Intermolecular potentials of the methane dimer calculated
with Møller-Plesset perturbation theory and density functional theory
Arvin Huang-Te Li and Sheng D. Chaoa兲
Institute of Applied Mechanics, National Taiwan University, Taipei, 106 Taiwan, Republic of China 共Received 15 May 2006; accepted 2 August 2006; published online 6 September 2006兲
We have calculated the intermolecular interaction potentials of the methane dimer at the minimum-energy D3d conformation using the Hartree-Fock 共HF兲 self-consistent theory, the correlation-corrected second-order Møller-Plesset 共MP2兲 perturbation theory, and the density functional theory 共DFT兲 with the Perdew-Wang 共PW91兲 functional as the exchange or the correlation part. The HF calculations yield unbound potentials largely due to the exchange-repulsion interaction. In the MP2 calculations, the basis set effects on the repulsion exponent, the equilibrium bond length, the binding energy, and the asymptotic behavior of the calculated intermolecular potentials have been thoroughly studied. We have employed basis sets from the Slater-type orbitals fitted with Gaussian functions共STO-nG兲 共n=3–6兲 关Quantum Theory of Molecular and Solids: The
Self-Consistent Field for Molecular and Solids 共McGraw-Hill, New York, 1974兲, Vol. 4兴, Pople’s
medium size basis sets of Krishnan et al. 关J. Chem. Phys. 72, 650 共1980兲兴 关up to 6-311+ + G共3df ,3pd兲兴 to Dunning’s correlation consistent basis sets 关J. Chem. Phys. 90, 1007 共1989兲兴 共cc-pVXZ and aug-cc-pVXZ兲 共X=D, T, and Q兲. With increasing basis size, the repulsion exponent and the equilibrium bond length converge at the 6-31G**basis set and the 6-311+ + G共2d,2p兲 basis set, respectively, while a large basis set共aug-cc-pVTZ兲 is required to converge the binding energy at a chemical accuracy共⬃0.01 kcal/mol兲. Up to the largest basis set used, the asymptotic dispersion coefficient has not converged to the destined C6value from molecular polarizability calculations. The slow convergence could indicate the inefficacy of using the MP2 calculations with Gaussian-type functions to model the asymptotic behavior. Both the basis set superposition error 共BSSE兲 corrected and uncorrected results are presented to emphasize the importance of including such corrections. Only the BSSE corrected results systematically converge to the destined potential curve with increasing basis size. The DFT calculations generate a wide range of interaction patterns, from purely unbound to strongly bound, underestimating or overestimating the binding energy. The binding energy calculated using the PW91PW91 functional and the equilibrium bond length calculated using the PW91VP86 functional are close to the MP2 results at the basis set limit. © 2006 American Institute of Physics.关DOI:10.1063/1.2345198兴
I. INTRODUCTION
Intermolecular interaction potentials, or van der Waals interactions, or non-covalent-bonded interactions, play an es-sential role in condensed matter physics, materials chemistry, and structural biology. While these interactions are normally one or two orders of magnitude weaker than typical covalent bonds, they are crucial in determining the thermodynamic properties of molecular liquids and solids,1the energy trans-fers among molecular complexes,2 and the conformational tertiary structures of macromolecules such as protein and DNA.3Unlike intramolecular covalent bonds, intermolecular bonds do not originate from sharing of electrons but rather arise from simultaneous electron correlation of the separated subsystems.4 Different from stiff covalent bonds, they are relatively soft and nonrigid. Early studies of intermolecular interactions can be traced back to one century ago,5 while measurements of these interactions are still challenging in the present time.6 The main difficulty in determining
inter-molecular interactions experimentally resides at limited sam-plings of the potential energy surface. For example, experi-ments using the x-ray crystallography or the laser spectroscopy mainly explore the equilibrium regions of the potential, while thermodynamic measurements in the gas or liquid phase often yield isotropic potential data without the desired stereochemical responses. In addition, the measured potentials sensitively depend on the thermodynamic condi-tions. Usually two measurements carried out in different con-ditions cannot be compared directly but rely on auxiliary theoretical modeling.
Alternatively intermolecular potentials can be calculated in terms of correlation-corrected quantum chemistry methods7–9 or density functional theory 共DFT兲.10,11 These quantum mechanics based potentials are requested by ab
ini-tio molecular dynamics simulaini-tions12 and by classical
mo-lecular simulations using force field constructions.13Among the components of an intermolecular interaction, the London dispersion force is the most difficult to calculate. The reason is that dispersion interactions arise from the nonlocal “dy-namic” correlations.14This nonlocality demands full explo-a兲Author to whom correspondence should be addressed; electronic mail:
ration of the tidependent perspective of quantum me-chanics. Often an electron correlation-corrected method and a large basis set are required to obtain accurate dispersion forces.15 Also a technical note is in order. Most present implementations of quantum chemistry programs utilize Gaussian-type functions to fasten the calculations of Cou-lomb repulsion integrals. Because Gaussian-type functions are local functions, a large basis set is indispensable to per-form a correlation energy calculation. Moreover, these func-tions do not have the correct asymptotic behavior as the in-termolecular separation becomes large. Therefore, the basis set limit of the calculated potential must be estimated so as to be consistent with the conventional perturbation theory.
For general polar molecular systems, the relatively weak dispersion energy is masked by the competing electrostatic energies and hydrogen bond interactions. Nonpolar atomic and molecular dimers are usually taken as a prototype case to study the dispersion energy. Many previous studies on dis-persion forces have focused on atomic inert gas dimers and several important conclusions have been drawn from the calculations.16 There are, however, comparatively fewer de-tailed studies on whether similar conclusions can be applied to molecular dimers. Because of the extra degrees of freedom and the stereochemical responses, the conclusions about atomic dimers may need to be extended or modified in deal-ing with molecular dimers. Methane is a nonpolar molecule with vanishing dipole and quadrupole moments and the first nonvanishing electrostatic interaction is the octopole-octopole interaction. The higher order electrostatic interac-tions are rather weak and decay fast at large intermolecular separation. The dominant long-range attraction for the meth-ane dimer is thus due to the London dispersion force. On the other hand, the strong repulsive force almost comes from the exchange-repulsion interaction due to the overlapping of electron clouds. Because the exchange-repulsion interactions have been incorporated in the Hatree-Fock 共HF兲 self-consistent theory, post-HF methods such as the Møller-Plesset 共MP兲 perturbation theory and the coupled cluster 共CC兲 theory are often used to calculate the correlation effect. Contrasting both sets of calculation helps to delineate the relative importance of the dispersion energy in the overall intermolecular interaction. In addition, the interaction poten-tials of alkanes, among them methane being the smallest model, are very crucial in determining the packing morphol-ogy in solids and liquids and in lipid bilayers.17–19The po-tentials are also requested for mesoscale simulations for macromolecules20 because many polymers contain alkyl groups as their backbone units or side chains. Therefore, the calculation of intermolecular interactions of the methane dimer is a “must” and serves as a prototype example to start to investigate the various factors affecting the calculations of these interactions.
There have been many quantum chemistry studies of in-termolecular potentials of the methane dimer using correlation-corrected methods.21–32 Szczesniak et al.21 have used the MP2 method with the Sadlej basis to calculate the interaction energies for the six conformers of the methane dimer. They found that the minimum-energy conformation is the D3dconformer and the dimer structure is determined by
minimizing the steric repulsion between hydrogen atoms be-longing to opposite subsystems. Tsuzuki and Tanabe24 have studied the basis set effects using basis sets from 6-31G* to 6-311+ + G共2d,2p兲 to calculate the interaction energies for two conformers of the methane dimer 共but none of them is the D3d conformer兲. They observed little basis set effect on the HF calculations as long as one uses a basis set larger than the 6-31G* one. The basis set effect is significant for the MP2 interaction energies and the basis set superposition er-ror共BSSE兲. The dispersion energy can be seriously underes-timated if a smaller 共than 6-31G*兲 basis set has been used. They found that the BSSE uncorrected interaction energies do not systematically converge to a destined value, in con-trast to those with BSSE corrections. Tsuzuki et al.27,31 stud-ied 12 conformers and verifstud-ied that the D3d conformer cor-responds to the minimum-energy geometry. They observed that a large basis set with multiple polarization functions is necessary to evaluate the dispersion energy accurately. They found that augmentation of the diffuse d and p functions to the 6-311G**basis set more efficiently yields the dispersion energy. Tsuzuki and Luthi28and Tsuzuki et al.31explored the effect of the choice of the correlation-correction methods on the calculations of intermolecular interactions. They demon-strated that the MP2 and MP3 energies are not too far away from the higher level MP4共SDTQ兲 calculations, while the latter is not less expensive than the CCSD共T兲 calculation. They tested the DFT using the BLYP, BPW91, and B3LYP functionals but found unbound interactions while the PW91 functional underestimates only 8% of the potential well depth. They suggest that DFT with the PW91 functional could be an alternative to the ab initio methods. Recently, Tsuzuki et al.32have estimated the MP2 and CCSD共T兲 inter-action energies of the n-alkane dimers at the basis set limit using Dunning’s correlation consistent basis sets.
Many previous studies mainly focused on the equilib-rium region of the potentials with relatively few discussions of the full potential curves. Nevertheless, to construct a reli-able force field model for molecular simulations, the full intermolecular potential surfaces are required. In this paper we perform a comprehensive up-to-date study on interaction potentials of the prototype methane dimer in terms of the HF, MP2, and DFT methods to gain more understanding of this system. With current computational powers, a detailed edit-ing of the potential database can be obtained for small size molecular clusters. It is thus so important to obtain general features of the calculations that we can follow to explore large scale molecular simulations via similar procedures. The purpose of this paper is twofold. First, we use the state-of-the-art methodology to obtain accurate potential energies for the methane dimer. We would like to verify or modify the previous conclusions about the basis set effects and the effect of including the BSSE on the calculation details of the inter-molecular interactions. The basis set effects on repulsion ex-ponents, equilibrium bond lengths, binding energies, and asymptotic coefficients of the calculated intermolecular po-tentials are thoroughly studied. This is achieved using basis sets from STO-3G共Ref.35兲 to aug-cc-pVQZ.37The full po-tential curves are presented in order to see the overall scope of the potential. In particular, both the BSSE corrected and
uncorrected results are presented to emphasize the impor-tance of these corrections. Second, this paper attempts to reassess the utilities of using the available implementation of the density functional theory in determining the intermolecu-lar interactions. From the studies of atomic dimers, it has been found that conventional DFT based on the local density approximation 共LDA兲 and generalized gradient approxima-tion 共GGA兲 cannot calculate the intermolecular interactions to a satisfying level of accuracy.52 The popular BLYP and B3LYP functionals actually yield unbound potentials for the methane dimer.28It is thus desirable to investigate other pos-sible combinations of available functionals to serve as alter-natives for ab initio calculations.
The paper has been organized as follows. In Sec. II, we describe the details of these calculations. In Sec. III the re-sults are presented and discussed. A summary and a brief perspective are given in Sec. IV.
II. METHODS AND CALCULATIONS
In the case of methane dimers, a large part of the exchange-repulsion interactions can be calculated by the HF method. The calculation of electron correlation energies de-pends on the level of the correlation-corrected method, the size of the basis set, and the correction of the BSSE. The state-of-the-art choice of the correlation-corrected method is either the Møller-Plesset 共MPx,x=2,3,4兲 perturbation method33or the coupled cluster method with iterative single and double substitutions and with noniterative triple excita-tions 关CCSD共T兲兴 method.34 It has been found that the MP2 results for the methane dimer are not too much different from those calculated by the much more expensive CCSD共T兲 as long as a large basis set has been used.31To study the basis set effects, we have employed comprehensive basis sets from the Slater-type orbitals fitted with Gaussian function
共STO-nG兲 共n=3–6兲,35 medium size basis sets of Krishnan et al. 关up to 6-311+ +G共3df ,3pd兲兴 共Ref. 36兲 to Dunning’s
corre-lation consistent basis sets 共aug-cc-pVXZ兲 共X=D, T, and Q兲.37
The BSSE was corrected by the counterpoise 共CP兲 method of Boys and Bernardi.38The MP2 interaction poten-tials at the basis set limit have been estimated using the methods of Helgaker et al.39 and Feller40 and a numerical extrapolation scheme based on the Lagrangian formula.41
All the HF, MP2, and DFT calculations are performed using the GAUSSIAN 03program package42 on a single node two-processor AMD 250 personal computer 共PC兲 cluster with distributed memory. The equilibrium geometry of a single methane molecule was first optimized at the MP2 / 6 -31G*level of theory. To obtain the most stable intermolecu-lar geometry, the methane dimer has been modeled by first fixing the carbon-carbon 共C–C兲 distance while letting the two monomers to rotate freely. By approaching the mono-mers from the far side with several initial choices of mutual orientation, we found the minimum-energy conformation corresponds to the D3dsymmetry conformer. This optimized conformer has been reached through the interplay of the steric stabilization of repulsive hydrogens in opposite monomers.21Subsequently the C–C distance was sampled in step 0.1 Å for a quite large range of intermolecular
separa-tion 共normally 3–9 Å兲. During the scan we allow the indi-vidual methane molecule to be fully relaxed. This means that we do not fix the monomer geometry and the methane mol-ecule is not assumed to be rigid. Although it is not expected to see much deviation from the rigid molecule approxima-tion, in the real condensed phase environment, stretching, bending and torsional relaxations could be important for many subtle thermodynamic properties. The inclusion of in-termolecular relaxation is especially relevant to the construc-tion of force fields for use in molecular dynamics simulaconstruc-tions where flexible models often work better than rigid models.43 III. RESULTS AND DISCUSSIONS
The intermolecular interaction potentials of the D3d con-former of the methane dimer have been calculated with the HF, the MP2, and the DFT methods. We present the respec-tive results along with discussions and make comparisons among the results.
A. Hartree-Fock self-consistent field calculations The BSSE corrected HF interaction potentials of the methane dimer using several basis sets are shown in Fig.1. All the HF calculations yield purely repulsive potentials without minima for all the basis sets used. This can be attrib-uted to the rather weak electrostatic interaction for the meth-ane dimer. In the short range, the strong exchange-repulsion interaction dominates with little alternation from electrostatic and induction attractions. The HF potential is insensitive to the basis size as long as the 6-31G**basis set has been used. We can model the HF potential using the repulsive Bucking-ham function,44
VHF共R兲 = Ae−␣R,
where R is the C–C distance, A and ␣共the repulsion expo-nent兲 are the fitting parameters. The dependence of the repul-sion exponent on the basis size is shown in TableI. It is seen that the repulsion exponent converges quickly after the 6-31G**basis set being used.
FIG. 1. The BSSE corrected HF interaction potentials of the methane dimer using several basis sets.
B. MP2 calculations
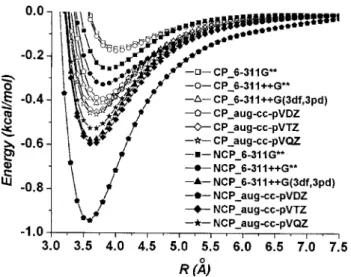
Unlike the HF potentials, the MP2 potentials shown in Fig.2display clear minima and long-range attractive poten-tial tails. Because the contributions from the octopole-octople interactions are small, the dispersion energy is mainly responsible for the attractions. Sharp differences be-tween the HF calculations and the MP2 calculations indicate the importance of including the correlation corrections in the
wave function calculations. The HF method in principle does not include the correlation effect so the attraction forces are due exclusively to the correlation effect.
In Fig.2, we compare the MP2 potentials with and with-out the BSSE corrections 共denoted as CP and NCP, respec-tively兲. We see very strong dependence of the interaction potentials on the BSSE corrections. The potentials without the BSSE corrections fluctuate with increasing basis size and do not systematically converge to the destined curve at the basis set limit. On the contrary, the BSSE corrected poten-tials systematically approach to the destined curve with in-creasing basis size. Therefore, it is important to consider the BSSE correction in calculating the intermolecular interac-tions, in particular, for small basis sets.
As shown in Table I, the basis set effect on the BSSE corrected interaction potentials is significant. The STO-3G basis set yields very small binding energy. The interaction energy becomes more accurate as one adds polarization func-tions and augments diffuse funcfunc-tions in Pople’s basis sets. Small cc-pVDZ and cc-pVTZ basis sets lead to underesti-mated binding energies and it requires cc-pVQZ to saturate the result. Augmentation of the diffuse functions has signifi-cant effect on optimizing the binding energy. The cc-pVTZ basis set underestimates the energy by 30%, while the aug-cc-pVTZ basis set underestimates only 5% of the binding energy. Some subtle basis set features can also be observed. For small basis sets, adding polarization functions to the TABLE I. The basis set dependence of important potential parameters using the BSSE corrected HF and MP2 intermolecular potentials. R0is the distance at
which the potential is zero and Rmis the equilibrium bond length. The CPU time of the MP2 calculation was recorded on a single node two-processor AMD 250 PC cluster with distributed memory.
Basis set MP2 HF MP2 Number of basis function CPU time 共h兲 Aa 共kcal/mol兲 ␣ a 共Å−1兲 R0 共Å兲 Rm 共Å兲 Eb 共kcal/mol兲 共cm−1兲 one termb C6 two termsc C6 C8 STO-3G 9 0.25 219 400 3.62 4.36 4.75 −0.009 42.99 189.03 136.04 1 596.61 3-21G 34 0.28 83 319 3.29 4.05 4.50 −0.047 78.39 641.57 419.58 6 477.70 6-31G 34 0.28 104 302 3.36 3.99 4.45 −0.053 73.33 676.57 438.12 6 957.93 6-311G 50 0.37 113 764 3.39 3.91 4.36 −0.064 83.45 747.23 486.94 8 437.88 3-21G** 58 0.38 83 602 3.30 3.85 4.31 −0.078 101.15 832.07 556.44 10 586.51 6-311G* 60 0.48 115 208 3.40 3.73 4.16 −0.104 108.73 951.51 454.57 8 356.77 cc-pVDZ 68 0.68 120 594 3.41 3.63 4.05 −0.150 130.17 1174.71 684.90 11 986.23 6-31G** 70 0.57 108 646 3.39 3.73 4.19 −0.101 108.73 941.68 544.89 11 028.72 6-311G** 84 1.17 115 208 3.40 3.59 4.02 −0.165 161.64 1207.04 640.12 14 790.89 6-311+ G** 92 1.57 115 057 3.40 3.58 4.01 −0.174 159.31 1245.15 673.37 15 081.38 6-311+ + G** 100 2.42 115 298 3.40 3.57 4.01 −0.176 159.31 1259.19 671.72 15 584.98 aug-cc-pVDZ 118 3.22 123 739 3.42 3.35 3.78 −0.395 369.19 1995.05 764.34 22 587.36 6-311+ + G共2d,2p兲 134 5.62 114 025 3.40 3.38 3.80 −0.315 217.47 1674.00 812.170 21 662.07 6-311+ + G共3d,3p兲 168 12.22 112 605 3.40 3.33 3.75 −0.401 278.15 1964.62 1456.53 9 860.58 cc-pVTZ 172 13.35 113 006 3.40 3.37 3.80 −0.317 215.83 1880.25 1446.17 10 119.20 6-311+ + G共2df ,2pd兲 188 20.35 113 940 3.40 3.36 3.78 −0.331 209.88 1697.49 1437.55 10 518.06 6-311+ + G共3df ,3pd兲 222 22.63 112 373 3.40 3.30 3.73 −0.415 270.57 1979.52 1593.49 7 987.79 aug-cc-pVTZ 276 82.70 112 320 3.40 3.27 3.70 −0.453 260.45 2048.04 1442.32 11 755.28 aug-cc-pVQZ 528 941.83 111 828 3.40 3.26 3.68 −0.464 250.34 2029.20 1053.08 18 021.33
Basis set limit 113 458 3.40 3.25 3.66 −0.470 256.54 ¯ ¯ ¯
aFit to the formula V
HF共R兲=Ae−␣R. bFit to the formula V
disp共R兲=−C6/ R6, C6in unit共kcal/mol Å6兲, using data R⬎5.0 Å. cFit to the formula V
disp共R兲=共C6/ R6兲−共C8/ R8兲, C8in unit共kcal/mol Å8兲, using data R⬎4.0 Å.
FIG. 2. The BSSE corrected共CP兲 and uncorrected 共NCP兲 MP2 potentials of the methane dimer using a series of basis sets.
basis set does not significantly change the potential. On the other hand, augmentation of the diffuse functions has pretty significant effect. For example, the aug-cc-pVDZ energy is very close to the high level 6-311+ + G共3df ,3pd兲 and the cc-pVQZ results. These paraphrase Tsuzuki et al.31 to con-struct a diffuse-function-augmented medium size 6-311G** basis set in their calculations. Together with diffuse func-tions, adding more polarization functions also improves the accuracy of the calculated potential. For example, the 6-311+ + G** underestimates the binding energy by 60%, while the 6-311+ + G共3df ,3pd兲 yields a binding energy by 12% lower than the MP2 energy at the basis set limit. It is understood that dispersion interactions arise from the nonlo-cal electron correlation effect so adding functions of exten-sive range help to optimize the potentials.
With the wide span of the basis sets used, the basis set dependence of important potential parameters can now be fully studied. In TableIwe present the BSSE corrected data for the equilibrium bond length, the binding energy, and the asymptotic behavior. R0is the distance at which the potential is zero and can be obtained from a two point interpolation of the calculated data. The bond length Rm, the binding energy
Eb, and the intermolecular vibration frequencycan be
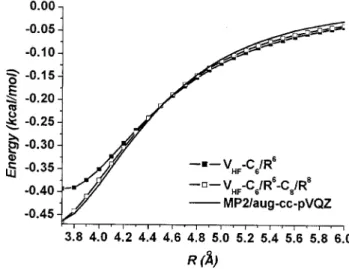
ob-tained through a harmonic modeling of the three lowest po-tential data near the equilibrium regions. C6 and C8 are the dispersion coefficients and can be obtained through a nonlin-ear fitting of the long-range potential data. With increasing basis size, the equilibrium bond length converges at the 6-311+ + G共2d,2p兲 basis set to a 0.1 Å accuracy, while a pretty large basis set 共aug-cc-pVTZ兲 is required to converge the binding energy at a chemical accuracy 共⬃0.01 kcal/mol兲. On the other hand, up to the largest basis set used, the asymptotic behavior has not yet converged to the destined C6value from the calculated monomer polariz-ability共⬃1784 kcal/mol Å6兲.45–47Inclusion of the C8term is important if shorter range data were used for the modeling. As shown in Fig.3, the long-range curve can be reproduced better when including the C8 term. The slow convergence could be an indication of the inefficacy of using the MP2
method with Gaussian functions to calculate long-range in-teractions. Because Gaussian-type functions are local func-tions, a large basis set is required to obtain converged corre-lation energy calcucorre-lations. Therefore, the basis set limit of the calculated potential must be estimated so as to be consis-tent with the conventional perturbation theory. Together with the nonlinear scaling of the computational cost with respect to the basis size, this is actually the main practical reason for the difficulty of obtaining dispersion interactions through ab
initio molecular orbital methods.
The strong basis set dependence and the slow conver-gence on the dispersion coefficients call for an estimation of the important potential features at the basis set limit in a calculated potential. Basis set limit of the binding energy can be approached using Dunning’s basis sets with an extrapola-tion scheme. We consider two analytical schemes39,40 and a numerical scheme41while the results are similar. The binding energies obtained at the basis set limits 共using Dunning’s basis sets, aug-cc-pVXZ, X = D, T, and Q兲 are 0.472, 0.467, and 0.470 kcal/ mol using the methods of Helgaker et al.,39 Feller,40and the numerical method,41respectively. These val-ues are very close to the results obtained by Tsuzuki et al.32 For the other potential parameters, we used the numerical extrapolation based on the vanishing inverse of the number of basis function.41 These results are shown in Table I for comparison.
C. Density functional theory
The density functionals used in the present work include two sets. The first set fixes the exchange functional as the PW91 and changes the correlation functionals. The correla-tion funccorrela-tionals include PL,48 VWN5,49 VWN,49 TPSS,50 PBE,51 PW91,52VP86,53 P86,53 V5LYP,54 LYP,54 HCTH93, and HCTH407.55–57 The second set fixes the correlation functional as the PW91 and changes the exchange function-als. The exchange functionals include HCTH93, HCTH147, HCTH407,55–57MPW,58MPW1,58OPTX or O,59B88 or B,60 PBE,51 Slater or S,61 and X␣ or XA.61The choice of these combinations is motivated by a recent suggestion that the PW91PW91 functional yield good binding energy of the methane dimer interaction.28However, the role of the PW91 as an exchange versus a correlation functional has not been systematically studied. Moreover, it is not clear if the func-tional is reliable for use in determining other potential parameters.
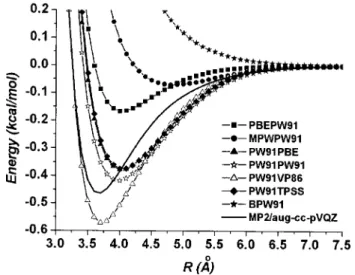
Figure4 presents the calculated intermolecular potential curves using the PW91 as the exchange or the correlation functional. We see that the DFT calculations generally gen-erate a diverse range of potential curves from purely un-bound 共BPW91兲 to strongly bound 共XAPW91兲. In TableII
we compare the bond lengths and the binding energies cal-culated using the several exchange-correlation functionals with the MP2 results. We see that the PW91PW91 combina-tion produces reasonable potential well depth while the bond length is estimated too long. The PW91VP86 functional yields good bond length and captures the attraction effect quite well, although it overestimates the binding energy by FIG. 3. Comparison of the BSSE corrected MP2 potential curve calculated
at the aug-cc-pVQZ basis set and the sum of the HF potential and the long range dispersion potentials.
20%. It is promising that available functionals do capture partly the correlation effects which are essential in calculat-ing the dispersion forces.
The results shown in Table II clearly demonstrate the relative roles played by the exchange and correlation func-tionals in the DFT calculations. By fixing the PW91 as the exchange functional, most correlation functionals yield bound potentials and the binding energies are around the MP2 result共except the HCTH correlation functionals, which
yield seriously overbound potentials兲. On the other hand, by fixing the PW91 as the correlation functional, the varying exchange functionals either overestimate共S and XA兲 or un-derestimate共HCTH, MPW, O, B, and PBE兲 the binding en-ergy. Previous studies on van der Waals systems78,79 have shown that the exchange functional plays an essential role in determining the dispersion energy while the correlation part of a density functional does not significantly affect the DFT calculations. Our results are basically consistent with these observations, although we see appreciable effects due to the choice of the correlation functional. The performance of varying exchange functionals for a fixed correlation func-tional can be understood in terms of the behavior of the GGA enhancement factor F共s兲 of the exchange functional at the large gradient-to-density ratio, denoted as s, region.80 Fol-lowing Lacks and Gordon,81 we plot the F共s兲 vs s curve in Fig. 5. We see in Fig. 5 that the order of the magnitude of
F共s兲 at large s is B88⬎HCTH⬎OPTX⬎PBE⬎MPW
⬎PW91. This order is essentially the order of the binding energies calculated by the corresponding functionals in Table
II. This correlation has been found in previous studies on van der Waals systems78,79 and serves as a useful tool in analyz-ing the DFT calculations. Another criterion for F共s兲 to be satisfied is the Lieb-Oxford condition82 which requires that
F共s兲艋2.273. From Fig. 5 we see that the PBE, MPW, and PW91 exchange functionals obey this condition. The above analyses, together with Perdew’s suggestion that the PW91 exchange functional should be used with the corresponding PW91 correlation functional,83explain why the PW91PW91 functional outperforms other combinations of the exchange and correlation functionals in the calculations of dispersion interactions.
IV. CONCLUSION
In this paper we have systematically studied the calcu-lated intermolecular potentials of the methane dimer at the most stable D3dconformation using the HF, MP2, and DFT methods. A wide selection of basis sets has been employed in order to determine the basis set effects on the repulsion ex-ponent, the binding energy, the equilibrium bond length, and the asymptotic behavior of the intermolecular potentials. BSSE corrections are considered as an important factor af-fecting the quality of the calculated potentials.
From this study we can draw several important conclu-sions about using the current theoretical methods to generate FIG. 4. The BSSE corrected DFT intermolecular potential curves using the
PW91 as the exchange or correlation functional.
TABLE II. Comparison of the bond lengths and the binding energies calcu-lated using the several exchange-correlation functionals with the MP2 results using the 6-311+ + G共3df ,3pd兲 basis set.
Functional 6-311+ + G共3df ,3pd兲 Bond length 共Å兲 Binding energy 共kcal/mol兲 PW91PL 4.19 −0.360 PW91VWN5 4.19 −0.358 PW91VWN 4.14 −0.387 PW91TPSS 4.05 −0.376 PW91PBE 4.04 −0.379 PW91PW91 3.99 −0.418 PW91VP86 3.73 −0.572 PW91P86 3.72 −0.577 PW91V5LYP 3.80 −0.669 PW91LYP 3.80 −0.669 PW91HCTH93 3.56 −1.444 PW91HCTH407 3.52 −3.367 HCTH93PW91 5.93 −0.003 HCTH147PW91 5.82 −0.006 HCTH407PW91 5.93 −0.004 MPWPW91 4.83 −0.071 MPW1PW91 4.80 −0.050 OPW91 5.23 −0.021 BPW91 Unbound ¯ PBEPW91 4.04 −0.167 SPW91 3.08 −2.483 XAPW91 2.97 −2.671 MP2 3.73 −0.415
FIG. 5. The GGA enhancement factor as a function of s for the HCTH, B88, OPTX, mPW, PBE, and PW91 exchange functionals.
the intermolecular potentials. Although only the methane dimer has been studied, these conclusions should also be useful to other molecular dimers.
共1兲 The HF calculations yield purely repulsive potentials for nonpolar molecular dimers. The basis size effect of the HF calculations is very small as long as the 6 -31G**basis set has been used.
共2兲 The van der Waals bond of the methane dimer is well produced using the MP2 method. BSSE corrections must be considered to yield systematic results. Basis set effects are significant for many important parameters such as bond lengths, binding energies, and dispersion coefficients. Small basis sets, especially without the augmentation of diffuse functions, could produce se-vere underestimation of the binding energy and ose-veres- overes-timation of the bond length. Addition of diffuse func-tions and polarization funcfunc-tions leads to reliable binding curves.
共3兲 The DFT potentials display a wide range of patterns of binding curves, underestimating or overestimating the binding energy. The binding energy calculated using the PW91PW91 functional and the equilibrium bond length calculated using the PW91VP86 functional are close to the MP2 results.
From the present study we see very clearly that the HF method captures the exchange-repulsion interaction and satu-rates the potential curves at small basis set. Inclusion of the correlation corrections using ab initio molecular orbital methods makes the calculations computationally demanding 共CPU time ⬃N4, where N is the number of basis function兲. The DFT calculations are comparatively cheaper but the re-sults are not systematic. Some functionals do capture partly the correlation effects. These observations justify the recent efforts in improving the calculations of the intermolecular interactions using the HF or the DFT method by including explicit empirical or nonempirical dispersion energy corrections.62–78More works are definitely required to make the two ends共accuracy versus efficiency兲 meet.
ACKNOWLEDGMENTS
This work was supported by the National Science Coun-cil of Taiwan, ROC共2113-M-002-016 and NSC-94-2120-M-002-014兲. We acknowledge the National Center for High-Performance Computing 共NCHC兲 for providing com-puting resources. We would like to thank J. C. Jiang and M. Hayashi for useful discussions.
1O. S. Tyagi, H. S. Bisht, and A. K. Chatterjee, J. Phys. Chem. B 108,
3010共2004兲.
2P. Hobza and R. Zahradnik, Intermolecular Complexes: The Role of van
der Waals Systems in Physical Chemistry and in the Biodisciplines 共Elsevier, New York, 1988兲.
3G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures
共Springer-Verlag, Berlin, 1991兲.
4A. J. Stone, The Theory of Intermolecular Forces 共Oxford University
Press, Oxford, 1996兲.
5H. Margenau, Rev. Mod. Phys. 11, 1共1939兲.
6A. van der Avoird, P. E. S. Wormer, and R. Moszynski, Chem. Rev.
共Washington, D.C.兲 94, 1931 共1994兲.
7A. K. Rappe and E. R. Bernstein, J. Phys. Chem. A 104, 6117共2000兲.
8G. Chalasinski and M. M. Szczensniak, Chem. Rev.共Washington, D.C.兲 100, 4227共2000兲.
9R. J. Wheatley, A. S. Tulegenov, and E. Bichoutskaia, Int. Rev. Phys.
Chem. 23, 151共2004兲.
10Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput. 1, 415共2005兲. 11S. Grimme, J. Comput. Chem. 25, 1463共2004兲.
12R. A. Friesner, Proc. Natl. Acad. Sci. U.S.A. 102, 6648共2005兲. 13D. Frenkel and B. Smit, Understanding Molecular Simulations
共Aca-demic, New York, 2002兲.
14M. Marques and E. Gross, Annu. Rev. Phys. Chem. 55, 427共2004兲. 15C. E. Dykstra, G. Frenking, K. S. Kim, and G. E. Scuseria, Theory and
Applications of Computational Chemistry: The First Forty Years 共Elsevier, Amsterdam, 2005兲.
16A. Ruzsinszky, J. P. Perdew, and G. I. Csonka, J. Phys. Chem. A 109,
11015共2005兲.
17M. D. Malinsky, K. L. Kelly, G. C. Schatz, and R. P. van Duyne, J. Am.
Chem. Soc. 123, 1471共2001兲.
18G. Y. Liu, S. Xie, and Y. L. Qian, Acc. Chem. Res. 23, 457共2000兲. 19W. Rawicz, K. C. Olbrich, T. McIntosh, D. Needham, and E. Evans,
Biophys. J. 79, 328共2000兲.
20S. D. Chao, J. D. Kress, and A. Redondo, J. Chem. Phys. 122, 234912
共2005兲.
21M. M. Szczesniak, G. Chalasinski, S. M. Cybulski, and S. Scheiner, J.
Chem. Phys. 93, 4243共1990兲.
22R. L. Rowley and T. Pakkanen, J. Chem. Phys. 110, 3368共1999兲. 23D. E. Williams and D. J. Craycroft, J. Phys. Chem. 91, 6365共1987兲. 24S. Tsuzuki and K. Tanabe, J. Phys. Chem. 95, 2272共1991兲.
25J. J. Novoa, M.-H. Whangho, and J. M. Williams, J. Chem. Phys. 94,
4835共1991兲.
26D. H. Gay, H. Dai, and D. R. Beck, J. Chem. Phys. 95, 9106共1991兲. 27S. Tsuzuki, T. Uchimaru, K. Tanabe, and S. Kuwajima, J. Phys. Chem.
98, 1830共1994兲.
28S. Tsuzuki and H. P. Luthi, J. Chem. Phys. 114, 3949共2001兲. 29J. Nagy, D. F. Weaver, and V. H. Smith, Mol. Phys. 85, 1179共1995兲. 30E. Fraschini and A. J. Stone, J. Comput. Chem. 19, 847共1998兲. 31S. Tsuzuki, T. Uchimaru, and K. Tanabe, Chem. Phys. Lett. 287, 202
共1998兲.
32S. Tsuzuki, K. Honda, T. Uchimaru, and M. Mikami, J. Chem. Phys. 124,
114304共2006兲.
33C. Møller and M. S. Plesset, Phys. Rev. 46, 618共1934兲.
34J. A. Pople, M. Head-Gordon, and K. Raghavachari, J. Chem. Phys. 87,
5968共1987兲.
35A. Szabo and N. S. Ostlund, Modern Quantum Chemistry: Introduction
to Advanced Electronic Structure Theory共Dover, New York, 1996兲.
36R. Krishnan, J. S. Binkley, R. Seeger, and J. A. Pople, J. Chem. Phys. 72,
650共1980兲.
37T. H. Dunning Jr., J. Chem. Phys. 90, 1007共1989兲. 38S. F. Boys and F. Bernardi, Mol. Phys. 19, 553共1970兲.
39T. Helgaker, W. Klopper, H. Koch, and J. Noga, J. Chem. Phys. 106,
9639共1997兲.
40D. Feller, J. Chem. Phys. 96, 6104共1992兲.
41W. H. Press, S. A. Teukolsky, W. T. Vetterling, and B. P. Flannery,
Nu-merical Recipe in C共Cambridge University Press, Cambridge, 1996兲.
42M. J. Frisch, G. W. Trucks, H. B. Schlegel et al.,
GAUSSIAN 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004.
43D. C. Rapaport, The Art of Molecular Dynamics Simulation共Cambridge
University Press, New York, 1995兲.
44F. Jensen, Introduction to Computational Chemistry共Wiley, New York,
1999兲.
45M. A. Spackman, J. Chem. Phys. 94, 1295共1991兲. 46Q. Wu and W. Yang, J. Chem. Phys. 116, 515共2002兲.
47A. D. Becke and E. R. Johnson, J. Chem. Phys. 122, 154104共2005兲. 48J. P. Perdew and A. Zunger, Phys. Rev. B 23, 5048共1981兲. 49S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys. 58, 1200共1980兲. 50J. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria, Phys. Rev.
Lett. 91, 146401共2003兲.
51J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865
共1996兲.
52K. Burke, J. P. Perdew, and Y. Wang, in Electronic Density Functional
Theory: Recent Progress and New Directions, edited by J. F. Dobson, G. Vignale, and M. P. Das共Plenum, New York, 1998兲.
53J. P. Perdew, Phys. Rev. B 33, 8822共1986兲. 54
C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B 37, 785共1988兲.
Phys. 109, 6264共1998兲.
56A. D. Boese, N. L. Doltsinis, N. C. Handy, and M. Sprik, J. Chem. Phys. 112, 1670共2000兲.
57A. D. Boese and N. C. Handy, J. Chem. Phys. 114, 5497; see EPAPS
Document No. E-JCPSA6-114-301111 for fit set of HCTH/407 func-tional. This document can be reached via a direct link in the online article’s HTML reference section or via the EPAPS homepage 共http:// www.aip.org/pubservs/epaps.html兲.
58C. Adamo and V. Barone, J. Chem. Phys. 108, 664共1998兲. 59N. C. Handy and A. J. Cohen, Mol. Phys. 99, 403共2001兲. 60A. D. Becke, Phys. Rev. A 38, 3098共1988兲.
61J. C. Slater, Quantum Theory of Molecular and Solids: The
Self-Consistent Field for Molecular and Solids 共McGraw-Hill, New York, 1974兲, Vol. 4.
62X. Wu, M. C. Vargas, S. Nayak, V. Lotrich, and G. Scoles, J. Chem. Phys. 115, 8748共2001兲.
63W. Kohn, Y. Meir, and D. E. Makarov, Phys. Rev. Lett. 80, 4153共1998兲. 64U. Zimmerli, M. Parrinello, and P. Koumoutsakos, J. Chem. Phys. 120,
2693共2004兲.
65A. D. Becke and E. R. Johnson, J. Chem. Phys. 123, 154101共2005兲. 66E. R. Johnson and A. D. Becke, J. Chem. Phys. 124, 174104共2006兲. 67X. Xu and W. A. Goddard III, Proc. Natl. Acad. Sci. U.S.A. 101, 2673
共2004兲.
68E. J. Meijer and M. Sprik, J. Chem. Phys. 105, 8684共1996兲.
69Q. Wu and W. Yang, J. Chem. Phys. 116, 515共2002兲.
70M. Lein, J. F. Dobson, and E. K. U. Gross, J. Comput. Chem. 20, 12
共1999兲.
71A. J. Misquitta, B. Jeziorski, and K. Szalewicz, Phys. Rev. Lett. 91,
033201共2003兲.
72J. F. Dobson and J. Wang, Phys. Rev. Lett. 82, 2123共1999兲.
73M. Dion, H. Rydberg, E. Schröder, D. C. Langreth, and B. I. Lundqvist,
Phys. Rev. Lett. 92, 246401共2004兲.
74V. F. Lotrich, R. J. Bartlett, and I. Grabowski, Chem. Phys. Lett. 405, 43
共2005兲.
75M. Kamiya, T. Tsuneda, and K. Hirao, J. Chem. Phys. 117, 6010共2002兲. 76T. Sato, T. Tsuneda, and K. Hirao, Mol. Phys. 103, 1151共2005兲. 77J. G. Ángyán, I. C. Gerber, A. Savin, and J. Toulouse, Phys. Rev. A 72,
012510共2005兲.
78T. A. Wesolowski, O. Parisel, Y. Ellinger, and J. Weber, J. Phys. Chem. A 101, 7818共1997兲.
79Y. Zhang, W. Pan, and W. Yang, J. Chem. Phys. 107, 7921共1997兲. 80G. E. Scuseria and V. N. Staroverov, in Theory and Applications of
Com-putational Chemistry: The First Forty Years, edited by C. E. Dykstra, G. Frenking, K. S. Kim, and G. E. Scuseria共Ref.15兲, Chap. 24.
81D. J. Lacks and R. G. Gordon, Phys. Rev. A 47, 4681共1993兲. 82E. H. Lieb and S. Oxford, Int. J. Quantum Chem. 19, 427共1981兲. 83J. Tao and J. P. Perdew, J. Chem. Phys. 122, 114102共2005兲.