular gene regulation agents for the control of protein expression in cells.
Figure 2.3...13
Confocal fluorescence microscopy images showing EGFP knockdown. (A) Untreated control cells (upper left, Cy5.5 emission, 706 to 717 nm; upper right, EGFP emission, 500 to 550 nm; lower left, transmission image of cells; lower right, composite overlay of all three channels) showed a significant amount of emission throughout the cell. (B) 1 mm sectioning images of control cells. (C and D) Cells treated with antisense particles showed a decrease in the amount of EGFP emission.
Figure 2.4...14
a) Molecular structure of α-chymotrypsin. b) Chemical structure of amino-acid-functionalized gold nanoparticles and substrates. c) Schematic representation of monolayer-controlled diffusion of the substrate into and the product away from the active pocket of nanoparticles-bound ChT.
Figure 2.5...15
a) The design of functionalized, cationic GNPs as a template for peptide ligation. b) Helicity of E1E2, E1, and E2 with added cationic GNPs. c) Initial rate of E1E2 versus the concentration of GNPs.
Figure 2.6...17
Structure of lipase in closed (A, C) and open form (B, D). A and B (side view): the catalytic triad (yellow) and secondary structure elements showing the α/β-hydrolase fold common to all lipases. Upon opening of the lid, the catalytic triad (yellow) becomes accessible (D).
Figure 2.7...18
Catalytic mechanism of lipases based on a “catalytic triad” of serine (nucleophile), histidine, and aspartate or glutamate.
Chapter 3: Experiments
Figure 3.1...23
The reactions in gold nanoparticles synthesis.
Figure 3.2...26
The self-assembly steps for enzyme-functionalized capped gold nanoparticles onto silicon oxide surface.
Chapter 4: Results and Discussion
Figure 4.1...27
Mechanism of the catalytic reaction mediated by the AuNP-bound lipase.
Figure 4.2...29
a) SEM image of AuNPs immobilized onto a silicon dioxide surface through an MPTMS linker. b) Particle size distribution of the AuNPs present in the SEM image, analyzed using a personal computer and Image-Pro Plus software.
Figure 4.3...30
XPS spectra of AuNPs measured in the presence (blue) and absence (red) of the enzyme on the silicon dioxide surface.
Figure 4.4...30
FTIR spectra of enzyme capped gold nanoparticles on the silicon wafer.
Figure 4.5...32
a) Determining the degree of enzyme immobilization on non-aggregated AuNPs. UV–Vis absorption spectra of AuNPs in DI water (black), AuNPs in NaCl solution (red), enzyme-capped AuNPs in DI water (green), and enzyme-capped AuNPs after adding NaCl solution (blue); b) Photographic image of solutions of (A) AuNPs in DI water, (B) AuNPs in NaCl solution, (C) AuNPs capped with lipase in DI water, and (D) AuNP-bound lipase in DI water after adding NaCl solution. The variation in color allowed discrimination between the aggregated and non-aggregated AuNPs in aqueous solution; i.e., a distinguishable color change from red (A, C, and D) to blue (B) occurred upon aggregation.
Figure 4.6...34
a) The diagram of determining the optimal immobilization of lipase-functionalized on gold nanoparticles. Variation in b) photographic image and c) UV-vis absorbance spectra of the AuNPs (2.2 nM) at different concentrations of enzyme after adding NaCl solution.
Figure 4.7...36
Plots of changes in the time-dependent absorption ratio (A620/A520) in the presence of varying concentration of lipase after the addition of salt solution.
Figure 4.8...37
Plot of the absorption ratio (A520/A620) in equilibrium versus the lipase concentration.
Figure 4.9...40
a) Product (pNP) formation over time in the catalytic reactions of various concentrations of AuNP-bound (solid) and free lipase (hollow) solutions monitored at 405 nm. b) Initial velocities of pNP from pNPP plotted as a function of the concentration of the free enzyme (●) and the enzyme-capped AuNPs (○).
Figure 4.10...41
Initial rate of pNP production versus the concentration of AuNPs. Conditions : pH 7.4, 30 °C, [enzyme]=100 nM, [substrate]=22.22 μM.
Figure 4.11...44
Michaelis–Menten plots for the hydrolyses of pNPP mediated by the free enzyme (blue) and enzyme-capped AuNPs (red).
Figure 4.12...44
Arrhenius plots of ln(absorbance) versus the reciprocal of absolute temperature for the free (blue, inset) and AuNP-bound (red) lipase. The activity was measured for reaction mixtures containing 0.1 μM enzyme at pH 7.4.
Chapter 1: Introduction
1.1 General Introduction
An atom measures about 1 Ǻ, or 10-10 meters. The study of atoms and molecules is the conventional field of chemistry as was studied in the late 19th and 20th centuries. A nanometer (nm), or 10-9 meters, represent a collection of a few atoms or molecules. Properties of bulk substances of micrometer sizes or larger have been studied for years by solid state physicists and material scientists and are currently well understood. Materials on the 1–100 nm scale were not studied by either group in the past. It was just recently shown that on this size scale the properties of a material become dependent on its size and shape. Thus, the nanometer scale incorporates collections of atoms or molecules, whose properties are neither those of the individual constituents nor those of the bulk. On this scale, many of the atoms are still located on the surface, or one layer removed from the surface, as opposed to the interior. New properties are observed on this scale due to the interface that is not observed in the bulk or individual atoms. Since the properties depend on the size of the structure, instead of the nature of the material, reliable and continual change can be achieved using a single material.[1]
The major publications in this area have appeared in the Journal of Physical Chemistry B, Journal of the American Chemical Society and Langmuir. Nano Letters has also published a large number of letters in this field since its conception in 2001. The number of patent applications and symposium articles for the Materials Research Society (MRS), the American Chemical Society (ACS) and Advanced Materials also represent a large number of the publications every year on noble metal nanoparticles.
This increase in the available nanoparticles generates an increase in the number of applications, driving the potential for great advances in every day life due to nanotechnology. One of the hottest areas for nanoparticles use is in biological systems, owing to their potential application in medicine. Since structures can be accurately designed on the nanometer scale they can be incorporated into biological systems, due to the similar size scales. Biological systems are complex, with synthesis, structure, and function all rarely understood in detail. The ability to rationally design structures on the same size as biological molecules generates the ability to probe and modify biological systems. Furthermore, biological systems are used to build up nanomaterials of specific shape and function. Nanostructures are being used as drug delivery agents, labeling agents, sensors, and to enhance electromagnetic fields. The field of nanotechnology has received increasing attention over the last 20 years, and the number of publications of nanoparticles has grown exponentially, as shown in Figure1.1.
1.1.1 Why Nanomaterials?
In the last 10 years the field of molecular diagnostics has witnessed an explosion of interest in the use of nanomaterials in assays for gases, metal ions, and DNA and protein markers for many diseases.[2] Intense research has been fueled by the need for practical, robust, and highly sensitive and selective detection agents that can address the deficiencies of conventional technologies. Not all molecular fluorophores make for suitable probes in biodiagnostic assays nor do all nanomaterials offer advantages in biodetection. Certain nanomaterials are attractive probe candidates because of their (1) small size (1-100 nm) and correspondingly large surface-to-volume ratio, (2) chemically tailor-able physical properties, which directly relate to size, composition, and shape (Figure 1.2), (3) unusual target binding properties, and (4) overall structural robustness. The size of a nanomaterial can be an advantage over a bulk structure, simply because a target binding event involving the nanomaterial can have a significant effect on its physical and chemical properties, thereby providing a mode of signal transduction not necessarily available with a bulk structure made of the same material. Indeed, in this regard, nanomaterials and biology have a long history as nanoparticles have been used in bioconjugation and as cellular labeling agents for the past four decades. However, new synthesis, fabrication, and characterization methods for nanomaterials have evolved to the point that deliberate modulation of their size, shape, and composition is possible, thereby allowing exquisite control of their properties. Additionally, tools and techniques for surface modification and patterning have advanced to a point that now allows generation of nanoscale arrays of biomacromolecules and small molecules on surfaces.[3-5] Along with synthetic advances for varying the size, shape, and composition of nanostructured materials has come the ability to tailor their binding affinities for various biomolecules through surface modification and engineering.[6-7]
Figure 1.2: Sizes, shapes, and compositions of metal nanoparticles can be
systematically varied to produce materials with distinct light-scattering properties.[2]
1.1.2 Nanobiotechnology
In Figure 1.3, three main disciplines including chemistry, materials science and biotechnology are presented.[8] Merging these disciplines will allow us to take advantage of the improved evolutionary biological components to generate new smart materials and to apply today’s advanced materials and physicochemical techniques to solve biological problems. Both biotechnology and materials science meet at the same length scale (Figure 1.4). On the one hand, biomolecular components have typical size dimensions in the range of about 5 to 200 nm. On the other hand, commercial requirements to produce increasingly miniaturized microelectronic devices strongly motivate the elaboration of nanoscale systems. Today’s nanotechnology research puts a great emphasis on the development of bottom-up strategies, which concern the self-assembly of (macro) molecular and colloidal building blocks to create larger, functional devices.[9]
Figure 1.3: Chemistry is the central science for the development of applied
disciplines such as materials research and biotechnology. Materials science, which is based on classic chemical research fields and engineering technologies, has led to enormous advances in tailoring advanced modern materials.[8]
Figure 1.4: A gap currently exists in the engineering of small-scale devices. Whereas
conventional top-down processes hardly allow the production of structures smaller than about 100-200 nm, the limits of regular bottom-up processes are in the range of about 2-5 nm.[8]
The convergence of biotechnology and nanotechnology has led to the development of hybrid nanomaterials that incorporate the highly selective catalytic and recognition properties of biomaterials, such as proteins/enzymes and DNA, with the unique electronic, photonic, and catalytic features of nanoparticles. Novel nanomaterials for use in bioassay applications represent a rapidly advancing field. Various nanostructures have been investigated to determine their properties and possible applications in biotechnology. These structures include nanoparticles, nanowires, nanotubes, and thin films. Functional nanoparticles (electronic, optical and magnetic) bound to biological molecules have been developed for use in biosensors to detect and amplify various signals.[10-12] The interdisciplinary cooperation of various techniques can solve the human-related diseases. The conjugation of NPs and other nanoobjects (e.g. nanorods and carbon nanotubes) with biomolecules is an attractive area of research within nanobiotechnology.[13-16] Biomolecules are fascinating macromolecular structures in terms of their unique recognition, transport, and catalytic properties. The conjugation of NPs with biomolecules could provide electronic or optical transduction of biological phenomena in the development of novel biosensors.[17] Enzymes, antigens and antibodies, and biomolecular receptors have dimensions in the range of 2–20 nm, similar to those of NPs, thus the two classes of materials are structurally compatible.
Because of several fundamental features, biomaterials are important future building blocks for NP architectures: 1) Biomaterials display specific and strong complementary recognition interactions, for example, antigen–antibody, nucleic acid–DNA, and hormone–receptor interactions. The functionalization of nanoparticles with biomolecules could lead to biomolecule–nanoparticle recognition interactions and thus to self-assembly. 2) Various biomolecules contain several binding sites, for example, antibodies exhibit two Fab (antigen-binding fragment) sites, whereas
streptavidin or concanavalin A each display four binding domains. This allows the multidirectional growth of NP structures. 3) Proteins may be genetically engineered and modified with specific anchoring groups. This facilitates their aligned binding to NPs or the site-specific linkage of the biomaterial to surfaces. Consequently, the directional growth of NP structures may be dictated. Furthermore, other biomaterials, such as double-stranded DNA, may be synthetically prepared in complexrigidified structures that act as templates for the assembly of nanoparticles by intercalation, electrostatic binding to phosphate groups, or association to functionalities tethered to the DNA. 4) Enzymes are catalytic tools for the manipulation of biomaterials. For example, the ligation or the endonuclease scission processes of nucleic acids provide effective tools for controlling the shape and structure of biomolecule–NP hybrid systems. In this context, it is important to note that Mother Nature has developed unique biocatalytic replication processes. The use of biocatalysts for the replication of biomolecule–NP conjugates may provide an effective system for the formation of nanostructures of predesigned shapes and compositions.[18-19]
Biomolecule-functionalized NPs and nanorods could be exploited for numerous applications in biomolecular electronics,[20-21] biosensors,[22-23] immunoassays,[24] and medicine,[25] namely in photodynamic anticancer therapy, targeted delivery of radioisotopes, drug delivery,[26] electronic DNA sequencing, nanotechnology of gene-delivery systems, and gene therapy in Figure 1.5.[18] Novel fascinating areas of technologies are feasible with the use of bionanomaterials. A combination of the unique properties of nanoobjects and biomaterials provides a unique opportunity for physicists, chemists, biologists, and material scientists to mold the new area of nanobiotechnology.[27] Based on recent advances in the field, exciting new science and novel systems can be anticipated from this interdisciplinary effort. Future advances will require continued innovations by nanotechnology in close collaboration with experts in medical and biological fields.
Chapter 2: Literatures Review
2.1 The Application of Gold Nanoparticles on Nanobiotechnology
In this section, we will review some important properties in the nanobiotechnology field with gold nanoparticles (AuNPs). Although gold is the subject of one of the most ancient themes of investigation in science, its renaissance now leads to an exponentially increasing number of publications, especially in the context of emerging nanoscience and nanotechnology with nanoparticles and self-assembled monolayers (SAMs). AuNPs, also called gold colloids, are the most stable metal nanoparticles, and they present fascinating aspects such as their assembly of multiple types involving materials science, the behavior of the individual particles, size-related electronic, magnetic and optical properties (quantum size effect), and their applications to catalysis and biology.[1] Colloidal gold was used to make ruby glass and for coloring ceramics and these applications are still continuing now. It is ruby red in transmitted light and green in reflected light, due to the presence of gold colloids. In the 20th century, various methods for the preparation of gold colloids were reported.[2-4] In the past decade, gold colloids have been the subject of a considerably increased number of books and reviews.[5-10] AuNPs, which have been known for 2500 years, are the subject of an exponentially increasing number of reports and biomedical applications in the 21st century, using the “bottom-up” approach with the hybrid organic-inorganic and biological-inorganic building blocks derived wherefrom.
2.1.1 Oligonucleotides-Modified Gold Nanoparticles
An early indication of the potential of nanomaterials as biodetection agents was reported in 1996 with the observation that oligonucleotide-modified nanoparticles and sequence-specific particle assembly events, induced by target DNA, could be used to
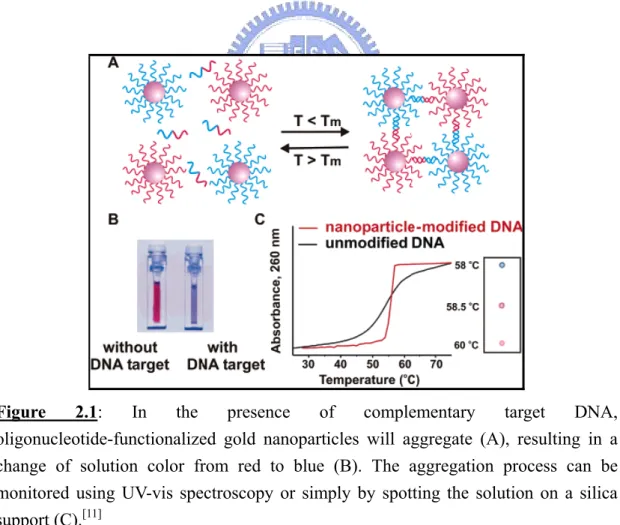
generate materials with unusual optical and melting properties.[11] Specifically, when 13-nm gold particles were used in the assay, the color of the solution changed from red to blue upon the analyte-directed aggregation of gold nanoparticles, a consequence of interacting particle surface plasmons and aggregate scattering properties. Further studies indicated that the melting profiles of the nanoparticle-labeled DNA aggregates were extraordinarily sharp, occurring over a temperature range much narrower than the transition for unlabeled or conventional fluorophore-labeled DNA (Figure 2.1). These two observations, both consequences of the high surface area and unique optical activity of the gold nanoparticles, created worldwide interest in exploring the potential for designer nanomaterials in biodiagnostic applications.
Figure 2.1: In the presence of complementary target DNA,
oligonucleotide-functionalized gold nanoparticles will aggregate (A), resulting in a change of solution color from red to blue (B). The aggregation process can be monitored using UV-vis spectroscopy or simply by spotting the solution on a silica support (C).[11]
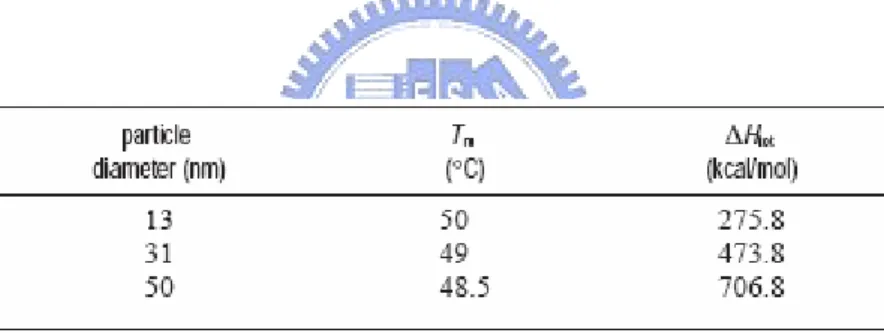
After discovering this property, the group of Chad A. Mirkin have quantitatively determined and compared the thermodynamic values of oligonucleotide functionalized gold nanoparticle probes and molecular fluorophore probes of the same sequence. Between 2003 to 2005,[12-14] they report a series of experiments and a theoretical model designed to systematically define and evaluate the relative importance of nanoparticle, oligonucleotide, and environmental variables that contribute to the observed sharp melting transitions associated with DNA-linked nanoparticle structures. These variables include the size of the nanoparticles, the surface density of the oligonucleotides on the nanoparticles, the dielectric constant of the surrounding medium, target concentration, and the position of the nanoparticles with respect to one another within the aggregate (Table 2.1 & 2.2).
Table 2.1: Melting temperatures and enthalpies for different sized nanoparticles in the
solution aggregate system.[14]
Table 2.2: Melting temperatures and enthalpies for different surface densities of probe
oligonucleotides on the nanoparticles in the solution aggregate system.[14]
Melting data were analyzed according to literature procedures for molecular systems by graphing 1/Tm as a function of concentration according to the following equation:
Where Tm is the melting temperature, R is the gas constant, and CT is the total concentration of nanoparticles plus fluorophore or quencher plus fluorophore.
In 2006 on the journal of Science, they describe the use of gold nanoparticle-oligonucleotide complexes as intracellular gene regulation agents for the control of protein expression in cells (Figure 2.2).[15] These oligonucleotide-modified nanoparticles have affinity constants for complementary nucleic acids that are higher than their unmodified oligonucleotide counterparts, are less susceptible to degradation by nuclease activity, exhibit greater than 99% cellular uptake, can introduce oligonucleotides at a higher effective concentration than conventional transfection agents, and are nontoxic to the cells under the conditions studied. By chemically tailoring the density of DNA bound to the surface of gold nanoparticles, they demonstrated a tunable gene knockdown (Figure 2.3), thus opening the door for new possibilities in the study of gene function and nanotherapies.
Figure 2.2: The diagram of using of gold nanoparticle-oligonucleotide complexes as
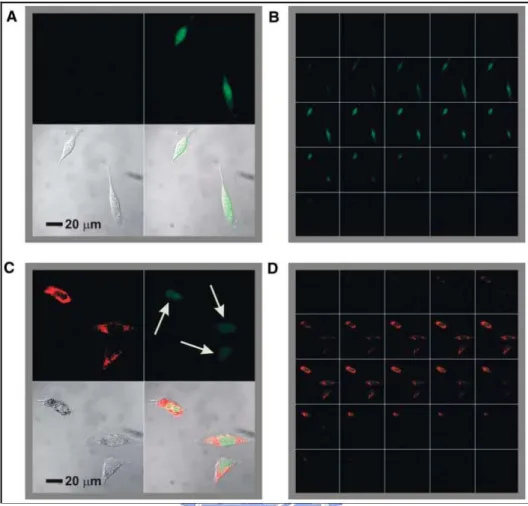
Figure 2.3: Confocal fluorescence microscopy images showing EGFP knockdown. (A)
Untreated control cells (upper left, Cy5.5 emission, 706 to 717 nm; upper right, EGFP emission, 500 to 550 nm; lower left, transmission image of cells; lower right, composite overlay of all three channels) showed a significant amount of emission throughout the cell. (B) 1 mm sectioning images of control cells. (C and D) Cells treated with antisense particles showed a decrease in the amount of EGFP emission.[15]
2.1.2 Enzyme-Modified Gold Nanoparticles
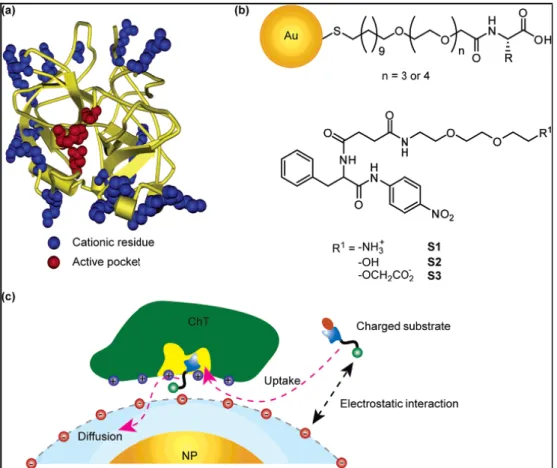
Modulation of enzyme activity provides a potent means to gain control over cellular processes such as signal transduction, DNA replication, and metabolism. On the other hand, aberrant activities of some enzymes can lead to the development of numerous diseases and disorders including cancers. In Figure 2.4,[16] V. M. Rotello et al. have systematically investigated the enzymatic kinetics of α-chymotrypsin (ChT) upon binding to amino-acid-functionalized gold nanoparticles toward different substrates and demonstrated that the complex formation provides a powerful tool to
tune the enzyme specificity in 2006 at the Journal of American Chemical Society (JACS).
Figure 2.4: a) Molecular structure of α-chymotrypsin. b) Chemical structure of
amino-acid-functionalized gold nanoparticles and substrates. c) Schematic representation of monolayer-controlled diffusion of the substrate into and the product away from the active pocket of nanoparticles-bound ChT.[16]
The association of ChT with anionic nanoparticles leads to the increase of specificity to the positively charged substrate and the decrease of specificity to the negatively charged substrate. Such enhanced substrate selectivity originates from the electrostatic interaction as well as the steric repulsion of the substrates with the nanoparticles monolayer. Monolayer-functionalized nanoparticles provide a potent scaffold for creation of an enzyme modulator based on surface recognition. In the following year (2007) at JACS, this group has successfully designed a system
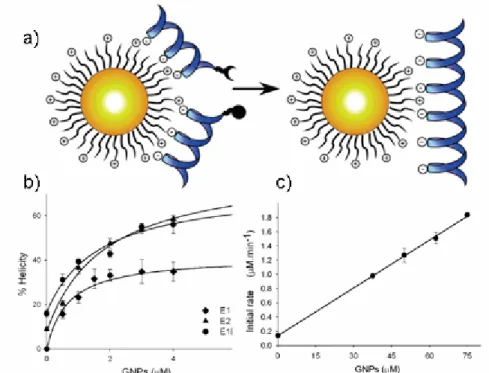
whereby functionalized gold particles promote the association and ligation of peptide fragments E1E2 (Figure 2.5).[17] The Figure 2.5 demonstrate a clear increase in the rate of production with addition of cationic AuNPs as compared to the control reaction with no AuNPs. Increasing the amount of AuNPs induced a faster production rate, thereby demonstrating the ability of the cationic AuNPs to act as a template for ligation.
Figure 2.5: a) The design of functionalized, cationic GNPs as a template for peptide
ligation. b) Helicity of E1E2, E1, and E2 with added cationic GNPs. c) Initial rate of E1E2 versus the concentration of GNPs.[17]
2.2 Lipases
Lipases are used industrially as detergent enzymes, in paper and food technology, in the preparation of specialty fats, and as biocatalysts for the synthesis of organic intermediates, and in various clinical studies and drug delivery.[18-20] The kinetic model of lipases is based on the so-called ping-pong mechanism, which also applies to many other enzymes, such as glucose oxidase, horseradish peroxidase, and alkaline phosphatase. As a representative esterase, lipase is an excellent model for studying the enhanced activity of AuNP-bound enzymes because of its well-defined structure, properties, and applications. In this section, we will review some literatures to introduce lipases with the applications in the biocatalysts, structure, and reaction mechanism.
2.2.1 Interfacial Enzymes with Attractive Applications
Lipids are key elements in the chemistry of life. Most organisms use the supramolecular chemistry inherent to phospholipids to form their exterior and compartmental membranes. Many plants and animals store chemical energy in the form of triglycerides, which are sparingly soluble in water. For the metabolic turnover of these and other biochemicals, they produce esterases, enzymes which can hydrolyze bonds of water-soluble esters. Esterases which can hydrolyze triglycerides at the water/oil boundary are termed lipases and those which attack phospholipids are termed phospholipases.[21] Both types of enzymes have recently received considerable attention. Whereas phospholipases are involved in key metabolic events such as membrane turnover and signal transduction, lipases have diverse functions in the degradation of food and fat; they have qualified as valuable drugs against digestive disorders and diseases of the pancreas. They also find applications in biotechnology and as catalysts for the manufacture of specialty chemicals and for organic synthesis.
Furthermore, lipases play an important physiological role in the digestion of fat by mammals and humans, and lipase inhibitors may have a potential as antiobesity drugs.
2.2.2 Structure and Reaction Mechanism of Esterase
The protein structure underlying these observations remained a mystery until a few years ago. Only in 1990 were the first two lipase structures solved by X-ray crystallography. They revealed a unique mechanism, unlike that of any other enzyme: Their three-dimensional structures suggested that interfacial activation might be due to the presence of an amphiphilic peptide loop covering the active site of the enzyme in solution, just like a lid or flap.[22-23] From the X-ray structure of cocrystals between lipases and substrate analogues, there is strong indirect evidence that, when contact occurs with a lipid/water interface, this lid undergoes a conformational rearrangement which renders the active site accessible to the substrate (Figure 2.6).[24]
Figure 2.6: Structure of lipase in closed (A, C) and open form (B, D). A and B (side
view): the catalytic triad (yellow) and secondary structure elements showing the α/ β-hydrolase fold common to all lipases. Upon opening of the lid, the catalytic triad (yellow) becomes accessible (D).[24]
All lipases whose structure has hitherto been elucidated are members of the “α/ β-hydrolase fold” family with a common architecture composed of a specific sequence of α helices and β strands.[25-26] They hydrolyze ester bonds by means of a “catalytic triad”, composed of a nucleophilic serine residue activated by a hydrogen bond in relay with histidine and aspartate or glutamate (Figure 2.7).
Figure 2.7: Catalytic mechanism of lipases based on a “catalytic triad” of serine
(nucleophile), histidine, and aspartate or glutamate.[24]
As pointed out already, a unique structural feature common to most lipases is a lid or flap composed of an amphiphili α helix peptide sequence, which in its closed conformation prevents access of the substrate to the catalytic triad. After the lid has opened, a large hydrophobic surface is created to which the hydrophobic supersubstrate binds. This presumed mechanism is supported by the X-ray structures of lipases covalently complexed with hydrophobic inhibitors such as alkyl phosphonates, cycloalkyl phosphonates, or alkyl sulfonates.[27]
2.3 Motivation
Gold nanoparticles, which have high affinity for biomolecules, have been used as biosensors, immunoassays, therapeutic agents, and gene and drug delivery agents; thus, the conjugation of AuNPs and biomolecules has become a major area of research for advancing the use of nanotechnology in biomedical applications.[1] Indeed, proteins, enzymes, DNA, and oligonucleotides have all been immobilized on AuNPs; the physicochemical characteristics of these functionalized AuNPs have been investigated in a variety of academic studies.[11-15] Several techniques have been used to immobilize enzymes on variety nanostructures in attempts to improve the enzymatic activity and stability.[28-29] Most of these kinetic investigations need steps such as modified biomolecules onto the AuNPs surface and separating the modified AuNPs from the unmodified AuNPs or surplus molecules. These steps, firstly, led to complication and relatively high cost of the experiments. In addition, long-time course (covalent bond) also led to activation lost of enzyme. What’s more, the target binding sites and conformational changes of the enzyme after binding were not all known precisely, so labeling sites were not only difficult to design, but labeling also could weaken the affinity between the reactant and the enzyme. Although some enzyme-functionalized AuNPs with modified linkers exhibit enhanced catalytic activity, which has been supposed to favorable conformational changes and electrostatic interactions,[16-17] there have been no detailed studies aimed at quantifying the differences between the rate constants of AuNP-immobilized and free enzymes. To gain insight into the mechanisms of enzyme catalysis in the presence of functionalized AuNPs, we must obtain kinetic data using linker-free immobilization and suitable analytical systems. Therefore, developing modification-free AuNPs kinetic assays to simplify the detection process would be important and attractive.
Chapter 3: Experiments
3.1 General Introduction
All the experiments were preceded in National Chiao Tung University (NCTU). All the equipments were also conducted in our laboratories in NCTU. The reagents were purchased commercially and used by following with the directions unless specially mentioned.
All the reagents were listed alphabetically in the form of “Name {abbreviation; chemical formula; purity; manufacturer}”. Some information will be omitted if not available or not necessary. The following text will use the abbreviation of the reagent.
Deionized and distilled water {DI water, ddH2O}
The water we used was purified with filters, reverse osmosis, and deionized system until the resistance was more than 18 MΩ. DI water was used to clean, wash, and be a solvent.
Dimethyl sulfoxide {DMSO; Sigma}
It was a solvent for dissolving many hydrophobic organic compounds like 3-mercaptopropyltrimethoxysilane in the experiments.
Hydrogen chloride {HCl; ≥99% purity; Sigma}
1 M HCl in DI water was used for pH adjustment.
Hydrogen peroxide {H2O2; ≥30% purity; Sigma}
Hydrogen peroxide was mixed with sulfuric acid to form piranha solution which cleaned the wafer surface.
Hydrogen tetrachloroaurate trihydrate {HAuCl4·3H2O; Sigma}
The chemical provided Au3+ ion for the mechanism of gold nanoparticles synthesis. The detailed information described in the next section.
Lipase (EC 3.1.1.3) from Candida rugosa {Sigma}
The enzyme played the leading role in this thesis. We immobilized lipase onto the nanoparticles’ surfaces, without any surface modification, provided colloidal stability that allowed us to determine the coverage of enzyme onto the AuNPs and assay the enzymatic activity.
3-Mercaptopropyltrimethoxysilane {MPTMS; 99%; Sigma}
MPTES was used to modify the property of the SiO2 surface. The ethoxy functional groups of APTES were displaced through formation of covalent bond between the hydrophilic hydroxyl groups of the SiO2 surface and the silicon atom of MPTES. This process leads to the formation of a molecular layer of thiol groups for labeling with gold nanoparticles on SiO2 surface. Prior to use, the powder needed to dissolve in the dimethyl sulfoxide (DMSO) solvent.
Phosphate-buffered saline tablets {PBS, 1X; CALBIOCHEM}
PBS, a biological buffer solution, was used for all kinetic assay reaction. This chemical is dissolved in one liter ddH2O to yield 10 mM phosphate buffer, pH 7.4, 140 mM NaCl, 3 mM KCl.
Propan-2-ol {IPA}
It was an organic solvent for dissolving our substrate 4-nitrophenyl palmitate (pNPP).
4-Nitrophenyl palmitate {pNPP; Sigma}
This chemical was a substrate of lipase. Passing through the catalytic reaction, it was converted to the product 4-nitrophenol (pNP). Prior to use, the powder needed to dissolve in the propan-2-ol (IPA) solvent.
Sodium chloride {NaCl; ≥ 99.5%; Sigma}
Sulfuric acid {H2SO4; 98% purity; Sigma}
Sulfuric acid was mixed with hydrogen peroxide in a 3:1 ratio to remove impurities on the surface of the SiO2 surface. This material was very corrosive and dangerous. We must handle it with carefulness and patience.
Trisodium citrate dehydrate {HAuCl4·3H2O; SHOWA}
We used a previously reported chemical reduction method to prepare gold nanoparticles in aqueous solution. This chemical was the reductant of the reaction. Prior to use, the powder needed to dissolve in the DI water.
3.2 Experimental Methods
3.2.1 Preparation of Gold Nanoparticles
The AuNPs were prepared in aqueous solution using a previously reported chemical reduction method.[1] Aqueous solutions of 1 mM HAuCl4 (10 mL) and 38.8 mM trisodium citrate dehydrate (1 mL) were mixed and then heated under reflux for 15 min with vigorous stirring. The color of the solution gradually changed from yellow to purplish-red. After cooling to room temperature, colloidal AuNPs were formed in the solution. SEM images and absorption spectra confirmed the size and shape of these AuNPs. During the synthesis process we had to put much emphasize on the experimental environment, including container cleaning process, adding reducing agent as soon as possible, and continuously stirring during the synthesis step to prevent aggregation and prevent changing the size uniformity.
The mechanism of gold nanoparticles synthesis had two main steps, the first step was nucleation and the second step was growth. In the beginning of reaction the reducing agent transferred Au (III) to many small gold particles. The small gold particles served as seeds. Once the seeds were accomplished, the amount of the seeds could not change. Then the growth mechanism started and the oxidation-reduction
reaction on the surface of seeds making the gold particles to growth. When Au (III) was completely consumed, the growth mechanism was end up. Trisodium citrate was used as reducing agent; the carboxylic group (COO-) converted Au+3 to Au+1 and formed acetone dicarboxylate. But how Au+1 were transferred to Au was still not confirmed so far. There were two possible arguments about how the Au+1 changed to Au atom. One was carboxylic group (COO-) continuously reacted with Au+1 to become Au atom. And other argument said that three Au+1 ions may form ion-clusters, and performed self-redox reaction to become two gold atoms and one Au+3 ion. The reaction was description as below:
Figure 3.1: The reactions in gold nanoparticles synthesis.
3.2.2 AuNPs-Bound Lipase
:Prior to labeling the AuNPs with the enzyme, the gold colloid was separated through centrifugation (13,000 rpm, 20 min)[2] to exchange the buffer solution while maintaining the temperature at 30 °C. Lipase was added to a solution of 13 (± 1)-nm AuNPs (1.5 μM enzyme per 1 mL of 2.2 nM colloidal gold) and the mixture incubated for 10 min. Lipase concentration was determined using UV-Vis spectroscopy (molar extinction coefficient 3.7 × 104 M-1cm-1at 280 nm). In the experiment of determining the coverage of enzyme onto gold nanoparticles, the different concentrations of lipase was added into the colloidal gold to make the corresponding solution. After the addition of salt solution, we displayed the plots of changes in the time-dependent absorption ratio (A620/A520) in the presence of varying concentration of lipase.
3.2.3 Assaying the Free and AuNPs-Bound Lipase
The reaction solution was prepared by mixing PBS solution (140 mM NaCl, 2.7 mM KCl, and 10 mM phosphate buffer; pH 7.4; 30 °C; 1.3 mL), 0.5 mM pNPP in IPA (0.1 mL), and various concentrations of the free or AuNP-bound lipase (0.1 mL).[3] After mixing the catalytic substance with the reactants, the initial product release at the onset of the reaction was measured using a personal computer and a Hitachi UV-Vis-3310 enzyme reaction measurement system (a UV–Vis spectrophotometer possessing a temperature-controlled thermostatted cell holder; Hitachi, Tokyo, Japan) monitored at 405 nm. In the experiments of varying gold nanoparticles concentration, we recorded the initial reaction rates from the slopes of the changes in absorption over time. The absorbance was converted to a concentration scale by a molar extinction coefficient of 12,800 M-1 cm-1 for 4-nitrophenol (pNP).
3.2.4 Michaelis–Menten Kinetics
The substrate was dissolved in the assay buffer (at concentrations of 5.55, 8.33, 11.11, 16.67, 22.22, 33.33, 44.45, 66.67, 88.89, and 133.33 μM). The free or immobilized enzyme (1.5 μM) was added to the various concentrations of the substrate in the assay buffer as described above. The initial reaction velocity of the change in absorbance at 405 nm was recorded. The plot of the initial velocity of the production of 4-nitrophenol (pNP) versus the pNPP concentration was fitted to a hyperbolic curve. The values of KM and Vmax were obtained through nonlinear regression analysis using SigmaPlot 2001 (v. 7.0) and Enzyme Kinetics Module (v. 1.1, SPSS, Chicago, IL USA) software.[4] The assays were obtained in triplicate; average values were reported.
3.2.5 Immobilization of gold nanoparticles with Lipase on Silicon
wafer
Prior to immobilization, the silicon wafer should be carefully cleaned by the solution of H2SO4 and H2O2 (volume ratio was 3:1). The temperature of the mixture must be maintained above 85 oC to possess the oxidative power. If the temperature was dropped, hydrogen peroxide was required to replenish. After pure water rinsing and drying, the sample was immersed in the 1 mM MPTMS/DMSO solution for 12 hour in room temperature. Then, the sample was rinsed with DMSO and DI water for 10 min, respectively. The succeeding step was to bake the sample at 120 oC for 30 min. At last, the wafer was placed in the gold nanoparticles solution for 2 hours. Then, we dropped a few drops of the lipase solution onto a silicon wafer modified with the AuNPs for 10 min. Finally, we washed the wafer with DI water and followed by nitrogen purge.
Self-assembled monolayers of enzyme functionalized AuNPs onto silicon wafer were characterized by Absorption spectra, scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), and fourier transform infrared spectroscopy (FTIR) spectra. Details of these measurements were described in the following experimented results.
Figure 3.2: The self-assembly steps for enzyme-functionalized capped gold
Chapter 4: Results and Discussion
4.1 Mechanism of the catalytic reaction by the AuNP-bound lipase:
Figure 4.1: Mechanism of the catalytic reaction mediated by the AuNP-bound lipase.
A linker-free AuNP-bound lipase was developed to study the kinetics of the immobilized enzyme. Our kinetic and structural studies led us to propose the mechanism depicted above. There are four independent rate constants (k1, k2, k3, and
k4) in this system.[1] Initially, the substrate (S) is bound to the active site of the enzyme
(E) with a rate constant k1, forming an intermediate complex (ES). Passing through
the transition state, the ES complex is converted into the acyl enzyme (E*) with a rate constant k3, releasing the product (P), in this case pNP. The acyl bond is then cleaved,
with a rate constant k4, upon nucleophilic attack, returning the enzyme to its initial
[E0] is the total enzyme concentration; the kinetic parameters kcat (turnover number), KM (apparent dissociation constant), and kcat/KM (apparent specificity constant) are defined as follows:
4 3 4 3 cat k k k k k + = 4 3 4 1 3 2 M k k k k k k K + + = 3 2 3 1 M cat k k k k K k + =
The rate constants in Figure 4.1 are related to the experimental values of KM and
kcat through the following equations:
] [ ] [ ] [ S K S E k v M 0 cat + = ] [ 0 cat max k E V =
The value of KM for the kinetic studies represents the affinity of the enzyme toward the substrate; the rate-limiting step (kcat), which occurred prior to formation of the acyl enzyme intermediate, was determined by monitoring the product’s release.[2] According to these equations, we observed almost the same value of Vmax for the reactions of the bound and free enzymes, indicating no significant difference in their rate-limiting steps (kcat) at identical values of [E0]. Therefore, the lower value of KM for the AuNP-bound lipase reflected its increased activity (higher v) at each concentration of the substrate.
4.2 Preparation of Gold Nanoparticles:
Figure 4.2: a) SEM image of AuNPs immobilized onto a silicon dioxide surface
through an MPTMS linker. b) Particle size distribution of the AuNPs present in the SEM image, analyzed using a personal computer and Image-Pro Plus software.
The SEM image was recorded using a JEOL JSM-6700F electron microscope operated at 20 kV. The sample for SEM analysis was prepared by placing a few drops of the AuNPs solution onto a silicon wafer modified with the MPTMS linker as described above. The sample was rinsed with DI water for 10 min and then dried prior to viewing under the electron microscope. Figure 4.2.a showed that the gold nanoparticles were immobilized perfectly on the silicon oxide surface. The uniformity and the size distribution of gold nanoparticles were both great. The average size of gold nanoparticles was 13 nm. Figure 4.2.b was analyzed by the software named Image-pro plus (IPP), and the analysis region was the full SEM image.
4.3 Determining the immobilization of Lipase-functionalized on gold
nanoparticles
Figure 4.3: XPS spectra of AuNPs measured in the presence (blue) and absence (red)
of the enzyme on the silicon dioxide surface.
The XPS spectroscopy (VG Scientific Microlab F310) was used to verify the attachment of the enzymes onto the surfaces of the AuNPs. For the AuNPs modified with lipase, the marked peak indicates the presence of NH2 groups (N1s binding energy = 399.6 eV) under X-ray irradiation initiated by the photoelectrons and secondary electrons emitted from the surface.[3] The result is in agreement with the existing literature for proteins and enzymes bound to the surface of AuNPs without surface modification.[4] The FTIR spectroscopy was used to verify the attachment of proteins on the surface of the gold nanoparticles. For gold nanoparticles modified with lipase, the peaks at 1550 cm-1 indicate the presence of the primary amine on the nanoparticles surface. Broad band 3300 cm-1 was an indication of the bonded NH or NH2 groups on the surface.[4] These data suggested that the surface of the gold nanoparticles was modified by nonspecific adsorption of proteins. The addition of a protein to gold colloid resulted in spontaneous adsorption on the surface of the NPs as a result of electrostatic, hydrophobic, and van der Waals interactions. This adsorption process appeared to be instantaneous.
Figure 4.5: a) Determining the degree of enzyme immobilization on non-aggregated
AuNPs. UV–Vis absorption spectra of AuNPs in DI water (black), AuNPs in NaCl solution (red), enzyme-capped AuNPs in DI water (green), and enzyme-capped AuNPs after adding NaCl solution (blue).
Figure 4.5: b) Photographic image of solutions of (A) AuNPs in DI water, (B) AuNPs
in NaCl solution, (C) AuNPs capped with lipase in DI water, and (D) AuNP-bound lipase in DI water after adding NaCl solution. The variation in color allowed discrimination between the aggregated and non-aggregated AuNPs in aqueous solution; i.e., a distinguishable color change from red (A, C, and D) to blue (B) occurred upon aggregation.
The UV–Vis absorption spectrum of the AuNP solution (black trace) exhibited a strong surface plasmon resonance (SPR) at ca. 520 nm; adsorption of the enzyme onto the AuNPs caused a slight shift (green trace) in the SPR peak (Figure 4.5.a); previous studies showed proteins adsorbed onto AuNPs could cause similar shifts in the SPR.[5] The gold colloids aggregated at a high concentration of electrolytes in the absence of a protecting coating layer (red trace). After adding NaCl solution to the enzyme-capped AuNPs, the absorption signal remained sharp (blue trace) and the solution retained the color of the enzyme-capped AuNPs (Figure 4.5.b).
4.4 Determining the coverage of the gold nanoparticles with the
Lipase
Figure 4.6: a) The diagram of determining the optimal immobilization of
lipase-functionalized on gold nanoparticles. Variation in b) photographic image and c) UV-vis absorbance spectra of the AuNPs (2.2 nM) at different concentrations of enzyme after adding NaCl solution.
Prior to each experiment, the amount of enzyme required to stabilize the colloidal gold was titrated using NaCl solution to determine the coverage of protein coating the NP’s surface.[4] As for any conjugation procedure, optimization of the ratios of reactants ensures that the enzyme molecules were occupied on the AuNPs’ surfaces to maintain the stability protected. This is an essential method to discriminate between aggregated and nonaggregated AuNPs in aqueous solution, based on a distinguishable color change from red to blue upon aggregation.[6] As shown in Figure 4.6, by exploiting interactions between the AuNPs and lipase, color changes of the AuNPs could sensitively differentiate the concentration used of enzyme after titrating with thesalt solution.We monitored the stability of the AuNPs solution by its color or by the absorbance spectra. If the enzyme did not cap the AuNPs completely, we obtained spectra exhibiting aggregation of AuNPs. As long as the colloid continued to turn blue, and thus formed gold aggregates, with addition of electrolyte, the amount of enzyme added is not sufficient to stabilize the suspension (A, B, C, and D traces). When the concentration of enzyme added was enough to stabilize the colloid, the solution no longer changed color and the absorbance spectra exhibited a strong surface plasmon resonance (SPR) at ca. 520 nm (E and F traces).[7]
Figure 4.7: Plots of changes in the time-dependent absorption ratio (A620/A520) in
the presence of varying concentration of lipase after the addition of salt solution.
As shown in Figure 4.7, once the salt was added, the time-dependent absorption ratio A620/A520 values of solutions with insufficient enzyme displayed an increasing, which illustrated that the AuNPs/lipase system gradually lost the stability protected. On the other hand, the sufficient enzyme could stabilize the AuNPs in the presence of a given high concentration of salt. The destabilization trend was obviously dependent on the concentration of lipase. We just made use of this property to determine the coverage of the particles with the enzyme and the optimal ratio of AuNPs to enzyme.[8]
Figure 4.8: Plot of the absorption ratio (A520/A620) in equilibrium versus the lipase
concentration.
Figure 4.8 displayed a plot for the reciprocal of the ratio (A620/A520) of 1.3 mL 13 nm AuNPs stabilized by the different lipase concentrations after the addition of salt (0.1 mL 10% NaCl solution). When the concentration of lipase added was enough to provide the stability protected for the AuNPs/lipase system, the value of ratio kept constant with time and was almost the same as the nonaggregated AuNPs. On the contrary, there was a significant decrease in the value of ratio with addition of insufficient enzyme. This result suggested that the coverage of the AuNPs can be defined by the ratio which was dependent on the concentration of lipase. According to the property, we obtained the dissociation constant Kd and the concentration of lipase
that we used for kinetic assays can be up to 80% coverage of enzyme onto the gold nanoparticles.
In general, the binding between two substances can be described by a simple equilibrium expression.
PE E
P + ↔
The reaction was characterized by equilibrium constant, Ka, such as:
[P][E] [PE]
a=
K
Where
P: binding site of particles E: enzyme
Ka: association constant Kd: dissociation constant
So, Kd = 1/Ka
Define fraction, Y = the fractional occupancy of the gold nanoparticles-binding sites:
d [E] [E] [P] [PE] [PE] sites binding total occupied sites binding Y K + = + = = , At Y = 1/2, Kd = [E] = 0.37 μM
Y versus the concentration of enzyme [E] = enzyme dissociation curve:
The concentration of enzyme [E] that we prepared for kinetic assays is 1.5 μM. The degree of coverage can be determined as this hyperbolic curve.
In the simple equilibrium expression, lipase was added to a solution of the AuNPs to form the AuNPs-enzyme complex. Actually, it is a system with mixture of particle-bound and unbound enzyme for the catalytic reaction in our manuscript. In fact, the AuNPs-enzyme complex system has reached chemical equilibrium, the state in which the concentration of the AuNPs and enzyme remain constant. According to Le Châtelier’s principle, if the unbound enzyme was removed in the process of purifying nanoconjugates, the particle-bound enzyme desorbs from the surface of the AuNPs and the chemical equilibrium could be broken. It would result in the aggregation of the AuNPs because of the insufficient amount of enzyme. Therefore, we develop linker-free AuNPs kinetic assays to simplify the detection at equilibrium for using the same amount of enzyme in the compared systems.
Since the surface modification of linkers protected the AuNPs from aggregation, it would be perplexed to determine the enzyme coverage and the optimal ratio of AuNPs to enzyme because less or much enzyme can not be identified by salt titration. In our study, we use the same concentration of enzyme in the compared systems for ensuring that accurate parameters presented and then we can understand the mechanism of enhanced catalytic activity. However, we used the system of the direct binding onto AuNPs without surface modification for avoiding the congregation and determining the enzyme coverage with minimum unbound enzyme. This method suggested that the lipase-AuNPs complex, with the mixture of particle-bound and unbound enzyme in catalytic reaction system, has been optimized with regard to its colloidal stability and retention of activity for kinetic assays.
4.5 Kinetic Assays:
4.5.1 Activity Assays of the Free and Gold Nanoparticles-Bound
Lipase
Figure 4.9: a) Product (pNP) formation over time in the catalytic reactions of various
concentrations of AuNP-bound (solid) and free lipase (hollow) solutions monitored at 405 nm. b) Initial velocities of pNP from pNPP plotted as a function of the concentration of the free enzyme (●) and the enzyme-capped AuNPs (○).
Prior to kinetic experiments, we dissolve 4-nitrophenyl palmitate (pNPP) in isopropyl alcohol solution, and then mix the reactants in reaction buffer. We measured the activities of the free and immobilized enzymes by monitoring (at 405 nm; Figure 4.9.a) the initial velocity of 4-nitrophenol (pNP) production from 4-nitrophenyl palmitate (pNPP) at various concentrations of the enzyme solutions in l-cm-pathlength cuvettes and the changes in absorbance were recorded as a function of the reaction time. In Figure 4.9.b, we observed that the initial release of pNP was proportional to the concentrations of both the free and bound enzymes—with a significant increase in the catalytic activity of the enzyme-capped AuNPs. This result also suggested that the catalytic mechanism and the analytic method observed the enzymatic kinetics equations in both cases, allowing us to perform accurate kinetic assays.
4.5.2 The Effect of the Concentration of Gold Nanoparticles on
Catalysis
Figure 4.10: Initial rate of pNP production versus the concentration of AuNPs.
To optimize the supreme efficiency of enhanced catalytic activity, the ratios of AuNPs to enzyme were studies under fixed enzyme and substrate concentration. Figure 4.10 showed the quantitative relationship between the production rate change at 405 nm and the different concentrations of gold nanoparticles. The non-aggregated enzyme–AuNP complex mixed with assay buffer showed an increase in the absorbance. The initial reaction rates were calculated from the slopes of the changes in absorption over time (the absorbance was converted to a concentration scale using a value of 12,800 M–1 cm-1 for the molar absorbance coefficient of pNP).[2] The largest change in reaction rate, which was regarded as optimal condition from dispersed to fully aggregated nanoparticles, was observed when the concentration of AuNPs was approximately 2.2 nM in common with condition E in Figure 4.6.c. There was a significant increase in the rate of production with addition of AuNPs as compared to the control reaction with no AuNPs. Each increase in concentration of 25% resulted in an increase in reaction rate by about 0.14 μM s-1, thereby demonstrating the ability of the AuNPs to act as a factor for enhancing activity. Over optimal condition, reaction rate decreased because the redundant AuNPs aggregated without sufficient coverage, and this phenomenon can be confirmed by the shifted peak of the absorbance spectra.[9] Therefore, it was estimated that catalytic activity can be manipulated by the ratios of AuNPs to enzyme.
4.5.3 Michaelis–Menten and Arrhenius plots
We designed an accurate analysis of enzyme kinetics in order to understand the property of the significantly enhanced activity for enzyme functionalized gold nanoparticles. Figure 4.11 displays Michaelis–Menten plots for the hydrolysis of
pNPP (5.55–133.33 μM) by the free enzyme and enzyme-capped AuNPs at pH 7.4
and presence of the AuNPs, but the Michaelis constant (KM) was obviously smaller when using the lipase-capped AuNPs. According to the Michaelis–Menten equation, the values of Vmax can represent the values of kcat, which indicated that the degrees of
product formation and release from the active site were identical in both systems when the concentration of the enzyme was the same. Therefore, the value of kcat of the bound enzyme was not significantly different from that of the free enzyme. We infer that the enzymes capped on the AuNPs retained their ability to perform nucleophilic attack via the formation of an acyl enzyme; i.e., the presence of the AuNPs had no influence on the release of product in the rate-limiting step. The values of the Michaelis constant were 23.91 and 9.10 μM (p < 0.05) for the free and bound enzymes, respectively. A smaller value of KM represented a higher affinity of an enzyme toward a substrate; i.e., the presence of the AuNPs enhanced the selectivity of lipase toward the substrate. Because the binding of lipase on the surface of the AuNPs affected only the value of KM, the addition of the AuNPs was an efficacious means of tuning the enzyme–substrate association. The active site of an enzyme not only recognizes the substrate(s) to create an intermediate complex (ES) but also complemented and stabilized the transition state. The higher affinity of the substrate toward the enzyme-bound AuNPs resulted in a lower-energy transition state; indeed, the linear Arrhenius plots for the hydrolyses of pNPP mediated by the AuNP-bound and free enzymes at 25–50 °C (Figure 4.12) provided activation energies of 5.9 and 12.2 kJ mol–1, respectively. Thus, the enzymes immobilized onto the surfaces of the AuNPs exhibited higher catalytic activity through more ready formation of the ES complex and reduction of the activation energy.
Figure 4.11: Michaelis–Menten plots for the hydrolyses of pNPP mediated by the free
enzyme (blue) and enzyme-capped AuNPs (red).
Figure 4.12: Arrhenius plots of ln(absorbance) versus the reciprocal of absolute
temperature for the free (blue, inset) and AuNP-bound (red) lipase. The activity was measured for reaction mixtures containing 0.1 μM enzyme at pH 7.4.
Chapter 5: Conclusions
In summary, we have investigated the kinetics of the significantly enhanced activity of an AuNP-bound enzyme. The immobilization of lipase onto the nanoparticles’ surfaces, without any surface modification, provided colloidal stability that allowed us to determine the coverage of the particles with the enzyme and assay the enzymatic activity. The efficiency of enhanced catalytic activity with colloidal stability has been optimized by regularizing the ratios of AuNPs to enzyme. The lipase capping the AuNPs catalyzed the hydrolysis of pNPP through the same kinetic model as that of the free enzyme, with the product’s release being the rate-limiting step, but with increased catalytic activity, as evidenced by lower values of the activation energy and KM. We ascribe this behavior to the enhanced substrate selectivity of the AuNP-bound, thereby decreasing the activation energy through enhancing the rate constants leading to the formation of the ES complex. Our findings suggest that the ability of AuNPs to manipulate catalytic activity might become an important nanobiotechnological tool for optimizing clinical studies and improving drug delivery.
Reference
Chapter 1
[1] Eustis, S.; El-Sayed, M. A., Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes.
Chemical Society Reviews 2006, 35, (3), 209-217.
[2] Rosi, N. L.; Mirkin, C. A., Nanostructures in biodiagnostics. Chemical
Reviews 2005, 105, (4), 1547-1562.
[3] Ginger, D. S.; Zhang, H.; Mirkin, C. A., The evolution of dip-pen nanolithography. Angewandte Chemie-International Edition 2004, 43, (1), 30-45.
[4] Liu, M. Z.; Amro, N. A.; Chow, C. S.; Liu, G. Y., Production of nanostructures of DNA on surfaces. Nano Letters 2002, 2, (8), 863-867.
[5] Kenseth, J. R.; Harnisch, J. A.; Jones, V. W.; Porter, M. D., Investigation of approaches for the fabrication of protein patterns by scanning probe lithography. Langmuir 2001, 17, (13), 4105-4112.
[6] Alivisatos, A. P.; Johnsson, K. P.; Peng, X. G.; Wilson, T. E.; Loweth, C. J.; Bruchez, M. P.; Schultz, P. G., Organization of 'nanocrystal molecules' using DNA. Nature 1996, 382, (6592), 609-611.
[7] Liu, G. L.; Yin, Y. D.; Kunchakarra, S.; Mukherjee, B.; Gerion, D.; Jett, S. D.; Bear, D. G.; Gray, J. W.; Alivisatos, A. P.; Lee, L. P.; Chen, F. Q. F., A nanoplasmonic molecular ruler for measuring nuclease activity and DNA footprinting. Nature Nanotechnology 2006, 1, (1), 47-52.
[8] Niemeyer, C. M., Nanoparticles, proteins, and nucleic acids: Biotechnology meets materials science. Angewandte Chemie-International Edition 2001, 40, (22), 4128-4158.
[9] Whitesides, G. M.; Mathias, J. P.; Seto, C. T., Molecular self-assembly and nanochemistry: a chemical strategy for the synthesis of nanostructures.
![Figure 1.1: Plot of the number of articles published on nanoparticles since 1990. [1]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8260369.172129/15.892.141.741.488.1086/figure-plot-number-articles-published-nanoparticles.webp)
![Figure 1.5: Integrated nanoparticle–biomolecule hybrid systems. [18]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8260369.172129/20.892.270.623.760.1093/figure-integrated-nanoparticle-biomolecule-hybrid-systems.webp)