The Role of Transesterification on the Miscibility in Blends of
Polycarbonate and Liquid Crystalline Copolyester
Kung-Hwa Wei* and Jia-Chong Ho
Institute of Material Science & Engineering, National Chiao Tung University, Hsinchu, Taiwan 30049, ROC
Received October 16, 1996; Revised Manuscript Received January 3, 1997X
ABSTRACT: Transesterification mechanisms and rate in blends of polycarbonate (PC) and random liquid crystalline polyester copoly(oxybenzoate-p-terephthalate) at molar ratio 40/60 (P46) were studied through a five-component diad analysis and with13C and1H
-13C nuclear magnetic resonance spectroscopy. It was found that the ester-ester interchange in the two polymers took place within 15 min when the blend was annealed at 260 °C in vacuum. In the annealed blend, the Bisphenol A unit in polycarbonate reacted first with the terephthalate unit and then with the oxybenzoate unit in copoly(oxybenzoate- p-terephthalate). As the transesterification in the blend continued for about 1 h, the forming of diad Bisphenol A-oxybenzoate exceeded that of diad Bisphenol A-terephthalate. This large loss (57%) of the diad oxybenzoate-oxybenzoate caused the disappearance of the liquid crystalline phase in the blend. In sharp contrast to the originally immiscible blend of PC and P46 (two distinctive glass transition temperatures), the large loss of the liquid crystalline diad resulted in complete miscibility in the annealed blend, as evidenced by the appearance of a single glass transition temperature in the differential scanning calorimetry measurement.
Introduction
Thermotropic liquid crystalline polymers (TLCPs) had attracted a great deal of attention since their arrival. These TLCPs usually contained rigid molecular struc-tures and therefore possessed unique properties such as low melt viscosity and high modulus in the oriented direction in the solid form. The major drawback for these materials in the market place is the high cost of the monomers. From a practical point of view, blending TLCPs with other flexible polymers can lower the blend’s viscosity and form organic polymer composites in situ, as described by previous workers.1-5 However,
liquid crystalline polymer chains are very stiff and of rigid-rod nature. The enthalpy of mixing a rigid-rod polymer with a flexible-chain polymer was mostly positive. The small increase in entropy due to the mixing in these two polymers was not able to compen-sate for the enthalpy effect. The free energy of mixing is therefore mostly positive. In other words, the mis-cibility of TLCPs and flexible-chain polymers is not favorable in thermodynamics. Phase separation of TLCP blends occurred where high-stress and -temper-ature conditions were encountered.6,7 Therefore, the
miscibility of TLCP blends is a major obstacle in their applications. Introducing some kind of interaction between TLCPs and a flexible-coil polymer is necessary to improve the miscibility of the two polymers. In this respect, some TLCPs contained unique chemical struc-tures that resulted in reaction with flexible chain polymers. A case in point is the most studied thermo-tropic copolyesters of oxybenzoate and ethylene tereph-thalate (POB-PET).
8 A well-known feature for
poly-esters is that it can have alcoholysis by a hydroxyl end group, acidolysis by an acid end group, and midchain ester-ester interchange (transesterification) with other polymers at high temperatures9,10(generally above 200
°C). For high molecular weight polymers, the prob-ability of having transesterification is much higher than
that of alcoholysis or acidolysis because of low end group concentrations. Hence, transesterification usually domi-nated the reaction process in relatively high molecular weight polyesters. Both the solution and melt blends of POB-PET copolyester with poly(butylene terephtha-late) (PBT), polycarbonate (PC), and poly(hexamethyl-ene terephthalate) (PHMT) have been studied with differential scanning calorimetry by various research groups.11-13 They have reported that the miscibility of
blends of PC/PET, PBT/POB-PET, PC/POB-PET, and PC/poly(p-oxybenzoate-co-p-phenylene isophthalate) was found to increase with transesterification.11,12,14-16 As
transesterification in blends continues, new amorphous compatible blends will form. This brings about the fact that the liquid crystalline character in blends is lost, and the purpose of using TLCP blends is destroyed. We are therefore motivated to study the process of trans-esterification in TLCP blends. We would like to know if there is a point during transesterification where the miscibility of the blends increased while maintaining the liquid crystalline character. Specifically, since two kinds of esters existed in the PET and POB segments of POB-PET copolymers, we wondered if ester-ester interchange takes place randomly or in a preferential manner. Since transesterification between PC and PET has been well studied, we chose a TLCP copolymer of high molar ratio of PET to POB (60/40) for this study. This POB-PET (40/60) copolymer is termed P46. For a theoretical analysis of transesterification in three-component copolycondensates, Yamadera17studied the
average sequence length and the degree of randomness in blends of poly(ethylene terephthalate) (PET) and poly(ethylene sebacate) (PES). For four-component copolycondensates, Devaux18investigated the sequence
length and the randomness in blends of Bisphenol A polycarbonate and poly(butylene terephthalate). Ad-ditionally, Kollodge19studied both alcoholysis and
trans-esterification on the phase behavior of blends of PC and lower molecular weight poly(2-ethyl-2-methylpropylene terephthalate) (PEMPT). In this P46/PC blend case, we did a theoretical diad analysis of transesterification of five-component copolycondensates. In order to pinpoint
* 886-3-573-1871 (Tel). 886-3-572-4727 (Fax).
XAbstract published in Advance ACS Abstracts, March 1, 1997.
the chemical structure changes during transesterifica-tion in the P46/PC blend, a model compound study of blends of PC/hydroxybenzoic acid (HBA) and PC/tereph-thalic acid (TA) at a weight ratio of 80 to 20 has been carried out. This model compound study is similar to that for blends of polyarylate and poly(butylene tereph-thalate) by previous workers.20
Experimental Section
The copolymer of POB-PET at mole ratio 40/60 (P46) was synthesized in our Laboratory following the method by Kuh-fuss21and was termed P46 in this study. Polycarbonate was purchased from General Electric Co. The trade name of PC is Lexan 121, and its molecular weight is Mw)158 900. The intrinsic viscosity of P46 and PC were 0.53 and 1.15 dL/g, respectively, when they were prepared in mixed solvent of phenol and tetrachloroethane (50/50 by weight). The solution blending of PC and P46 was carried out by dissolving both polymers in 25 mL of a mixed solvent of phenol and tetrachlo-roethane (50/50). The concentration of the solution containing PC and P46 is 2% by weight. The weight ratio of PC to P46 in the solution was 40/60. The solution was stirred and heated to 70 °C. After the polymers were completely dissolved and became a one-phase solution for 0.5 h, the solutions were precipitated in a 10-fold volume of methanol. The blends were then washed five times, each time with 200 mL methanol. The blends were then dried in a vacuum oven at 100 °C for 4 days. The thermal gravimetric analysis of the dried blends showed no appreciable weight loss up to 350 °C, indicating a complete removal of the solvent. The 60/40 P46/PC blends were put into a high-temperature vacuum tube at 260 °C and were annealed for different times. The thermal analysis of the blends was carried out with a DuPont 2910 differential scanning calorimetry (DSC). The samples were heated from 30 to 260 °C, at a heating rate of 20 °C/min. Then, the samples were cooled quickly to 170 °C and annealed for 10 min. Subsequently, the samples were quenched to 25 °C. The samples were heated again from 25 to 260 °C at the same heating rate. The DSC curves of the samples were taken during the second heating. The midpoint of the glass transi-tion was taken as the glass transitransi-tion temperature (Tg). Without annealing, the DSC curves of the blends did not exhibit separate crystallization (by P46) and glass transition (by PC). Annealing at 170 °C for 10 min avoided the problem by allowing the samples to crystallize. Melt-blending of PC/ HBA and PC/TA at a weight ratio of 80/20 was performed in a Brabender mixer at 220 and 300 °C for 30 min, respectively. HBA can be dissolved in trifluoroacetic acid, while TA cannot. Both PC/HBA and PC/TA reacted blends were dissolved in trifluoroacetic acid and then were filtered with a filter of 0.45 µm in pore size. The soluble fractions of these solutions were analyzed with nuclear magnetic resonance (NMR).
For nuclear magnetic resonance (NMR) analysis of the 60/ 40 P46/PC blends, 10% solutions were prepared by dissolving the polymers completely in a mixed solvent of deuterated trifluoroacetic acid and deuterated chloroform, 15/85 by weight. The NMR spectra were obtained on a Bruker DMX-600 spectrometer. Two-dimensional NMR heteronuclear multiple bond correlation (HMBC) spectra for the blends’ solutions were obtained.
Diad Statistical Analysis
Similar to the analysis in PES/PET by Yamadera17
and in PC/PBT by Devaux,18 we divided P46 and PC
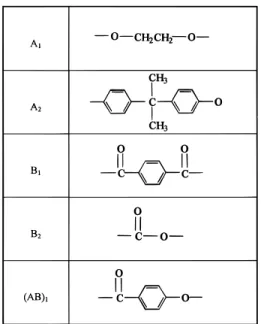
into five components. Monomer units of different chemical structure but of the same functionality were represented by Ai, i)1, 2 (or Bj, j)1, 2). We used the notations (AB)1, B1, and A1 standing for oxybenzoate,
terephthalate, and ethylene in P46, respectively. Bisphe-nol A and carbonate in PC were noted as A2and B2, as
listed in Table 1. Before the reaction started, we have A1(AB)1, A1B1, (AB)1B1, (AB)1(AB)1, and A2B2. The
degree of polymerization is high enough to neglect chain
end reaction in this analysis. When the transesterifi-cation went on, we had the probability of producing new species A2B1, A2(AB)1, A1B2, and (AB)1B2.
Before the reaction started, the mole fractions of Bisphenol A (A2), ethylene (A1), terephthalate (B1),
carbonate (B2), and oxybenzoate ((AB)1) were defined
by
where brackets stand for the mole concentration of each component in the blend.)
The diad mole fraction of the blend of 60/40 P46/PC is defined by
or
Table 1. Codes Used in Diads of P46 and PC
FAi) [Ai]
∑
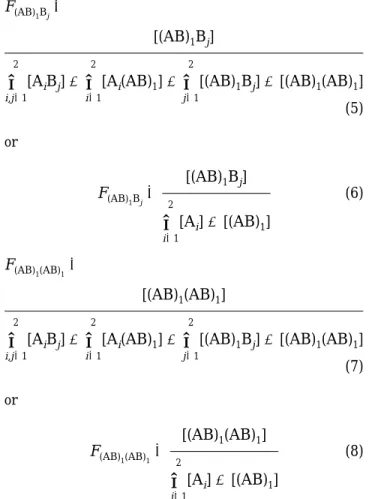
i)1 2 [Ai]+[(AB)1] (1A) FB j ) [Bj]∑
j)1 2 [Bj]+[(AB)1] (1B) F(AB)1) [(AB)1]∑
i)1 2 [Ai]+[(AB)1] (2) FA iBj ) [AiBj]∑
i,j)1 2 [AiBj]+∑
i)1 2 [Ai(AB)1]+∑
j)1 2[(AB)1Bj]+[(AB)1(AB)1] (3) FA iBj ) [AiBj]
∑
i)1 2 [Ai]+[(AB)1] (4)or
or
The probability of finding an Aigroup followed by a Bj unit, PAiBj,would be
PAiBj*PB
jAi(i*j) (except an equimolar blend)
From the above equation we obtained the following relation:
The integrals under the resonance peak of 13C NMR
spectra of 60/40 P46/PC are proportional to the copoly-condensate diad probabilities.
Results and Discussion
The DSC curves and chemical structure of P46 and PC are shown in Figure 1. The glass transition tem-peratures (Tg’s) of PC and P46 are 147.5 and 59.9 °C,
respectively. The crystallization temperature (Tc) for
P46 and is 98.7 °C. The broad endothermic peak is the melting point of crystalline PET. In this study, the Tg
of P46 belonged to the PET-rich phase, as cited in the literature.22 For the 60/40 P46/PC blends, the low T
g
belonged to P46 and the high Tg was due to PC, as
illustrated in Figure 2. In Figure 2, only the portion of the DSC curve containing glass transition temperatures of P46 and PC was displayed. The two Tg’s were
separated by 56 deg after the blend was annealed at F(AB) 1Bj ) [(AB)1Bj]
∑
i,j)1 2 [AiBj]+∑
i)1 2 [Ai(AB)1]+∑
j)1 2[(AB)1Bj]+[(AB)1(AB)1] (5) F(AB)1Bj) [(AB)1Bj]
∑
i)1 2 [Ai]+[(AB)1] (6) F(AB) 1(AB)1 ) [(AB)1(AB)1]∑
i,j)1 2 [AiBj]+∑
i)1 2 [Ai(AB)1]+∑
j)1 2[(AB)1Bj]+[(AB)1(AB)1] (7) F(AB)1(AB)1) [(AB)1(AB)1]
∑
i)1 2 [Ai]+[(AB)1] (8) PA iBj ) [AiBj]∑
j)1 2 [AiBj]+[Ai(AB)1] ) [AiBj] [Ai] (9) PA i(AB)1 ) [Ai(AB)1]∑
j)1 2 [AiBj]+[Ai(AB)1] ) [Ai(AB)1] [Ai] (10) P(AB)1Bj) [(AB)1Bj]∑
j)1 2[(AB)1Bj]+[(AB)1(AB)1] ) [(AB)1Bj] [(AB)1] (11) P(AB)1(AB)1) [(AB)1(AB)1]
∑
j)1 2[(AB)1Bj]+[(AB)1(AB)1] )
[(AB)1(AB)1] [(AB)1] (12)
Figure 1. Differential scanning curves and chemical
struc-tures of P46 and PC.
Figure 2. Partial differential scanning curves of the 60/40
P46/PC solution blend annealed at 260 °C for different times. FAiBj)FA iPAiBj )FB jPBjAi (13) FA i(AB)1 )FA iPAi(AB)1 )F(AB) 1P(AB)1Ai (14) F(AB)1Bj)F(AB) 1P(AB)1Bj )FB jPBj(AB)1 (15) F(AB) 1(AB)1 )F(AB) 1P(AB)1(AB)1 (16)
260 °C for 15 min. As the annealing time increases, the two Tg’s converged on each other. There was only
one Tgappearing at 109.5 °C after 60 min of annealing
at 260 °C. Near the single Tg, there appears to remain
some small structure at 90-100 °C that was possibly due to the residual crystallization of traces of PET microdomain in P46 after allowing the blend to crystal-lize at 170 °C for 10 min, as described in the Experi-mental Section, whereas the small structure at 120 -130 °C is likely due to traces of PC microdomain since the onset point of the PC glass transition is 125 °C (see Figure 1). Despite these two small structures, the single Tgbehavior exhibited by the blend indicated an almost
complete miscibility between P46 and PC. This misci-bility result is similar to the homogenization results in the PET/PC blend obtained with solid state NMR.15In
a parallel measurement, the intrinsic viscosity of the 60/40 P46/PC blends decreased with increasing anneal-ing time at 260 °C, as illustrated in Figure 3. After 60 min of annealing, the intrinsic viscosity of the blend dropped to almost half its original value. The reduction of the intrinsic viscosity of the annealed blend was apparently caused by a reaction between the polymers. The reaction possibilities included partial hydrolysis10
of the copolyester P46, decarboxylation in PC,23 and
ester-ester interchange
12 in P46 and PC. Since the
annealing of the blend was carried out in a vacuum tube at 260 °C under 10-1Torr, the possibility of hydrolysis
of P46 was reduced to a minimum. The decarboxylation resulting from acidolyis of PC by the end group of P46 existed but was not a dominant factor because the concentration of the end group of P46 was not particu-larly high. It seemed that the ester-ester interchange between the two polymers only averaged the molecular sizes. In fact, the transesterification would also cause P46 to lose its rigid rod nature, and therefore reduced
Figure 3. Intrinsic viscosity of the 60/40 P46/PC blend after
being annealed at 260 °C for different times.
Figure 4. 13C 600 MHz NMR spectra of (a) hydroxybenzoic
acid (HBA), (b) the soluble fraction of PC/HBA (80/20) mixed at 220 °C for 30 min (3* and 5* were new peaks), (c) the soluble fraction of PC/TA (80/20) mixed at 300 °C for 30 min (3** and 5** were new peaks).
Figure 5. (a) Proton 600 MHz NMR spectrum of the P46
copolymer. (b) Peak integrals of POB-POB and POB-PET peaks.
its intrinsic viscosity (IV) dependency on molecular weight. This is clarified by the Mark-Kuhn- Hou-wink-Sakurada equation IV ) KM
R, where K is a
constant and M is the molecular weight. For flexible-chain polymers theRvalue is between 0.5 and 1, and the Rvalue may exceed unity in the case of rigid-rod polymers.24 Hence, the crucial reduction in the intrinsic
viscosity of the annealed blend most likely came from some decarboxylation and transesterification. From the single Tgand a large decrease in intrinsic viscosity, one
deduced that the ester exchange reaction had taken place between P46 and PC. The actual confirmation of
the chemical structure change in these polymers result-ing from ester exchange has been carried out with NMR analysis. In the model compound study, the aromatic regions of the13C NMR spectra of hydroxybenzoic acid
(HBA), PC/HBA (80/20), and PC/terephthalic acid (TA) (80/20) are shown in Figure 4a-c, respectively. The NMR spectrum of HBA in Figure 4a was used for comparison later. The two resultant peaks from diad Bisphenol A-oxybenzoate (A2(AB)1) were identified as 3* and 5*, as indicated in Figure 4b, and their 13C
chemical shifts were 148.47 and 121.30 ppm, respec-tively, whereas for the TA/PC case, the 13C chemical
Figure 6. 1H
-13C 600 MHz NMR spectra of the 60/40 P46/PC blend: (a) freshly prepared; (b) after being annealed at 260 °C for 30 min; (c) after being annealed at 260 °C for 60 min.
Table 2. Chemical Shifts of Aromatic13C in Blends of HBA/PC and TA/PC
compound or polymer chemical shift δ (ppm)
hydroxybenzoic acid (HBA) 160.5 (Ca) 115.6 (Cb) 121.0 (Cc) 133.3 (Cd) 172.5 (Ce)
soluble fraction of reacted HBA/PC (20/80) at 220 °C for 30 min 148.6 (C3) 148.47 (C3*) 120.34 (C5) 121.3 (C5*) 148.92 (C6)
soluble fraction of reacted Terephthalic acid/PC (20/80) at 300 °C for 30 min
shifts for the new resultant peaks were 148.46 ppm (3**) and 120.88 ppm (5**), as shown in Figure 4c. This indicated formation of Bisphenol A-terephthalate (A2B1). The detailed chemical shifts in the NMR spectra of these model compounds are listed in Table 2. Probably due to the fact that the concentration of oxybenzoate is low, the peaks in the ester region of the13C NMR spectra
were much weaker than can be observed. The structure change caused by the transesterification in the ester region of the blend was directly analyzed with two-dimensional 1H
-13C HMBC NMR spectra. Since the
most change occurred in the aromatic and ester region of the P46/PC blend, we focused on these two regions of the NMR spectra. Before going on to the blends, we analyzed P46. The1H NMR spectrum of synthesized
P46 is shown in Figure 5a. The fraction of the POB -POB peak area in both the -POB-POB and PET-PET peak area is 0.269, as displayed in Figure 5b. On the basis of a probability calculation,25 a fully random
copolymer of P46 should have a value of 0.25. There-fore, P46 is very close to a random copolymer. The partial aromatic and carbonyl regions of the 1H
-13C
NMR spectra of the 60/40 P46/PC blend are shown in Figure 6. In Figure 6a, the resonance peaks of the freshly prepared 60/40 P46/PC blend indicated that the blend is a mechanical mixing of two polymers. When the 60/40 P46/PC blend was annealed at 260 °C for 30 min, a small new peak appeared at 8.3-166 ppm in the
1H
-13C spectra of the blend, as presented in Figure 6b.
Then, two full new cross peaks located at 8.3-166 ppm and 8.37-166 ppm showed up in the NMR spectra of the blend after 60 min of annealing, as shown in Figure 6c. From reading the relative position of1H to13C, we
knew it was the aromatic Hc in the POB segment
attached to the carbonyl Cein the POB segment. Hence,
we deduced that these new peaks in the ester region were caused by the new structure A2(AB)1or Bisphenol
A attached to oxybenzoate. To quantify the chemical structures corresponding to these resonance peaks, a one-dimensional 13C NMR spectral analysis of the
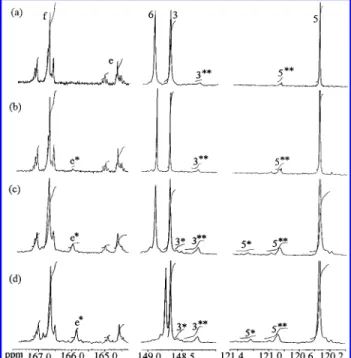
annealed blends was carried out and presented in Figure 7. After the blend was annealed for 15 min,
there were two new peaks 3** and 5** appearing at 120.9 and 148.38 ppm, respectively as shown in Figure 7a. These two new peaks represented the structure Bisphenol A-terephthalate (A2B1), judging from the model compound study. When the annealing time increased to 30 min, an additional new peak e* showed up at 165.9 ppm and was identified as Bisphenol A-oxybenzoate (A2(AB)1), as shown in Figure 7b. Al-though the new peaks 3*and 5* in the aromatic region
of the NMR spectra of the blend appeared after 45 min of annealing, we still recognized the e* peak as the first appearing evidence of Bisphenol A-oxybenzoate. For calculating the transient mole fraction of diads, the initial mole fractions of Bisphenol A (A2), ethylene (A1),
and oxybenzoate ((AB)1) in this 60/40 P46/PC blend were
determined from eqs 1A, 1B, and 2. The values of FA2,
FA1, and F(AB)1 obtained were 0.45, 0.33, and 0.22,
respectively. The diad existing probabilities were pro-portional to the integrals under the resonance peaks. Taking the integral values under the resonance peaks in Figure 7 and substituting them into eqs 13-16, the transient mole fraction of diads in the 60/40 P46/PC blend is obtained and presented in Figure 8. In Figure 8, the formation of the diad Bisphenol A-oxybenzoate (A2(AB)1) increased markedly and exceeded that of the
diad Bisphenol A-terephthalate (A2B1) after 55 min of annealing time. The increase of the diad Bisphenol A-terephthalate (A2B1) became saturated at 0.062 mole fraction after 60 min of annealing. The loss of diad mole fractions of ethylene-oxybenzoate (A1(AB)1), Bisphenol A-carbonate (A2B2), and oxybenzoate-oxybenzoate ((AB)1(AB)1) were 36%, 33.5%, and 57%, respectively,
after 60 min of annealing at 260 °C. The transient mole fraction of diad A2B2is determined by subtracting FA2B1
and FA2(AB)1from the initial FA2B2. This calculation
is at 1 h of annealing time when the annealed blend was miscible but not yet purely randomized. For a purely randomized blend, the loss of diad mole fraction A2B2 would be 84.7% based upon the initial P46 and
PC composition in the blend. The large loss of the diad oxybenzoate-oxybenzoate ((AB)1(AB)1) after 60 min of annealing resulted in the disappearance of the liquid crystalline phase. This certainly caused an improve-ment in the miscibility of the originally immiscible 60/ 40 P46/PC blends (two distinctive Tg’s). This result
corresponded very well to the complete miscibility
Figure 7. 13C 600 MHz NMR spectra of 60/40 P46/PC blends
after being annealed at 260 °C for (a) 15 min, (b) 30 min, (c) 45 min, and (d) 60 min.
Figure 8. Transient diad mole fraction in 60/40 P46/PC
blends annealed at 260 °C obtained from nuclear magnetic resonance analysis.
behavior (a single glass transition temperature) in the annealed 60/40 P46/PC blend obtained by the DSC measurement.
Conclusion
From the diad analysis and the NMR results, we concluded that ester-ester interchange in the blends of random liquid crystalline copolyester P46 and poly-carbonate took place within 15 min when the blend was annealed at 260 °C under vacuum. In the annealed blend, the Bisphenol A unit in polycarbonate reacted first with the terephthalate unit and then with the oxybenzoate unit in copoly(oxybenzoate- p-terephtha-late). As the reaction in the blend continued for about 1 h, the production of diad Bisphenol A-oxybenzoate sped up and exceeded that of diad Bisphenol A- tereph-thalate. This caused a large loss (57%) of diad oxyben-zoate-oxybenzoate and led to the disappearance of the liquid crystalline phase in the annealed blend. In sharp contrast to the originally immiscible blend of PC and P46 (two distinctive glass transition temperatures), the large loss in the liquid crystalline diad resulted in complete miscibility (a single glass transition temper-ature) in the annealed blend, as evidenced by the differential scanning calorimetry measurement.
Acknowledgment. The authors appreciate the fi-nancial support provided by the National Science Coun-cil through Project NSC85-2216-E-009-005. Addition-ally, we thank Ms. Chu-Lan Peng for the help with nuclear magnetic resonance experiments.
References and Notes
(1) Kiss, G. Polym. Eng. Sci. 1987, 27, 410.
(2) Kolhi, A.; Chung, N.; Weiss, R. A. Polym. Eng. Sci. 1989, 29, 573.
(3) Brostow, W. Polymer 1990, 31, 979.
(4) Wei, K. H.; Kiss, G. Polym. Eng. Sci. 1996, 36, 713. (5) Wei, K. H.; Hwang, W. J.; Tyan, H. L. Polymer 1996, 37, 2087. (6) Tang, P.; Reimer, J. A.; Denn, M. M. Macromolecules 1993,
26, 4269.
(7) Bafna, S. S.; Sun, T.; de Souza, J. P.; Baird, D. G. Polymer 1995, 36, 259.
(8) Economy, J.; Goranov, K. in Advances in Polymer Science; Hergenrother, P. M., Ed.; Springer-Verlag: Berlin, 1994. (9) Kotliar, A. M. J. Polym. Sci., Macromol. Rev. 1981, 16, 367. (10) Pilati, F.; Marianucci, E.; Berti, C. J. J. Appl. Polym. Sci.
1985, 30, 1267.
(11) Kimura, M.; Porter, R. S. J. Polym. Sci., Polym. Phys. Ed. 1983, 21, 367.
(12) Su, K. F.; Wei, K. H. J. Appl. Polym. Sci. 1995, 56, 79. (13) Laivins, G. V. Macromolecules 1989, 22, 3974.
(14) Godard, P.; Dekoninck, J. M.; Devlesaver, V.; Devaux, J. J. Polym. Sci.: Part A: Polym. Chem. 1986, 24, 3301. (15) Henrichs, P. M.; Tribone J.; Massa, D. J.; Hewitt, J. M.
Macromolecules 1988, 21, 1282.
(16) Wei, K. H.; Jang, C. J.; Ho, J. C. Polymer, in press. (17) Yamadera, R.; Murano, M. J. J. Polym. Sci., Polym. Chem.
Ed. 1967, 5, 2259.
(18) Devaux, J.; Godard, P.; Mercier, P. J. Polym. Sci., Polym. Phys. Ed. 1982, 20, 1875.
(19) Kollodge, J. S.; Porter, R. S. Macromolecules 1995, 28, 4106. (20) Espinosa, E.; Fernandez-Berridi, M. J.; Inaki, M.; Valero, M.
Polymer 1994, 34, 382.
(21) Jackson, J. R.; Kuhfuss, H. F. J. Polym. Sci., Polym. Chem. Ed. 1976, 14, 2043.
(22) Porter, R. S.; Wang, L. H. Polymer, 1992, 33, 2019. (23) Schilling, F. C.; Ringo, W. M., Jr.; Sloane, N. J. A.; Bovey, F.
A. Macromolecules 1981, 14, 532.
(24) Vinogradov, C. V.; Malkin, A. Y. Rheology of Polymers; Springer-Verlag: New York, 1980.
(25) Vollmert, B. Polymer Chemistry; Springer-Verlag: New York, 1973.