以固相微萃取建立水中三鹵甲烷及鹵乙酸之同步分析方法; Analysis of trihalomethanes and haloacetic acids in water by solid-phase microextraction
82
0
0
全文
(2) 致謝 能有這篇論文的誕生,至今我仍有點無法相信,從當初是如此的笨 拙,過程中經歷不斷的錯誤與學習,終有些成長。當中,想感謝的人太 多,而想說的話一時之間卻無法用筆墨描述。 感謝我的指導教授蔡詩偉老師,無論是學業或其他方面遇到的問 題,總是一語點醒夢中人,笑笑的說「It’s okay」,能讓我無懼的作自己 想做的事情,讓我覺得自己真是幸運的學生。感謝職安所王文析、趙克 平及各位老師的教導;感謝賴俊雄、林嘉明及陳美蓮老師在論文上的指 導與建議;感謝小胖、高國源及阿仔學長,總是不厭其煩的給予我解答 及鼓勵,沒有你們不會有我今天的成長;感謝同窗兩年的同學以及同研 究室的伙伴玫雯,祝大夥們鴻圖大展;感謝大學姊妹們芳如、舒婷、瑜 伶、小瞇、阿蓉等,當我需要幫助,妳們總是不吝惜伸出手;感謝可愛 的學弟妹,總是提供笑點,調劑苦悶的實驗時間;當然,還要感謝我的 摯友阿跩、小春及仲儀,謝謝你們的陪伴,還要當我的情緒垃圾桶,共 同分享苦樂。 最後,感謝我最親愛的家人們,長久以來對我的支持和無條件的付 出,我才得以順利畢業,以及有機會在此寫出我滿滿的感謝,我愛你們! 感謝上天讓我遇見如此多貴人,再次說聲謝謝大家! 謹以此論文獻給關心我的親友、同學,願將畢業的喜悅分享給大家。. I.
(3) 摘 要 飲用水加氯消毒可能產生消毒副產物(disinfection by-products, DBPs ),最常見的為三鹵甲烷(THMs)及鹵乙酸(HAAs)。為了改善現有 DBPs分析方法繁複萃取的缺失,本研究應用固相微萃取技術,探討水中 三鹵甲烷與鹵乙酸同步分析的可行性。 首先配製已知濃度之三鹵甲烷及鹵乙酸樣本,但頂空吸附之前,需 先以鹽酸及甲醇將鹵乙酸衍生為甲酯類,在100℃水浴中衍生20min,冷 卻至40℃ 之後,再利用固相微萃取技術進行頂空吸附,並以GC/MS 分 析。影響固相微萃取吸附之因素,包括: 纖維種類、萃取溫度、電磁攪拌 速度及含鹽量。 65μm PDMS/DVB纖維最符合研究所需;當纖維在注射口溫度180℃ 停留2min,其脫附效率>99.7%。以頂空吸附30min, 轉速為1000 rpm 的條 件下,以纖維頂空吸附再配合GC/MS 分析可以直接偵測三鹵甲烷及鹵乙 酸甲酯化物。 本研究發現65μm PDMS/DVB 固相微萃纖維可分別應用於水中三 鹵甲烷與鹵乙酸之分析,因此應有可能依此建立其共同存在時的同步偵 測技術。. 關鍵字:消毒副產物、三鹵甲烷、鹵乙酸、固相微萃取. II.
(4) Abstract Application of disinfection processes during water treatment could lead to the formation of disinfection by-products (DBPs)。The major categories of DBPs ,were trihalomethanes (THMs) and haloacetic acids (HAAs). A new analytical technique called solid-phase microextraction (SPME) has been developed which is favorable in terms of solvent-free and convenience. Therefore the purpose of this study was to develop a new simultaneous analytical method for trihalomethanes (THMs) and haloacetic acids (HAAs) in drinking water based on SPME. Known concentrations of trihalomethanes and haloacetic acids were prepared in spiked water samples. For the analysis of haloacetic acids, the derivatization of the acids to their ethyl esters using hydrochloric acid and methanol in 100℃ water bath for 20 minutes, followed by headspace SPME with a PDMS/DVB fiber. Headspace adsorptions were performed followed by GC/MS analysis. Affects factor of SPME included the types of fiber, temperatures, speeds of magnetic stirring, and salts were all the factors . The analysis of trihalomethanes (THMs) and haloacetic acids (HAAs) in water by 65μmPolydimethylsiloxane /Divinylbenzene (PDMS/DVB) SPME fiber were found to be possible. For future work, efforts on the tests of simultaneous analysis of trihalomethanes (THMs) and haloacetic acids (HAAs) are needed.. Key words:Disinfection by-products, trihalomethanes, haloacetic acids, SPME III.
(5) 目 錄 致謝……………………………………….………Ⅰ 摘要………………………………………….....…Ⅱ Abstract…………………………………...………III 目錄…………………………………….………... IV 表目錄…………………..………………………VIII 圖目錄……………………………………….……IX 附錄………………………………………………..X. 第一章、緒論……………………….……..……..……..…1 第一節. 研究背景……………………………………….....1. 第二節. 研究之重要性………………………………….....3. 第三節. 研究之目的………………………………….…....4. 第四節. 名詞界定…………………………………….……5. 第二章、文獻探討……………………………...…..…..…6 第一節. 三鹵甲烷及鹵乙酸的介紹……………….…..…..6. 2-1.1 基本特性………………………………………………….…..6 2-1.2 來源及分佈…..……………………………………..………..7 2-1.3 健康危害………………………………………………….…..8 IV.
(6) 第二節. 水中三鹵甲烷之檢測分析方法……………….....10. 2-2.1 行政院環保署-環境檢驗所..……………………….…….…10 2-2.2 美國環保署………………………………………….……….11 2-2.3 現行常用三鹵甲烷之分析方法…………………………….11. 第三節. 水中鹵乙酸之檢測分析方法………………….....14. 2-2.1 前處理的方法..…………………………….……………….14 2-2.2 衍生方法的介紹…………………………………………….16 2-2.3 分析方法的介紹……………....…………………………….18. 第四節. 固相微萃取技術………………………….…..…21. 2-4.1 固相微萃取之裝置………………………………………….21 2-4.2 固相微萃取之理論………………………………………….23 2-3.3 固相微萃取之應用………………………………………….28. 第五節. 研究架構……………………………………….....29. 第三章、材料與方法………………..………..………….30 第一節. 實驗器材………………….…………..…………30. 3-1.1 藥品試劑……………………………………….……………30 3-1.2 儀器設備………………………………………….…………30. V.
(7) 第二節. 實驗步驟……...………………………..…………32. 3-2.1 實驗衍生步驟….……………………………….……………32 3-2.2 固相微萃取方法建立.……………………………………….32. 第三節. 實驗分析方法……………..……………..……….35. 3-3.1 儀器分析條件……………………………………………….35 3-3.2 檢量線的配製……………………………………………….36 3-3.3 品保品管…………………………………………………….36. 第四章、結果與討論………………………....……….…38 第一節. 固相微萃取方法建立……………...……..………38. 4-1.1 固相微萃取纖維的選擇……….…………………………….38 4-1.2 萃取時間探討……………………………………………….38 4-1.3 萃取溫度探討……………………………………………….38 4-1.4 攪拌速度對萃取的影響…………………………………….39 4-1.5 添加鹽類的影響…………………………………………….40 4-1.6 熱脫附……………………………………………………….41 4-1.7 樣本體積…………………………………………………….41. 第二節. 採樣分析方法……………..……….…………….42 VI.
(8) 4-2.1 品保品管執行結果..…………………………………...…….42 4-2.2 與其他方法之比較………………………………………….43. 第五章、結論與建議…………………..………..……….44 參考文獻……………………………………………………..…...46 表列…………………………………………………………..…...51 圖列…………………………………………………………..…...63 附錄列………………………………………………………..…...71. VII.
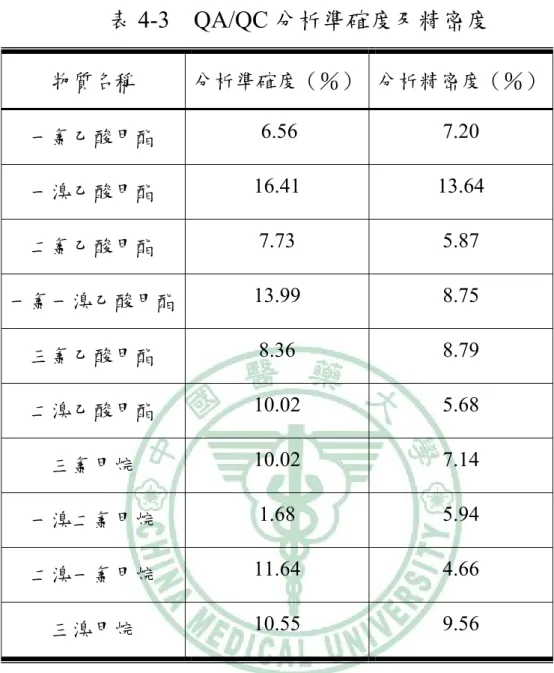
(9) 表目錄 表 2.1 三鹵甲烷的物理性質……………………………...….…..52 表 2.2 鹵乙酸及其衍生甲酯化物之物理性質…………………..53 表 2-3 三鹵甲烷的毒化性質…………….. ……………………...54 表 2-4 鹵乙酸的毒化性質…………………………………...…...55 表 2-5 市售固相微萃取纖維……………………………......……56 表 2-6 測試纖維的耐受溫度……………………………………..57 表 3-1 三鹵甲烷及鹵乙酸甲酯化物 SIM mode 設定……………58 表 4-1 熱脫附效率結果…………………………..………….…...59 表 4-2 QA/QC 檢量線、相關係數及偵測極限…………………60 表 4-3 QA/Q 分析準確度及精密度……………………………….61 表 4-4 本研究方法與已公告分析鹵乙酸方法之比較…………..62. VIII.
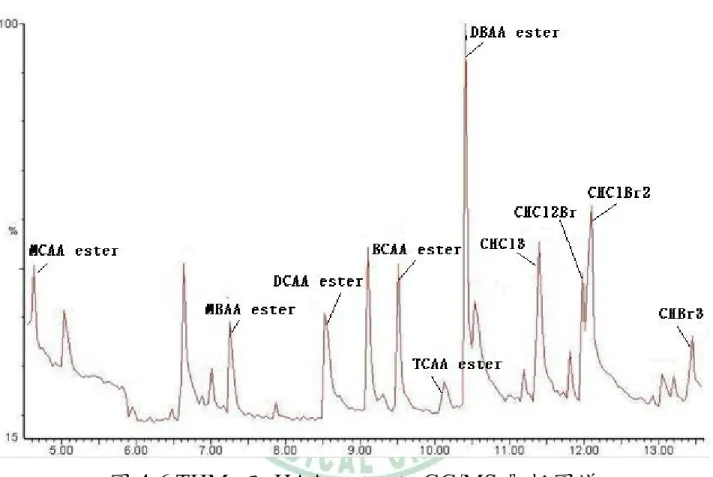
(10) 圖目錄 圖 2-1 固相微萃取手動裝置………...... ………………………..63 圖 2-2 固相微萃取操作圖解……………..…..………………….64 圖 4-1 固相微萃取纖維吸附 THMs 與 HAAs ester 吸附量之比較 …………………………………………………………...65 圖 4-2 THMs 及 HAAs ester 萃取時間與吸附量之關係….......66 圖 4-3 THMs 及 HAAs ester 萃取溫度與吸附量之關係….......67 圖 4-4 THMs 及 HAAs ester 轉速與吸附量關係圖…...............68 圖 4-5 THMs 及 HAAs ester 添加鹽類與吸附量比較結果.......69 圖 4-6 THMs 及 HAAs ester 之 GC/MS 層析圖譜.....................70. IX.
(11) 附 錄 附錄一 固相微萃取之萃取纖維前處理(Condition)……………………71. X.
(12) 第一章 緒論 第一節 研究背景 水是維持生命的重要要素,人類生活對於自然水源依賴甚深。隨 著人們對飲用水的重視,於是開始對飲用水進行過濾及消毒處理,並 提倡國人飲用煮沸過的水。. 目前國內飲用水所使用的消毒法大部份採用氯氣或次氯酸鹽,而 有些歐美國家則用臭氧進行消毒。消毒副產物最早在1970 年初期, 由美國環保署(U.S. Environmental Protection Agency;US EPA)於New Orlean淨水廠之處理後清水中(Chlorinated Water)發現含有原本並不 存在於原水中的多項有機化合物[1];後來經Rook[2] 等人發現加氯消毒 後之自來水含有如氯仿等有毒物質而引起各方面的關注,並認為此類 化合物是在消毒過程中所產生,因此將該類化合物定義為消毒副產 物,而近二十年相繼證實包括:氯仿、溴仿、一氯二溴甲烷、及二氯 一溴甲烷等通稱為三鹵甲烷( Trihalomethanes,THMs)的消毒副產物 存在。八十年代隨著儀器精進及分析方法的開發,亦陸續證實有包括 一氯乙酸、二氯乙酸、三氯乙酸、一溴乙酸、二溴乙酸及一溴一氯乙 酸等鹵乙酸(Haloacetic Acids,HAAs)氯化消毒副產物的存在;而這些 物質易經由吸入、食入或呼吸暴露等途徑進入人體,且經證實會對人. 1.
(13) 體造成一定程度的致癌性及致突變性[3, 4]。 我國環保署於日前修正發布飲用水水質標準,增列管制項目溴酸 鹽,而其管制標準為0.01毫克/公升;此外,亦將可能致癌的揮發性消 毒副產物總三鹵甲烷管制標準,由現行0.10毫克/公升加嚴至0.08毫克 /公升[5]。另一方面,雖然環保署已將鹵乙酸列為未來飲用水管理條例 之列管項目,不過目前國內仍無標準值[6]。國外方面,美國環保署 (EPA)[7] 對DBPs (Disinfection By-Products)之規定在stage 1中將總三 鹵甲烷的最大許可容量(MCL)定為80μg/L,五種含鹵有機乙酸 (MCAA、DCAA、TCAA、MBAA、DBAA) 的最大許可容量(MCL) 定為60μg/L;stage 2的法令則將總三鹵甲烷的最大許可容量(MCL)定 為80μg/L,五種含鹵有機乙酸(MCAA、DCAA、TCAA、MBAA、DBAA) 的最大許可容量(MCL)定為60μg/L[7]。. 2.
(14) 第二節. 研究之重要性. 目前國內水中總三鹵甲烷檢測方法為吹氣捕捉氣相層析法(Purge and Trap Method)[8]、水中鹵乙酸檢測方法為固相萃取濃縮/氣相層析 儀/電子捕捉偵測器法[9]。傳統上偵測水中消毒副產物-三鹵甲烷及鹵 乙酸的方法為各別檢測,雖然其兩種檢測方法在分離及監測上有良好 的效果,但由於整個樣品從前處理、採樣、進行前處理、及實驗分析 的程序較為複雜,且分析時間較長,且需使用到大量有機溶劑,因此 並不方便。. 相較於上述複雜的樣品前處理過程,固相微萃取技術 (Solid-phase Microextration;SPME)的操作則顯得簡單、容易,而 且不需要使用大量的溶劑,也因此近年來被廣泛應用於環境污染物的 分析[7, 8]。因此,本研究為簡化三鹵甲烷及鹵乙酸的分析流程,嘗試 以固相微萃取技術建立飲水消毒副產物三鹵甲烷及鹵乙酸之同步偵 測分析方法。. 3.
(15) 第三節. 研究之目的. 本研究希望結合固相微萃取技術及衍生反應之優點,針對三鹵甲 烷及鹵乙酸發展新的水中消毒副產物同步偵測分析方法,以改進現有 方法需要繁複萃取之缺失。. 4.
(16) 第四節 名詞界定 1. 消毒副產物: Disinfection By-Products;本文所探討的消毒副產物為 加氯消毒而產生的副產物。 2. 三鹵甲烷::Trihalomethanes(THMs);為氯仿、溴仿、一氯二溴 甲烷、及二氯一溴甲烷所組成。 3. 鹵乙酸:Haloacetic Acids(HAAs);為一氯乙酸、二氯乙酸、三 氯乙酸、一溴乙酸、二溴乙酸、三溴乙酸、一溴一氯乙酸、二溴 一氯乙酸及一溴二氯乙酸所組成。 4. 固相微萃取:Solid Phase Microextraction(SPME);固相微萃取 是利用一根上面塗有特殊聚合物材質的纖維來吸附樣品中的待測 物。. 5.
(17) 第二章 文獻探討 第一節 三鹵甲烷及鹵乙酸的介紹 2-1.1 基本特性 三鹵甲烷(Trihalomethanes,THMs)其化學式為 CHX3,X 即為 鹵素(Cl、Br、I) 。在水中最常見為三氯甲烷(CHCl3,氯仿)、一氯 二溴甲烷(CHClBr2)、二氯一溴甲烷(CHCl2Br)、及三溴甲烷( 溴仿,CHBr3)四種物質。總三鹵甲烷(Total Trihalomethanes,TTHMs )定義為以上四種三鹵甲烷之濃度總和(表 2.1 為三鹵甲烷之物理性 質)。. 鹵乙酸(Haloacetic acids,HAAs)根據鍵結的鹵素種類及數目, 可分為: 單鹵乙酸:一氯乙酸(MCAA)、一溴乙酸(MBAA) 二鹵乙酸:二氯乙酸(DCAA)、二溴乙酸(DBAA)、 一氯一溴乙酸(BCAA) 三鹵乙酸:三氯乙酸(TCAA)、三溴乙酸(TBAA)、 二氯一溴乙酸(DCBAA)、一氯二溴乙酸(DBCAA ) 鹵乙酸為具有極性及親水性,且沸點皆在 190℃以上之非揮發性. 6.
(18) 物質。由於鹵乙酸之 pKa 值偏低(0.66-2.86),因此在自然水體( pH>6)中可達完全解離(>99.9%),且以離子的形式存在於水中. [10]. (表 2.2 為鹵乙酸之物裡性質)。. 2-1.2 來源及分佈 2-1.2.1 來源 為了去除給水中造成健康危害的微生物而加入消毒劑如氯時, 水中天然有機物(natural organic material, NOM)可能與其反應所產 生之鹵化物可多達九大類,包括:三鹵甲烷(trihalomethanes,THMs) 、 鹵乙酸(Haloacetic acids,HAAs)、鹵乙氰(HANs)、鹵化酮(HKs)、 氯酚類(Chlorophenols)、氯氰類(Cyanogen Chloride)、有機氯化 胺(N-Organochloroamines)、氯化醛(Chloral Hydrate)、及三氯硝 [11]. 基甲烷(Chloropicin)等副產物 ,而研究指出DBPs 中最主要的物 質為THMs、其次為HAAs,而THMs 中又以氯仿佔絕大多數. [12]. 。. 三鹵甲烷的生成,與其前驅物質「腐植酸」(humic acid)的多 少有關;亦即原水中如含有腐植酸等有機污染物,當加氯消毒時,即 可能生成三鹵甲烷[13]。. 7.
(19) 2-1.2.2 環境流佈 三鹵甲烷在空氣中的來源主要是工業排放或洩漏三氯乙烯在大 氣中經光化學反應(photochemical reaction)也可能產生三鹵甲烷; 至於水中三氯甲烷及鹵乙酸的主要來源則為飲用水的加氯消毒. [13]. 。. 2-1.3 健康危害 [2]. Rook. 等人於 1974 年首先證實自來水加氯消毒會產生多種. 致癌性消毒副產物。許多流行病學研究發現加氯消毒副產物在致癌性 方面可能導致膀胱癌和結腸癌,另外並可能導致懷孕婦女的早產及胎 兒體重過輕. [14]. 。. 三鹵甲烷暴露研究顯示每天以120或250 mg/Kg 的高劑量注入 老鼠體持續14天,三鹵甲烷(包括氯仿、一溴二氯甲烷、二溴一氯甲 烷、及溴仿)會引起老鼠肝腫大及其他肝方面的病變;而在50~125 mg/kg 較低劑量同樣為期14 天的暴露則會引起免疫系統方面的損 害。另外,在200 mg/kg 的氯仿及溴仿暴露下也可能會造成 DNA 組 合上的問題. [3, 15]. 。. 8.
(20) 三氯甲烷(俗名氯仿)為三鹵甲烷消毒副產物中所最常見,其 可藉由吸入或攝入而引起肝及腎的傷害,而經由動物實驗也證實三氯 甲烷具致癌性與並可能導致基因突變。三氯甲烷若經由呼吸進入體 內,會對人體的中樞神經產生麻醉作用;如果劑量高或長期低劑量的 吸入則會對肝臟與腎臟功能產生影響. [3, 4]. 。. 研究顯示加氯消毒副產物中為非揮發性的物質大部分為含鹵 [16]. 乙酸 ;而由鹵乙酸之暴露研究發現,二氯乙酸及三氯乙酸具有肝毒 性及致癌性,亦會引起腫瘤發生. [17]. 。另一方面,一氯乙酸會使老鼠. 引發癌症、二溴乙酸則會危害雄性老鼠的生殖系統,影響精細胞, 並使精子退化及萎縮. [4, 18]. 。. 9.
(21) 第二節 水中三鹵甲烷之檢測分析方法 2-2.1 行政院環保署-環境檢驗所[8] 行政院環保署除了於民國八十三年四月公佈水中總三鹵甲烷之 檢測方法–吹氣捕捉氣相層析法之外,亦於民國八十六年十一月公佈 兩項飲用水中揮發性有機化合物(VOCs)的檢測方法,分別為吹氣 捕捉/氣相層析質譜法(Purge and Trap/GC/MS)以及吹氣捕捉毛細管 柱氣相層析/串聯式光離子化偵測器法(Purge and Trap/GC/PID)。另 外,吹氣捕捉毛細管柱氣相層析法/串聯式光離子化偵測器及電解導 電感應偵測器檢測法(Purge and Trap/GC/ELCD)亦可用於三鹵甲烷 的檢測[19]。. 除了上述方法外,環檢所亦於民國八十九年三月公佈水中揮發性 有機化合物檢測方法-吹氣捕捉氣相層析質譜法[20],並適用於飲用水 水質、飲用水水源水質、地面水體、地下水、廢(污)水及放流水中 可被吹出之有機化合物;而其三鹵甲烷偵測極限較之前所提之方法更 低。. 10.
(22) 2-2.1 美國環保署(U.S. Environmental Protection Agency) 因為三鹵甲烷屬於VOCs,因此現今的研究大多參考US EPA Method 524.2, Measurement of Purgeable Organic Compounds in Water by Capillary Column Gas Chromatography/Mass Spectrometry及Method 624, Methods For Organic Chemical Analysis of Municipal and Industrial Wastewater。此兩種分析方法相似,只是前者針對飲用水,後者則針 對廢水;雖然兩種方法分析的VOCs 種類略有不同,但均利用曝氣的 方式將水中可吹洗類化合物吹洗後捕留於吸附物質上,再經加熱脫 附,最後導入氣相層析/質譜儀(GC/MS)中以偵測各化合物濃度。. 2-2.3 現行常用三鹵甲烷之分析方法 1. 吹氣捕捉法(Purge and Trap Method)[13] 在分析過程中,首先將含有揮發性有機物之待測水樣以針筒注入 吹氣玻璃樣品容器內,再以特定流速、溫度以及時間通入惰性氣體(例 如高純度氦氣或氮氣),使其中的揮發性有機物導入室溫下的吸附劑 捕捉管收集。捕捉完成後,以瞬間加熱脫附並使用氦氣逆向通過捕捉 管之方式,將有機物質導入氣相層析管柱中進行分離,再利用質譜儀 (MS)、光離子化偵測器(PID)、或電解導電感應偵測器(ELCD) 等常用的偵測器進行偵測。分析過程的最後一個步驟是升高捕捉管的 溫度,並以與吹氣時逆向之氣流進行捕捉管的回洗,準備下一個樣品 11.
(23) 的分析(此步驟通常與氣相層析分離同時進行)。不過,此方法的主要 缺點為設備價格較昂貴,及較不適合分析基質複雜的水樣等(可能會 使捕捉管柱遭受污染)。. 2. 液相萃取法(Liquid- liquid Extraction Method)[21] 此方法為利用有機溶劑將水樣中的欲測微量分析物萃取,經分離 及濃縮後,再以微量注射器取出有機相、注入氣相層析儀分析。液相 萃取優點在於適用範圍廣泛,對基質複雜的樣品皆可萃取;但缺點是 在兩不互溶液體之兩相介面層易產生乳化(Emulsion)現象而導致取 樣過程可能造成樣品流失,同時萃取過程中需要大量有機溶劑亦將造 成環境的污染。. 3. 頂空法(Headspace Method)[22] 頂空法是將水樣置於密閉容器中,使分析物達氣液相平衡之後, 再以氣密注射針抽取頂空層的氣相部分,注入儀器分析。此方法優點 為溶劑使用量非常小,非常快速且具有良好的重複性(repeatability); 但因為未經任何的濃縮步驟,故此方法的靈敏度通常不佳,較不適用 於微量分析且需精確校正。. 12.
(24) 4. 固相萃取法(Solid-phase Extraction,SPE)[22, 23] 固相萃取技術為一種應用固相結合相(Solid bond phase),於矽 膠的未反應矽醇基上接上各種官能團,利用液體溶劑與固體相之分配 能力來達成純化分析物之效果。通常於矽膠的未反應矽醇基上接上有 非極性(C8-C18)、極性相、離子交換等不同材質。此方法較傳統萃取 的操作簡單、可現場操作,只需使用極少的有機溶劑;但固相萃取法 仍有使用上的限制,如:分析物與樣品基質間之鍵結較強時,將導致 較低的回收率。. 5. 固相微萃取法(Solid-phase Microextraction,SPME)[22-24] 依不同的取樣方式可分為直接固相微萃取(Directly Immersion Solid-phase Microextraction)和頂空固相微萃取(Headspace Solid-phase Microextraction)兩種。直接固相微萃取是將萃取纖維直 接浸入樣本基質中;而頂空固相微萃取則是以萃取纖維吸附揮發之分 析物,待纖維塗佈物質、樣本、及樣本頂空達成固、液、氣三相平衡 後即完成萃取(其可避免樣本基質干擾以及保護纖維損傷)。. 13.
(25) 第三節 水中鹵乙酸檢測分析方法 由於鹵乙酸的結構特性,在一般自然環境水體中絕大部分(99 %以上)以解離之含鹵乙酸鹽離子形式存在;而鹵乙酸對層析方法而 言極性過大(特別是氣相層析儀),同時酸類的揮發性也較低,使其不 適宜直接注入氣相層析儀分析[10]。因此,目前鹵乙酸的分析大多會先 經過衍生酯化步驟[25],以產生極性較低且較具揮發性的酯類物質,並 得到良好的分析結果。. 2-3.1 前處理的方法 前處理的流程可包含:將待分析物由樣本基質中萃取出來、將非 待測物或干擾物質分離、及將萃取液進行濃縮等三個部分。早期的萃 取方法包括:索氏萃取(Soxhlet extraction)及液相-液相萃取 (Liquid-liquid extraction)等,但其缺點為耗時費事及需要大量有機 溶劑[23];而目前常用的分析方法[26-29],如:頂空取樣法(Headspace sampling)、吹氣捕捉法(Purge and trap)、薄膜萃取法(membrane extraction)、固相萃取法(Solid-phase extraction)、超臨界流體萃 取法(Supercritical fluid extraction)、及固相微萃取法(Solid-phase. microextraction)等,相對於索氏萃取及液相-液相萃取,則具有快速、 簡便、及免溶劑等優點。. 14.
(26) 1. 薄膜萃取法(membrane extraction)[23] 以薄膜萃取法萃取分析物,其過程包含兩個步驟:利用聚合物薄 膜萃取樣品基質中分析物,再以氣體或液體將分析物由聚合物薄膜萃 取出來。薄膜型態可分為半支撐中空狀薄膜與傳統式支撐平版型薄 膜;前者,有較大的表面積對體積比值,可增加質量轉移及萃取速率。 不過,因薄膜吸附反應太慢,因此其缺點為不能同時處理大量化學物 質與極性物質。. 2. 超臨界流體萃取(Supercritical Fluid Extraction, SFE) 超臨界流體萃取是利用某些物質或溶劑,在臨界點以上所具有 的特性來提取混合物中可溶性成分的一種物理分離的技術。超臨界流 體的密度約為0.2-0.9 g/cm3,其流體密度及黏度介於氣體與液體間, 故具有氣相物質的傳輸能力及液相的溶解力。此一特性有利於從待測 物質中萃取某些易分解的成分;其優點是萃取時間短、節省費用、萃 取徹底,且可進行熱敏感樣品及微量樣品的萃取,基本解決了溶劑對 環境的影響。不過,超臨界流體萃取法需要昂貴的高壓傳送系統以及 大量高純度氣體,減少了使用的普遍性。. 15.
(27) 3. 固相萃取法(Solid-phase Extraction,SPE)[30] 固相萃取技術為一種應用固相結合相(Solid bond phase),於矽 膠的未反應矽醇基上接上各種官能團,利用液體溶劑與固體相之分配 能力來達成純化分析物之效果。通常於矽膠的未反應矽醇基上接上非 極性(C8-C18)、極性相、離子交換等不同材質。此方法較傳統萃 取的操作簡單、可現場操作,只需使用極少的有機溶劑。不過,固相 萃取法仍有使用上的限制,如:分析物與樣品基質間之鍵結較強時, 將導致較低的回收率。. 2-3.2 衍生方法的介紹[31] 1. 衍生化的使用時機 氣相層析儀對於揮發性或非極性物質的分析有較好的結果,同時 也較容易偵測。另一方面,針對不易藉由氣相層析儀偵測的物質,則 常經由衍生的方式,改變物質的極性特性後,再予以分析。一般而言, 需考慮衍生化的情況如下: (1)在氣相層析儀中,對熱不穩定的物質 (2)增加揮發性及減少極性物質的極性 (3)改進物質在氣相層析儀中特定官能基的分離 (4)在未知的混合物中,得到特定官能基的種類及數目等資訊. 16.
(28) (5)依據物質的行為所選擇偵測器,如:電子捕捉或質譜儀 衍生化需注意的為衍生試劑是否可以完全移除或可能造成分析 物的干擾;當分析物經由衍生後,配合內標準品的使用將可對衍生化 過程中分析物的可能損失有補償作用,進而增加定量的準確度[27]。. 2.酯化衍生的介紹 本研究中所謂酯化反應,即鹵乙酸的甲基酯化,反應式如下: R-COOH+HO-R’→ R-COO-R’+H2O 由上式得知,酯化是酸中的carboxyl group及醇中的hydroxyl group反應而得,並伴隨水的生成。. 而除了上述的反應外,另一種甲基反應-偶氮甲烷 (diazomethane)添加,也是常見的方法,其反應式如下: R-COOH+CH2N2→ R-COO-CH3+N2 其優點在於反應極快且側反應很少(side reaction);但偶氮甲烷 為致癌物質,且需顧慮到偶氮甲烷的的保存及氮氣產生導致容器升壓 的問題。不過,由於偶氮甲烷是一種黃色物質,故當反應完全後,顏 色會完全消失,易於觀測。. 17.
(29) 2-3.3 分析方法介紹 1.行政院環保署-環境檢驗所[9] 環檢所於民國九十四年七月公佈水中鹵乙酸檢測方法(固相萃取 濃縮/氣相層析儀/電子捕捉偵測器法)。本方法首先調整水樣pH值,再 加入一定量之擬似標準品,經陰離子交換固相萃取膜(管柱)濃縮、 再以酸性甲醇溶液流洗後,所得之流洗液於 70 ℃ 下進行酯化反 應。反應完成後,以甲基第三丁基醚(Methyl-tertbutyl-ether)萃取各 鹵乙酸之甲酯衍生物,經氮氣吹除溶劑濃縮後,注入氣相層析儀以電 子捕捉偵測器分析之。. 2.美國環保署(U.S. Environmental Protection Agency) 針對水中HAAs 的偵測,可使用US EPA Method 552, Determination of Haloacetic Acids in Drinking Water by Liquid-Liquid Extraction, Derivatization, and Gas Chromatography with Electron Capture Detection(1990), 及552.2, Determination of Haloacetic Acids and Dalapon in Drinking Water by Liquid-Liquid Extraction, Derivatization and Gas Chromatography with Electron Capture Detection(1995)來分析。上述兩種分析方法在萃取濃縮上相似, 但在酯化方式則不同;Method 552 之酯化方式是以重氮甲烷 (Diazomethane)進行甲酯化(Methyl Esterification) ;而Method 552.2. 18.
(30) 則利用10%之硫酸甲醇溶液於50℃下水浴二小時使其甲酯化,且所 分析的HAAs 種類也略有差異(分析上則皆以GC-ECD進行檢測)。. 3.其他分析方法 一般鹵乙酸的分析需要使用氣相層析儀配合電子捕捉偵測器( ECD)或質譜儀(MS),但其缺點為需要衍生化的步驟、增加分析 時間,且易受無機物質的影響。因此,包括:毛細管電泳、液相層析 儀、及電化學分析等儀器亦被用於鹵乙酸的分析。. (1)毛細管電泳(capillary zone electrophoresis)[32] 毛細管電泳所配合的萃取方法,是採用離線的液相-液相萃取 法;不過,易造成毒性及不可燃性溶劑大量使用及費時的萃取時間等 缺點。因此,亦有研究發展利用電動力學注入配合具高交互性吸附物 的固相萃取技術來改進其缺點。. (2)液相層析儀(liquid chromatography)[21] 此法利用逆相離子對層析儀配合間接紫外線光譜儀來測定鹵 乙酸,並使用 C18 管柱和動相中添加一種可吸收紫外線離子的方式加 以測得;然而其缺點為對三溴乙酸的敏感度較差及三氯乙酸有較長的 流析時間。 19.
(31) (3)電化學分析(electrochemical analysis)[33] 藉由導電高分子做成的離子感應器,利用不同分析物有不同 剝除電位的特性,可對鹵乙酸進行定性分析;此外,所產生之電流 正比於分析濃度,因此亦可用電流量進行定量分析。. 20.
(32) 第四節 固相微萃取技術 2-4.1 固相微萃取之裝置[34, 35] 固相微萃取(Solid-phase microextraction;SPME)主要構造如圖 2-1 所示,由兩個部分組成,包括:固相微萃取纖維和固相微萃取手 動裝置。固相微萃取纖維主要以熔融之矽纖維組成(一般約長 1cm, 直徑 0.11mm) ,表面裹附具吸附性的聚合物後,此熔融矽纖維再附著 在一金屬絲狀物上,並在不使用時縮入金屬針管內保護之。為使用方 便,萃取纖維係裝入固相微萃取手動裝置(SPME Fiber Holder)中使 用;在裝置中,萃取纖維可以更換使用。固相微萃取之特點為簡化前 處理步驟、體積小、選擇性高、重複使用,並將取樣、萃取、濃縮及 樣本注入整合為一個步驟。. 2-4.1.1 手動型裝置 手動型裝置主要有推桿(plunger)及注射筒(barrel) 。推桿的作 用是將縮放於注射針頭內之纖維伸出,以推桿停止螺絲(plunger retaining screw)固定於 Z 型溝槽(Z-slot) ,使纖維伸出於針頭之長度 可被固定;此外,可調式針頭引導⁄深度計(adjustable needle guide⁄depth gauge)則可決定注射針頭露出的長度(圖 2-1)。. 21.
(33) 2-4.1.2 萃取纖維 目前已商業化之固相微萃取的萃取纖維與手動裝置有數種不同 的規格(如表 2-5 所示),其主要的差異在於萃取纖維表面之吸附性聚 合物(即萃取之披覆靜相)的種類以及裹附的厚度(可針對不同的萃 取樣本與目的來選擇,以滿足應用上的需要);基本上固定靜相的選 擇即參考“極性溶於極性;非極性溶於非極性”及與樣本之分配係數 較大的原則。固相微萃取纖維大致可分為極性纖維(Polyacrylate; Carbowax®/Divinylbenzene, CW/DVB)、非極性纖維( Polydimethylsiloxane, PDMS)、及雙極性纖維( Polydimethylsiloxane/Divinylbenzene, PDMS/DVB; CarboxenTM/Polydimethylsiloxane, CAR/PDMS; Divinylbenzene/Carboxen/PDMS , DVB/CAR/PDMS)等三種;而萃取 纖維可重複使用一百次或者更多。另外,因應不同樣本的需求,萃取 纖維可先吸附物質再以此物質與樣本反應,如利用 Polydimethylsiloxane/Divinylbenzene(PDMS/DVB)先裹附 PFBHA 再進行甲醛之吸附[36](附表 2-4 為測試纖維的耐受溫度)。. 22.
(34) 2-4.2 固相微萃取之理論[34, 35] 利用SPME 萃取待測物主要包含兩個步驟:(1)萃取過程: 當纖維置入欲分析物樣品瓶中時,待測物便會在樣品基質與固定微萃 取纖維靜相之間進行分配(partition),進而吸附於纖維上,達到濃縮萃 取待測物之目的;(2)脫附過程:將固相微萃取纖維由欲分析物樣品 瓶取出,直接進入分析儀器中進行脫附。 固相微萃取原理為樣本基質中之標的分析物與可吸附之披覆固 定靜相進行平衡分配(equilibrium partition) ;萃取方式可分為直接固 相微萃取(direct SPME)與頂空固相微萃取(headspace SPME)兩種 。. 2-4.2.1 直接固相微萃取[24, 34, 35] 直接固相微萃取為將萃取纖維完全浸入樣本基質中,當萃取系統 中的液相及披覆固定靜相達到平衡時,所得吸附量由下列方程式表示 : n=KfsVfVsCo /(KfsVf+Vs)……………….……….(1) n:萃取纖維吸附之樣品量 Kfs:樣品披覆在固定靜相與樣本間的分配係數(Kfs=Cf/Cs) Cf:披覆固定靜相上樣品之濃度. 23.
(35) Cs:樣本中樣品之濃度 Vf:萃取纖維披覆靜相之體積 Vs:樣本體積 Co:樣本中樣品之原始濃度 當樣本體積大於披覆固定靜相體積許多時(Vs>>KfsVf),此時所 吸附之樣品量便簡化成下列方程式: n=KfsVfCo………………………..………………..(2). 2-4.2.2 頂空固相微萃取 當樣本基質過於複雜、污穢,或樣本為固態而分析物為揮發性有 機物質時,可利用頂空固相微萃取。頂空固相微萃取是將固相微萃取 纖維暴露在樣本的頂空氣相中,當萃取系統中的液相、頂空氣相及披 覆固定靜相達到平衡時,所得吸附量可由下列方程式計算: n=KfsVfVsCo /(KfsVf+ KhsVh+Vs)….………….(3) Khs:樣品披覆在固定靜相與頂空氣相間的分配係數 Khs=Ch / Cs Ch:頂空氣相中樣品之濃度 Vh:頂空氣相之體積 頂空固相微萃取之平衡速度不僅較直接固相微萃取快,而且可避. 24.
(36) 免污穢之水樣本或血液在直接固相微萃取時其油質或大分子物質緊 緊吸附於萃取纖維上造成傷害,因此較能延長萃取纖維的使用壽命。. 2-4.2.3 影響固相微萃取萃取效率之因素 影響固相微萃取萃取效率的因素也會影響萃取之敏感度及偵測 極限,包括披覆固定靜相的種類與體積、樣本體積及頂空氣相體積等 ,皆屬影響萃取的因素;而萃取條件如吸附及脫附時間與溫度,環境 因素如樣本攪拌程度、溫度、酸鹼度及添加極性溶劑與鹽類等亦有影 響[34, 35]。此外,依據不同樣本性質,取樣時選擇直接萃取或頂空萃取 亦有不同。. (1)固定靜相的種類及厚度[37] 商業化的固定靜相纖維(如表 2-3 所示)是由不同的吸附劑所組 成,其特性皆不相同。固定靜相的厚度越厚代表分配係數越大,而可 提高分析物的吸附量並進而提升靈敏度。. (2)分析樣本體積 吸附量公式(2)的前提假設為:樣本體積需遠大於靜相體積才 不至於影響吸附在介質上的量;此外,以頂空固相微萃取法萃取分析 物時,瓶中樣本的體積量將影響頂部空間的體積,而由公式(3)可 25.
(37) 知針對揮發性高的分析物(Khs 高) ,其頂空的體積(Vh)應該要小, 如此才能增加固定靜相纖維上所吸附分析物的量。. (3)萃取時間 萃取時間主要是和所選擇吸附纖維的種類以及分析物的性質有 關;吸附纖維的分配係數越大、吸附量越多,其吸附平衡時間越長; 而膜越厚、吸附量越多,同樣的吸附平衡時間也越長。進行吸附時, 吸附量並不會隨著時間的增長而無限量增加;其吸附量增加的最大量 ,將由分析物在固相微萃取纖維的靜相與樣本之間質量的傳遞而決定 。當傳遞達平衡時,即表示達吸附平衡;即使吸附時間再增長,吸附 量並不會再增加。. (4)萃取溫度 固相微萃取纖維吸附分析物質會受到萃取時溫度的影響,並可從 兩分面來討論: (1)當增加吸附溫度時會使樣本擴散速率增加,進而 對分析物的吸附量也會增加,同時達平衡所需的時間也會縮短; (2)由於吸附屬於放熱反應,分配係數 K 會隨著溫度上升而降低, 故樣本吸附量將會下降。在吸附過程中上述兩個因素通常是互相拮抗 ,因而形成溫度與纖維吸附能力之間的複雜關係。 一般而言,在吸附未達平衡之前,當溫度較高時會增加纖維的吸 26.
(38) 附量;而達平衡之後增加吸附溫度使分配係數 K 下降,反而使纖維 上已吸附之分析物再度被脫附,故會使吸附量下降。. (5)攪拌之影響 當萃取時樣本處於靜止狀態,纖維萃取的吸附平衡時間會受到分 配係數 K 與固定靜相體積的影響;但若樣品處於攪動的狀態,由於 質量傳遞速度的改變,對於平衡時間、偵測極限、纖維吸附量及萃取 回收率皆有影響,因而攪拌的規律性也會影響分析的結果。若是不規 律的攪拌動作,反而會造成精密度變差。. (6)鹽類之影響 若在分析樣本中添加入鹽類,可增加樣本的離子強度,形成鹽析 現象(salting out) ,對萃取效率有提升的作用。所謂的鹽析現象是在 樣本內添加鹽類,當鹽類解離成陰離子或陽離子時,水分子會形成水 合層包圍在離子周圍形成水合現象,會致使不易解離的分析物溶解度 下降,因而增加了擴散到頂空的量,而提升吸附萃取量;另外添加鹽 量的多寡也會影響吸附量,因為鹽量多寡會影響鹽析現象的強弱[22, 23] 。 (7)pH 值 分析樣本之酸鹼值對萃取效率的影響取決於分析物質本身的種 27.
(39) 類。從文獻中發現,pH 值的改變對分析物萃取效率的影響並不一定 ,如:鹵化醚類、硝基苯類或芳香族碳氫化合物等,pH 值的改變對 於萃取效率並無明顯影響;另一方面,若將 pH 值調整至較低時會使 酚類解離而提升其萃取量。. (8)脫附溫度及脫附時間 熱脫附為最常用的脫附方法之一,當脫附溫度太低或脫附時間太 短時會使得吸附在靜相上的分析物無法完全脫出殘留而在纖維上,進 而產生定量上的誤差以及造成纖維損害、影響其使用壽命。此外,當 分析物質對熱不安定時,太高的脫附溫度可能會造成分析物的熱分解 ,而降低其可信度。. 2-4.3 固相微萃取之應用 固相微萃取技術為一整合取樣、萃取、濃縮及樣本注入等步驟, 已廣泛用於氣相基質(空氣中戊醛、空氣中環氧乙烷)[38, 39],液相基 質(水中醛類、醋中呋喃甲醛)[24, 36],及固相基質(土壤中多氯聯苯 (PCBs)、苯、甲苯)等,應用範圍包括:環境、工業衛生、臨床、 刑事科學、食物及藥品分析等;例如由尿液樣本中萃取安非他命或天 使塵等非法禁用之毒藥、咖啡或茶葉中萃取咖啡因、及血液樣本中萃 取酒精成分等[40],可見固相微萃取技術應用的領域已相當廣泛了。 28.
(40) 第五節 研究架構 決定纖維和衍 生試劑. 製備標準溶液. 最適萃取條件. 分析條件. GC/MS 分析. 建立標準曲線. 實驗室 QA/QC. 29. 1.選擇萃取纖維 2.樣本進行衍生反應 3.GC 熱脫附條件 4.萃取因素: 萃取溫度、萃取時 間、含鹽量及脫附溫 度與時間等.
(41) 第三章 材料與方法 第一節 實驗器材 3-1.1 藥品試劑 1. 總三鹵甲烷(TTHMs)標準品:2000mg/L(含氯仿、一溴二. 氯甲烷、二溴一氯甲烷、及溴仿),Supelco 2. 鹵乙酸(HAAs)標準品:2000mg/L(含一氯乙酸、二氯乙 酸、三氯乙酸、一溴乙酸、二溴乙酸及一氯一溴乙酸) ,Supelco 3. 硫酸鈉,Na2SO4,Sodium sulfate,Wako (SEM3503) ,Japan 4. 氯化鈉,NaCl,Sodium chloride,SHOWA(SL2351Z),Japan 5. 甲醇,CH3OH,Methanol,TEDIA®, USA 6. 鹽酸,HCl,Hydrochloric acid 7. 去離子水. 3-1.2 儀器設備 1. 茶色螺旋蓋樣本瓶 Vials:15×45 mm 4mL,glass,Kimble®, Mexico(60815-1545) (含 PTFE 中孔螺蓋與 Kimble® Septa (73812-13)) 2. 電子分析天平:METTLER TOLEDO 標準型, AG245 3. 震盪器:VORTEX-GWNIE2®Scientific Industries, USA 4. 玻璃滴管 Pasteur pipet:229mm, Kimble® 72050-900, USA(含 2mL 吸頭) 5. 微量吸管:100μL、1000μL, NICHIRYO® Digital Micropipet, Japan 6. Pipette:Transferpipette 100~1000 µl 30.
(42) 7. 定量瓶 volumetric flask:10mL, glass, IWAKI®, Japan 8. 固相微萃取萃取纖維 SPME fiber:100 µm PDMS, SUPELCO® 57310-U , PDMS/DVB 9. 固相微萃取萃取纖維 SPME fiber:60 µm PDMS/DVB , SUPELCO® 57300-U, PDMS 10. 固相微萃取手動裝置 SPME holder:SUPELCO® 57330-U 11. 固相微萃取採樣裝置 SPME sampling stand/4mL vial puck: SUPELCO® 57333-U,(含電磁攪拌器 stir plate) 12. 加熱攪拌器:Barnstead/Thermolyne, SP46925;(Dubuque, Iowa, 52001 USA) 13. 攪拌子:magnetic stirring-bar, 2 mm × 7 mm;Spinbar (made in USA) 14. 氣相層析儀 GC:Perkin Elmer Autosystem XL Chromatograph (層析管柱 column:DB-1) 15. 質譜儀 MS,Perkin Elmer - TurboMass. 31.
(43) 第二節 實驗步驟 3-2.1 實驗衍生步驟 根據參考研究文獻[9, 23, 27, 41, 42],首先以去離子水配製含鹵乙酸溶 液、及以甲醇配製三鹵甲烷溶液後,分別取 1.068 mL 及 0.66 mL 置 於 4 mL 樣本瓶中,再加入 45.4 μL HCl 及 226.6 μL methanol 於樣本 瓶中後,置於 100℃水浴並以磁石攪拌以進行衍生反應。此衍生反應 維持 20 分鐘後,將樣本瓶置於冷水中冷卻,並以固定轉速攪拌至恆 溫 40℃以利水相與頂空相之間的平衡。. 取裝有萃取纖維之固相微萃取手動裝置(萃取纖維需先前處理, 見附錄二),將針頭插入 4 mL 樣本瓶中,以推桿把萃取纖維完全推 出,採用頂空萃取法並結合磁石攪拌以吸附三鹵甲烷及含鹵乙酸甲 酯,之後以氣相層析質譜儀進行熱脫附與分析,觀察不同因素對吸附 量之影響。. 3-2.2 固相微萃取方法建立 (1)固相微萃取纖維之選擇 首先配製濃度分別為 0.033μg/mL 的三鹵甲烷及 5.34μg/mL 的 鹵乙酸甲酯化物,並依物質特性選定適用於雙極性(揮發性及半揮發 性物質)的 65μm Polydimethylsiloxane/Divinylbenzene(PDMS/DVB) 32.
(44) 以及非極性的 100μm Polydimethylsiloxane(PDMS)進行試驗,以 決定合適的萃取纖維。. (2)萃取時間探討 配製濃度分別為 0.033μg/mL 的三鹵甲烷及 0.534μg/mL 的鹵乙 酸甲酯化物於樣本瓶中,以經前述所選定之纖維分別進行 5、20、30 及 60 分鐘之頂空吸附,以決定合適之頂空萃取時間。. (3)萃取溫度探討 配製濃度分別為 0.033μg/mL 的三鹵甲烷及 0.534μg/mL 的鹵乙 酸於甲酯化物樣本瓶中,以經前述所選定之纖維及頂空吸附時間,分 別在 50、60、80、及 100℃下進行合適萃取溫度之探討。. (4)攪拌速度對萃取的影響 配製濃度分別為 0.033μg/mL 的三鹵甲烷及 0.534μg/mL 的鹵 乙酸甲酯化物於樣本瓶中,以經前述所選定之纖維、頂空吸附時間、 及萃取溫度,分別於 200、500、1000 及 1200 rpm 下測試不同攪拌速 度對吸附效果的影響。. 33.
(45) (5)添加鹽類的影響 配製濃度分別為 0.033μg/mL 的三鹵甲烷及 0.534μg/mL 的鹵乙 酸甲酯化物於樣本瓶中,以經前述所選定之纖維、頂空吸附時間、萃 取溫度、及攪拌速度,分別添加 NaCl 及 Na2SO4(添加濃度分別為 2.5 %、12.5%及 25%),以探討添加鹽類對吸附效果的影響。. (6)熱脫附 將已有吸附物質之萃取纖維,利用氣相層析儀注射口之高溫直接 進行熱脫附;脫附時,將固相微萃取裝置之注射針頭刺入注射口內, 以推桿把萃取纖維完全推出,在注射口 180℃的溫度下進行熱脫附。 脫附效率的決定是將同一樣本在完成第一次纖維脫附分析後,在同樣 分析條件下,直接將纖維再置入注射口進行第二次纖維脫附分析;脫 附效率定義為:第一次脫附分析的面積╱(第一次+第二次脫附分析 面積)。. 34.
(46) 第三節 實驗分析方法 3-3.1 儀器分析條件 建立 GC/MS 分析方法,參考 GC 及 GC/MS 為分析方法的文獻[22, 23, 43-45]. ,建立適當的分析方法。. 3-3.1.1 氣相層析儀 z Injector Temp:180℃ z Carrier gas:He z Column:DB-1,30 m ×0.25 mm z Flow rate:1.25 mL/min z Temp program:40℃ for 5min,12℃/min to 130℃ for 2min,25℃/min to 230℃ for 4min z Run time:22.5 min. 3-3.1.2 質譜儀 質譜儀之離子源為電子撞擊離子源(EI);首先以 Full Scan mode 模式(m/z 25~250)進行定性分析,再利用 selected ion recording(SIR) mode 來選擇特定的 m/z 以進行定量分析及得到較好的敏感度(m/z 之設定如表 3-1 所示) 。 z Interface Temp:250℃. 35.
(47) z Ion Source Temp:270℃ z Scan Model:Selected Ion Recording(SIR),表 3-1 所示 z Solvent Delay:4 min. 3-3.2 檢量線的配製 配製含不同濃度之三鹵甲烷(1、0.5、0.2、0.1 及 0.05 μg/mL) 及鹵乙酸(10、5、2、1、0.5μg/mL) ,經酯化衍生反應後,以前述經 研究所決定之萃取條件進行頂空吸附,最後插入 GC 注射口中熱脫附 並以 GC-MS 分析,獲得標準曲線。. 3-3.3 品保品管 3-3.3.1 檢量線 標準曲線之相關係數(R)應達 0.995 以上。. 3-3.3.2 方法偵測極限(method detection limit, MDL) 將固相微萃取之萃取纖維吸附濃度為 0.0165 μg/mL 的三鹵甲烷 及 0.267 μg/mL 的鹵乙酸酯化標準品,吸附時間為 30min,之後插入 GC 注射口中熱脫附分析,依此連續分析七次,將面積值代入標準曲 線求出濃度,計算七次的標準差。偵測極限的計算如下:[45,46]. 36.
(48) MDL=3×S=3×{(Σxi2-n×X2)/(n-1)} 其中,MDL:方法偵測極限(method detection limit) S(STD):標準差 Xi:分析所得之濃度,i 為分析樣本數 n:重複分析次數 X:重複分析所得濃度的平均值. 3-3.3.3 分析準確度 配製檢量線內一濃度樣本,連續分析 7 次,以下列公式計算分析 結果,分析準確度以相對誤差表示,應低於 10%。 相對誤差=(|量測濃度-配製濃度|/配製濃度)× 100%. 3-3.3.4 分析精密度 配製檢量線內一濃度樣本,連續分析 7 次,以測試分析精密度, 根據分析結果計算其變異係數(Coefficient of variation,CV) ,以 CV 表示,應低於 7%。. 37.
(49) 第四章 結果與討論 第一節 固相微萃取方法建立 4-1.1 固相微萃取纖維的選擇 在相同的濃度、相同的分析條件下分別以 100 μmPDMS 及 60 μm PDMS/DVB 兩種纖維來進行吸附測試,結果如圖 4-1 所示:100 μm PDMS 及 60 μm PDMS/DVB 皆能同時吸附三鹵甲烷及鹵乙酸甲酯化 物,但 60 μm PDMS/DVB 纖維對於三鹵甲烷及鹵乙酸甲酯化物的吸 附面積值,明顯大於 100 μm PDMS 纖維的吸附面積值;故選定 60 μm PDMS/DVB 纖維作為本研究中固相微萃取的吸附介質。. 4-1.2 萃取時間探討 本研究以 60 μm PDMS/DVB 纖維分別頂空吸附 5、20、30 及 60 分鐘,探討萃取時間的影響。如圖 4-2 所示,當吸附 30 分鐘時纖維 吸附值大約已呈現飽和狀態,而吸附至 60 分鐘面積值並無明顯的變 化趨勢;因此,本研究選定以 30 分鐘為萃取吸附時間。. 4-1.3 萃取溫度探討 隨著萃取溫度的提升,將導致分析物的蒸汽壓增加,進而縮短樣 本與頂空之間達到平衡的時間;然而因為吸附為放熱反應,因此增加. 38.
(50) 溫度反而會使纖維上已吸附之分析物再度被脫附、造成吸附量下降。 另一方面,由於本實驗所使用的衍生步驟需添加強酸性的鹽酸;而為 避免衍生或所添加的強酸破壞塗覆纖維的高分子構造,以延長纖維壽 命,選定過高的溫度是必須避免的。 本研究以去離子水配製濃度分別為 0.33μg/mL 的三鹵甲烷及 0.534μg/mL 的鹵乙酸甲酯化物於樣本瓶中,以經前述所選定之纖維 及頂空吸附時間,分別在 50、60、80、及 100℃下進行吸附。如圖 4-3 所示,分析物之面積值隨加熱的溫度上升至 60℃之後而明顯減 少;因此,本研究選擇 60℃為最適之纖維萃取溫度。. 4-1.4 攪拌速度對萃取的影響 在萃取過程中,持續攪拌增加萃取效率、減少萃取平衡時間。一 般水樣攪拌方法即使用電磁攪拌器,並固定攪拌磁石的形狀大小及轉 速。當使用頂空固相微萃取時,需注意攪拌速度若過大,將造成水樣 漩渦過大及磁石非貼平瓶底轉動;如此將生成不穩定的流動,反而更 容易造成萃取時的不穩定性。 本研究以 2 mm × 7 mm 之長形磁石置於樣本瓶中,分別於 200、 500、1000 及 1200 rpm 下測試不同攪拌速度對吸附效果的影響。如圖 4-4 所示,在 1000 rpm 時吸附面積趨於平緩;當轉速至 1200 rpm 時,. 39.
(51) 雖然纖維吸附面積值大部份為穩定狀態,但一氯乙酸甲酯化物的面積 值已有下降的趨勢。因此,本研究選定以 1000 rpm 為萃取時之攪拌 轉速。. 4-1.5 添加鹽類的影響 配製濃度分別為 0.33μg/mL 的三鹵甲烷及 0.534μg/mL 的鹵乙酸 於樣本瓶中,以經前述所選定之纖維、頂空吸附時間、萃取溫度、及 攪拌速度,分別添加 NaCl 及 Na2SO4,以探討添加鹽類對吸附效果的 影響。 研究中首先將每 2 毫升樣本各添加 0.05、0.25 及 0.5 克的氯化鈉 進行測試。攪拌之後,發現添加 0.5 克氯化鈉的樣本無法完全溶解, 且當添加量過多時會使吸附效果降低;如圖 4-5 所示,由未添加氯化 鈉及添加 0.05、0.25 克的樣本吸附結果發現,未添加氯化鈉的吸附面 積值最大。 在 Na2SO4 方面,分別將 0.05、0.25 及 0.5 克的硫酸鈉加入 2 毫 升的樣本中。攪拌之後,發現添加 0.5 克硫酸鈉的樣本無法完全溶解, 且當量過多時會使吸附效果降低;如圖 4-5 所示,由未添加硫酸鈉及 添加 0.05、0.25 克的樣本吸附結果發現,未添加硫酸鈉的吸附面積值 最大。. 40.
(52) 綜合上述,不論是添加 NaCl 或 Na2SO4,並不能獲得較大的吸附 量,因此本研究選擇不添加鹽類於樣本中,以得到較好的纖維吸附效 果。. 4-1.6 熱脫附 依據文獻[27],部分鹵乙酸及鹵乙酸酯在衍生及 GC/MS 分析的過 程中,會因其對熱不穩定的特性,導致在注射口的較高溫度中經由脫 溴或脫酸而生成其他物種;因此,本研究設定在注射口 180℃的溫度 下進行熱脫附。脫附效率的決定是將同一樣本在完成第一次纖維脫附 分析後,在同樣分析條件下,直接將纖維再置入注射口進行第二次纖 維脫附分析;結果如表 4-1 所示,脫附效率大於 99.7%以上。. 4-1.7 樣本體積 由第二章第四節中的公式(3)n=KfsVfVsCo /(KfsVf+ KhsVh+Vs) 可知,頂空氣相之體積(Vh)會影響到吸附於萃取纖維上之量,因此 如要增加方法偵測極限,則需設法降低頂空氣相之體積。 根據文獻(36)比較兩種不同樣本體積(將 1 mL 與 2 mL 樣本置於 4 mL 之 vial 中)的方法偵測極限, 2 mL 之樣本體積明顯比 1 mL 為 佳,所以本研究直接以 2 mL 之樣本體積進行分析。. 41.
(53) 第二節 採樣分析方法 4-2.1 品保品管執行結果 4-2.1.1 檢量線 配製含不同濃度之三鹵甲烷(1、0.5、0.2、0.1 及 0.05 μg/mL) 及鹵乙酸(10、5、2、1、0.5 μg/mL),經酯化衍生反應後,以前述 經研究所決定之萃取條件進行頂空吸附,最後插入 GC 注射口中熱脫 附並以 GC-MS 分析,獲得標準曲線。其結果於表 4-2,其檢量線相 關係數(R)皆於 0.995 以上;GC/MS 層析圖譜如 4-6 所示。. 4-2.1.2 方法偵測極限 將固相微萃取之萃取纖維吸附濃度為 0.0165 μg/mL 的三鹵甲烷 及 0.267 μg/mL 的鹵乙酸酯化標準品(吸附時間 30min),之後插入 GC 注射口中熱脫附分析,依此連續分析七次,將面積值代入標準曲線求 出濃度,計算三次的標準差,結果如表 4-2 所示。 本研究所發展的分析方法,對三鹵甲烷的偵測極限為 0.004~0.006ppm (低於國內飲用水水質標準及美國環保署規範值 80ppb);而鹵乙酸偵測極限為 0.017~0.194ppm (高於美國環保署的規 範值 60ppb)。本研究由於儀器的限制,並未使用如美國環保署公告方 法的建議以 GC-ECD 分析樣品,而是以 GC/MS 偵測樣本;而如果可. 42.
(54) 以利用 GC-ECD 分析,相信本研究所發展分析方法的偵測極限將可 符合美國環保署對水中鹵乙酸的偵測需求。. 4-2.1.3 分析準確度 配製檢量線內一濃度樣本(三鹵甲烷 0.0495 μg/mL 及鹵乙酸甲酯 化物 0.801 μg/mL),進行分析準確度測試。如表 4-3 所示,其相對偏 差為 88~107%。. 4-2.1.4 分析精密度 配製檢量線內一濃度樣本(三鹵甲烷 0.0495 μg/mL 及鹵乙酸甲 酯化物 0.801 μg/mL),進行分析精密度測試。如表 4-3 所示,所得 結果變異係數(Coefficient of variation,CV)介於 4~14%。. 4-2.2 與其他方法之比較 本研究所發展之分析方法與其他方法之比較,如表 4-4 所示;而 針對需要衍生反應的含鹵乙酸之分析,本研究的方法相較於原 US EPA Method 552.2,由於不需使用萃取溶劑、且以 SPME 提昇萃取效 率,因此大幅將衍生所需 2 小時的時間縮短為 30 分鐘。. 43.
(55) 第五章 結論與建議 固相微萃取技術,整合了以往分析時複雜的萃取、濃縮等前處理 步驟,並具有快速、操作簡單,及免用溶劑的優點;其中,頂空固相 微萃取的方式可使靜相塗覆纖維不會受到基質污染而影響到纖維的 使用壽命,故本研究使用頂空固相微萃取技術來進行各項實驗分析討 論。. 本研究以 SPME 配合 GC/MS 萃取分析水中三鹵甲烷及鹵乙酸之 條件為: (1)65μm PDMS/DVB 纖維,(2)熱脫附 180℃、2 分鐘, (3)頂空萃取溫度 60℃, (4)頂空萃取時間 30 分鐘, (5)轉速 1000 rpm,及(6)不添加鹽類等;頂空萃取後注入氣相層析質譜儀 (GC/MS),並以 SIM mode 設定分析,能夠順利同時分析水中三鹵 甲烷及鹵乙酸甲酯化物。. 本研究分析之鹵乙酸為 MCAA、MBAA、DCAA、DBAA、TCAA 及 BCAA,其中前五種為 US EPA 規定之 HAA5;而未來可再研究九 種鹵乙酸同時分析的可行性。. 本研究發現鹽類添加並不會增加對三鹵甲烷及鹵乙酸的萃取效 果。此結果與一般添加鹽類增加樣本離子強度而增加萃取效果不同。 44.
(56) 在衍生過程中需要添加鹽酸,而使分析物偏向酸性,而當加入鹽 類時也許改變了樣本的 pH 值,因而影響了纖維的吸附量。因此,未 來應再將樣本 pH 值列入影響萃取效果的探討。. 45.
(57) 參考文獻 1.. 張哲誠, 台灣地區飲用水中致癌性化合物暴露之評估及其與癌 症死亡率相關性之探討. 中山醫學院毒理學研究所, 2000.. 2.. Rook , J.J., Formation of haloforms during chlorination of natural Waters. Water Treat, 1974. 23(2).. 3.. Bull, R.J., Health Effect of Drinking Water Disinfectants and Disinfection By-Products. Env. Sci. Tech., 1982. 16: p. 554-561.. 4.. Bull, R.J., Kopfler F.C, Health Effects of Disinfectants and Disinfection By-Products. American Water Works Association Research Foundation, 1991.. 5.. 行政院環境保護署, 飲用水水質標準修訂. 2006.. 6.. 行政院環境保護署環境檢驗所, 台灣地區飲用水中鹵乙酸氯化 消毒副產物分析方法及調查之研究, 環檢新知第 36 期.. 7.. EPA, U., National Primary Drinking Water Regulations: Disinfectants and Disinfection Byproducts Rule; Final Rule. 2006.. 8.. 行政院環境保護署環境檢驗所, 水中總三鹵甲烷檢測方法─吹 氣捕捉氣相層析法 1995, 水質檢測方法.. 9.. 行政院環境保護署環境檢驗所, 水中鹵乙酸檢測方法─固相萃 取濃縮/氣相層析儀/電子捕捉偵測器法. 2005, 水質檢測方法.. 10.. 張慧嫻, 台灣地區飲用水中含鹵乙酸之分析與流佈調查. 台灣 大學環境衛生研究所碩士論文, 2004.. 46.
(58) 11.. Jolley, R.L., Suffet ,I.H., Concentration Techniques for Isolating Organic Constituents in Environmental Water Samples. Organic Pollutants in Water., 1987.. 12.. Shin, D., Chung,Y., Choi,Y., Kim,J., Park,Y., Kum,H., Assessment of Disinfection By-Products in Drinking Water in Korea. Exposure and Environmental Epidemiology, 1999. 9: p. 192-199.. 13.. 陳作青, 吹氣捕捉法與快速氣相層析質譜系統偵測水中消毒副 產物三鹵甲烷. 國立臺灣大學化學研究所碩士論文, 1999.. 14.. Marabini L, Frigerio S, Chiesara E, Radice S., Toxicity evaluation of surface water treated with different disinfectants in HepG2 cells. Water Research, 2006. 40 p. 267–272.. 15.. Carhman, R.A., Shalsky ,H., Borzcell ,J.F., The Effect of Selected Water Disinfectant Product on testicular DNA Metabolism. Intermational Symposium on the Health Effects of Drinking Water Disinfectant and Disinfection By-Products, 1981: p. 53-59.. 16.. Sarrion M.N., Santos F.J., Galceran M.T., Solid-phase microextraction coupled with gas chromatography–ion trap mass spectrometry for the analysis of haloacetic acids in water. Chromatography A,, 1999. 859 p. 159-171.. 17.. Lianhui T, P.M.K., Rongrong G., and Michael A.P. , Effect of Dichloroacetic Acid and Trichloroacetic Acid on DNA Methylation in Liver and Tumors of Female B6C3F1 Mice. Toxicological sciences, 1998. 43, 139–144.. 18.. DeAngelo, A.B., Daniel,F.B., Stober,J.A., Olson,G.R., Pane,N.P, Carcinogen Bioassays of Chloroacetic Acid in Fischer 344 rats. 47.
(59) Toxicologic Pathology., 1993. 19.. 行政院環境保護署環境檢驗所, 吹氣捕捉毛細管柱氣相層析法/ 串聯式光離子化偵測器及電解導電感應偵測器檢測法. 水中揮 發性有機化合物檢測方法, 2002.. 20.. 行政院環境保護署環境檢驗所, 吹氣捕捉/氣相層析質譜儀法. 水中揮發性有機化合物檢測方法, 2002.. 21.. C, H., James F. R., Analysis of haloacetic acid mixtures by HPLC using an electrochemical detector coated with a surfactant-nafion film. Talanta, 1999. 48.. 22.. 陳奕卉, 以頂空固相微萃取技術分析水中微量三鹵甲烷. 台灣 大學環境衛生研究所碩士論文, 2001.. 23.. 鄭易翔, 以頂空固相為萃取來偵測水中鹵乙酸之研究. 清華大 學化學研究所碩士論文, 2000.. 24.. 高國源, 呋喃甲醛暴露評估方法之建立─空氣及食物中呋喃甲 醛採樣分析方法之驗證. 中國醫藥大學環境醫學研究所碩士論 文, 2005.. 25.. Gary L. Emmert, G.C., Gija Geme,Neena Joshi,Mostafizur Rahman, Methods for real-time measurement of THMs and HAAs in distribution systems. Awwa Research Foundation, 2004.. 26.. Gary L. Emmert, G.C., et al., Methods for Real-Time Measurement of THMs and HAAs in Distribution Systems. Awwa Research Foundation, 2004. 48.
(60) 27.. Ma Y.C., Chiang C.Y., Evaluation of the effects of various gas chromatographic parameters on haloacetic acids disinfection by-products analysis. Chromatography A, 2005. 1076: p. 216–219. 28.. Xie, Y., Analyzing Haloacetic Acids using Gas Chromatography Mass Spectrometry Wat. Res., 2001. 35: p. 1599-1602.. 29.. Liu Y, Mou S, Chen D. , Dengyun Chenb, Determination of trace-level haloacetic acids in drinking water by ion chromatography–inductively coupled plasma mass spectrometry. Chromatography A, 2004. 1039: p. 89–95.. 30.. Agency, U.E.P., Method 552.1 Haloacetic Acids and Dalapon. 1992.. 31.. Blaugh, P.J., Gas Chromatography- A Practical Approach. 1995.. 32.. Martinez, D.J., Evaluatuion if different electrolyte systems and on-line proconcentrations for the analysis of haloacetic acid \s by capillary zone electrophoresis. chromatography A, 1999. 835.. 33.. Akhtar, P., Too, CO., Wallace, GG Anal Chim., Detection of haloacetic acids at conductive electroactive polymer-modified microelectrodes. Anal. Chem. Acta., 1997. 341.. 34.. 黃敬德.謝有容, 固相微萃取技術之原理及其應用. Chemistry(The Chinese Chem.Soc.,Taipei), 1998. 56(4): p. 16-25.. 35.. 黃敬德.謝有容, 固相微萃取法. 科儀新知, 1997. 18(4): p. 59-67.. 36.. 張峻鳴, 水中醛類檢測方法之改進. 中國醫藥大學環境醫學研 究所碩士論文, 2002.. 37.. Supelco, SPME application Guide. 1999. 49.
(61) 38.. 吳鎧光, 以固相為萃取技術發展空氣中環氧乙烷之被動式採樣 器. 中國醫藥學院環境醫學研究所碩士論文, 2002.. 39.. 張娣安, 以固相為萃取技術發展空氣中戊醛之被動式採樣器. 中國醫藥學院環境醫學研究所碩士論文, 2001.. 40.. Centini F, M.A.C.B., Quantitative and qualitative analysis of MDMA, MDEA, MA and amphetamine in urine by head-space/solid phase micro-extration (SPME) and GC/MS. Forensic Science International 1996. 83: p. 161-166.. 41.. Villanueva CM., Kogevinas M., Grimalt JO., Haloacetic acids and trihalomethanes in finished drinking waters from heterogeneous sources. Water Research 2003. 37: p. 953–958.. 42.. Anastasia D. Nikolaou, S.K.G., Maria N. Kostopoulou, Themistokles D. Lekkas, Determination of haloacetic acids in water by acidic methanol esterification–GC–ECD method. Water Research 2002. 36: p. 1089–1094. 43.. Cho DH., Kong SH., Oh SG., Analysis of trihalomethanes in drinking water using headspace-SPME technique with gas chromatography. Water Research, 2003. 37 p. 402–408.. 44.. Zhao R., Lao W., Xu X., Headspace liquid-phase microextraction of trihalomethanes in drinking water and their gas chromatographic determination. Talanta, 2004. 62: p. 751–756. 45.. 行政院環境保護署環境檢驗所, 環境檢驗方法偵測極限測定指 引. 2004.. 50.
(62) 46.. 行政院環境保護署環境檢驗所, 環境檢驗室儀器及方法偵測極 限測定指引. 2003.. 51.
(63) 表 2.1 三鹵甲烷的物理性質 物質名稱. M.W.. 比重(25℃). mp(℃). Bp(℃). 三氯甲烷(Trichloromethane). 119.37. 1.485. -63.5. 61.2. 一溴二氯甲烷(Bromodichloromethane). 163.82. 1.971. -57.1. 90.1. 二溴一氯甲烷(Dibromochloromethane). 208.27. 2.38. ---. 116. 三溴甲烷(Tribromomethane). 252.72. 2.887. 8.5. 151.2. 52.
(64) 表 2.2 鹵乙酸及其衍生甲酯化物之物理性質 鹵乙酸類. 對應酯類. 名稱. M.W.. mp(℃). bp(℃). bp(℃). 一氯乙酸(Chloroacetic acid;MCAA). 94.4. 61-63. 189. 130. 50. 208. 144-145. 128.9. 10. 194. 143. 217.8. 39-41. 250. 182-183. 173.3. 38. 215. 155. 163.3. 57-58. 196. 152-153. 296.7. 129-135. 245. 220-222. 207.8. 70.5-74.5. 215. 200-203. 252.2. 95-98. 232-234. 180-182. 一溴乙酸(Bromoacetic acid;MBAA) 138.9 二氯乙酸 (Dichloroacetic acid;DCAA) 二溴乙酸 (Dibromoacetic acid;DBAA) 一氯一溴乙酸 (Bromochloroacetic acid;BCAA) 三氯乙酸 (Trichloroacetic acid;TCAA) 三溴乙酸 (Tribromoacetic acid;TBAA) 二氯一溴乙酸 (Bromodichloroacetic acid;DCBAA) 一氯二溴乙酸 (Dibromochloroacetic acid:DBCAA). 53.
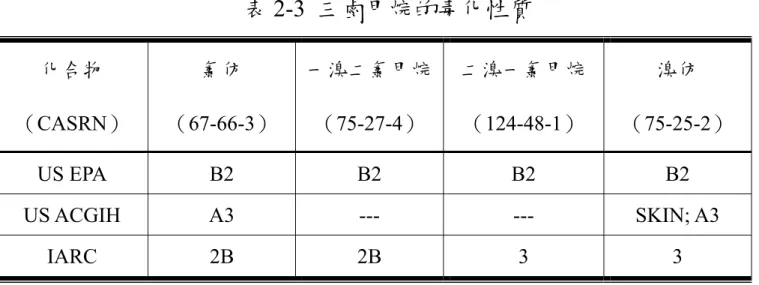
(65) 表 2-3 三鹵甲烷的毒化性質 化合物. 氯仿. 一溴二氯甲烷. 二溴一氯甲烷. 溴仿. (CASRN). (67-66-3). (75-27-4). (124-48-1). (75-25-2). US EPA. B2. B2. B2. B2. US ACGIH. A3. ---. ---. SKIN; A3. IARC. 2B. 2B. 3. 3. ※US EPA 的 B2 表示為對人類致癌資料沒有或不充足,但對動物 致癌有充足的數據 ※US ACGIH 的 A3 表示對動物致癌物 ※IARC 的 2B 為對人類可能致癌;3 表示對人類致癌無法分類. 54.
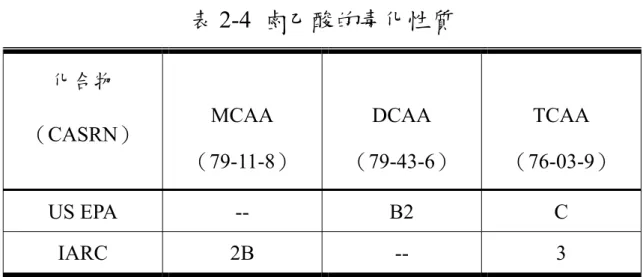
(66) 表 2-4 鹵乙酸的毒化性質 化合物 MCAA. DCAA. TCAA. (79-11-8). (79-43-6). (76-03-9). US EPA. --. B2. C. IARC. 2B. --. 3. (CASRN). ※US EPA 的 B2 表示為對人類致癌資料沒有或不充足,但對動物 致癌有充足的數據;C 為可能對人類致癌資料沒有或不充足, 且對動物致癌的證據不充足 ※IARC 的 2B 為對人類可能致癌,3 表示對人類致癌無法分類. 55.
(67) 表 2-5 市售固相微萃取纖維[37] Commercially available SPME fibers Stationary phase and film thickness. Abbreviation. General application ( analyte type ). Polydimethylsiloxane (100 µm). PDMS. Non-polar, volatile. Polydimethylsiloxane (30 µm). PDMS. Non-polar, volatile and semi-volatile. Polydimethylsiloxane (7 µm). PDMS. Non-polar, semi-and non-volatile. Polydimethylsiloxane-divinylbenzene (65 µm). PDMS-DVB. Polar. Polyacrylate (85 µm). PA. Polar, general use. Carboxen-polydimethylsiloxane (75、85 µm). CAR-PDMS. Volatile, gaseous, trace analysis. Carbowax-divinylbenzene (65、75 µm). CW-DVB. Polar, volatile (low temperature limit). Carbowax-templated resin (50 µm). CW-TPR. Polar, HPLC. Divinylbenzene-carboxen-PDMS (50/30 µm). DVB-CAR-PDMS. Broad range of polarities from C3 to C20. 56.
(68) 表 2-6 測試纖維的耐受溫度 Stationary Phase. Film Thickness. pH. Maximum. Recommended. Conditioning. Time. Temp. Operating Temp. Temp. (Hrs.). PDMS. 100µm. 2-10. 280 ℃. 200-280 ℃. 250 ℃. 0.5. PDMS/DVB. 65 µm. 2-11. 270 ℃. 200-270 ℃. 250 ℃. 0.5. 57.
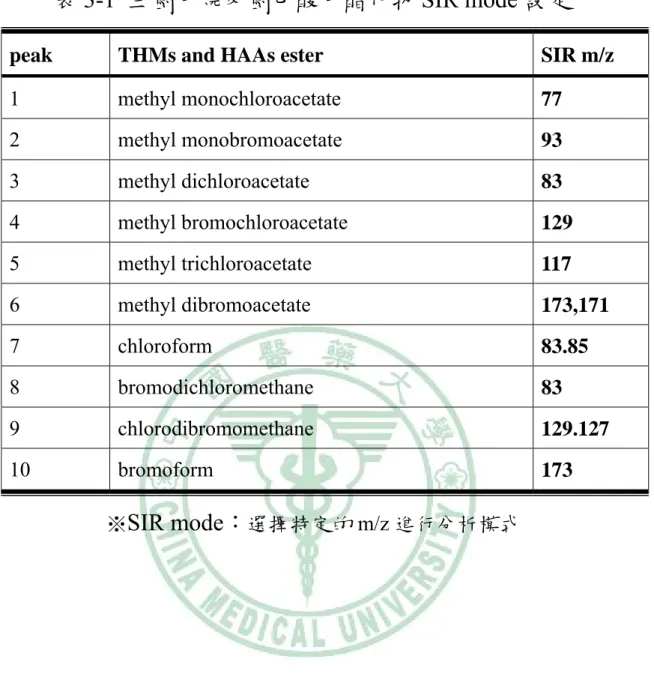
(69) 表 3-1 三鹵甲烷及鹵乙酸甲酯化物 SIR mode 設定 peak. THMs and HAAs ester. SIR m/z. 1. methyl monochloroacetate. 77. 2. methyl monobromoacetate. 93. 3. methyl dichloroacetate. 83. 4. methyl bromochloroacetate. 129. 5. methyl trichloroacetate. 117. 6. methyl dibromoacetate. 173,171. 7. chloroform. 83.85. 8. bromodichloromethane. 83. 9. chlorodibromomethane. 129.127. 10. bromoform. 173. ※SIR mode:選擇特定的 m/z 進行分析模式. 58.
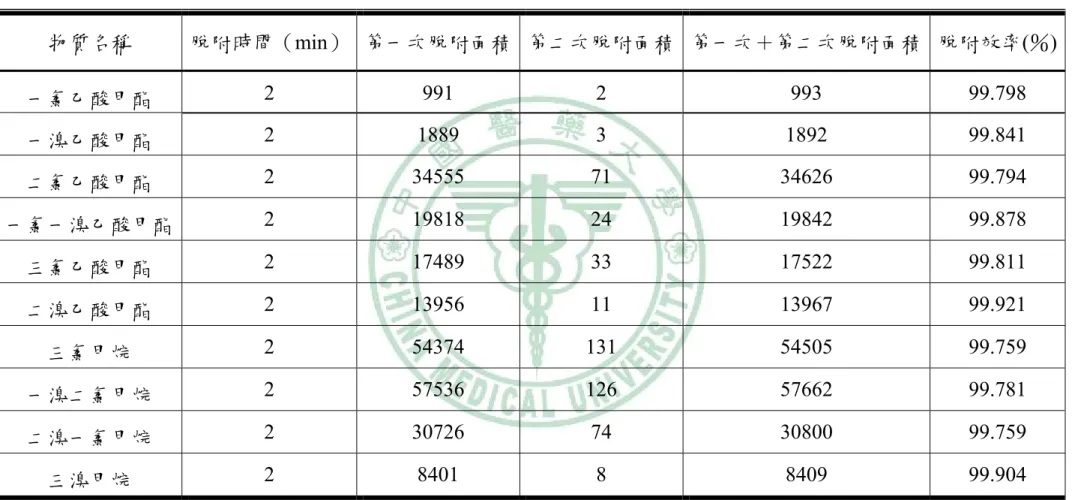
(70) 表 4-1 熱脫附效率結果 物質名稱. 脫附時間(min) 第一次脫附面積 第二次脫附面積 第一次+第二次脫附面積 脫附效率(%). 一氯乙酸甲酯. 2. 991. 2. 993. 99.798. 一溴乙酸甲酯. 2. 1889. 3. 1892. 99.841. 二氯乙酸甲酯. 2. 34555. 71. 34626. 99.794. 一氯一溴乙酸甲酯. 2. 19818. 24. 19842. 99.878. 三氯乙酸甲酯. 2. 17489. 33. 17522. 99.811. 二溴乙酸甲酯. 2. 13956. 11. 13967. 99.921. 三氯甲烷. 2. 54374. 131. 54505. 99.759. 一溴二氯甲烷. 2. 57536. 126. 57662. 99.781. 二溴一氯甲烷. 2. 30726. 74. 30800. 99.759. 三溴甲烷. 2. 8401. 8. 8409. 99.904. ※脫附效率=(第一次脫附分析面積/(第一次+第二次脫附分析面積))×100%. 59.
(71) 表 4-2 QA/QC 檢量線、相關係數及偵測極限 物質名稱. 線性方程式. 相關係數®. 偵測範圍. 方法偵測極限. (μg/mL). (μg/mL). 一氯乙酸甲酯. y = 304.42x - 8.6715. 0.997. 0.267-5.34. 0.089. 一溴乙酸甲酯. y = 70.658x + 26.594. 0.99. 0.267-5.34. 0.194. 二氯乙酸甲酯. y = 1644.2x - 32.576. 0.997. 0.267-5.34. 0.017. 一氯一溴乙酸甲酯. y = 357.68x + 283.5. 0.997. 0.267-5.34. 0.042. 三氯乙酸甲酯. y = 692.3x + 217.35. 0.997. 0.267-5.34. 0.165. 二溴乙酸甲酯. y = 69.631x + 16.023. 0.991. 0.267-5.34. 0.049. 三氯甲烷. y = 15436x + 27.305. 0.999. 0.0165-0.33. 0.005. 一溴二氯甲烷. y = 7213.5x + 41.426. 0.997. 0.0165-0.33. 0.005. 二溴一氯甲烷. y = 8127.2x + 32.07. 0.999. 0.0165-0.33. 0.004. 三溴甲烷. y = 6776.8x - 35.653. 0.995. 0.0165-0.33. 0.006. 60.
數據


+7
![表 2-5 市售固相微萃取纖維 [37]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8979278.281186/67.1263.109.1160.186.654/表25市售固相微萃取纖維37.webp)
![圖 2-1 固相微萃取手動裝置 [37]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8979278.281186/74.1263.218.1027.138.659/圖21固相微萃取手動裝置37.webp)
Outline
相關文件
Results of the analysis carried out by the Laboratory of the Civic and Municipal Affairs Bureau indicated that the quality of potable water of the distribution networks and
6 《中論·觀因緣品》,《佛藏要籍選刊》第 9 冊,上海古籍出版社 1994 年版,第 1
You are given the wavelength and total energy of a light pulse and asked to find the number of photons it
1.9 Chapters 3 to 7 cover the concerns and suggestions received and elaborate on our support measures covering the five proposed actions, including enhancing schools’
Wang, Solving pseudomonotone variational inequalities and pseudocon- vex optimization problems using the projection neural network, IEEE Transactions on Neural Networks 17
For pedagogical purposes, let us start consideration from a simple one-dimensional (1D) system, where electrons are confined to a chain parallel to the x axis. As it is well known
Define instead the imaginary.. potential, magnetic field, lattice…) Dirac-BdG Hamiltonian:. with small, and matrix
incapable to extract any quantities from QCD, nor to tackle the most interesting physics, namely, the spontaneously chiral symmetry breaking and the color confinement..
