Chemical Physics 162 (1992) 335-348 North-Holland
Theoretical study of the neutral and ionic states
of hypermetalated potassium compounds &OH and K2NH2
and potassium complexes KHz0 and KNH3
Y.-W. Hsiao, K.-M. Chang
Department of Chemistry, National Tarwan Umversrty. Talpel, Tatwan, ROC
and
T.-M. Su
Department of Chemrstry, National Tarwan University, Talper. Taiwan. ROC
and The Institute ofAtomic and Molecular Sciences, Academia Smlca, Tarpel, Tarwan, ROC Received 23 September 199 1
Ab uutio molecular orbital calculations are reported for potassium-contaimng hypermetalated compounds of K,OH and K2NH2, and potassium-Lewis-base complexes of KHZ0 and KNH,. the corresponding positive ions were also studled. The optimized equilibrium geometries, the harmonic vibrational frequencies, the bond dissociation energies. and the adiabatic ionization poten- tials are reported for these compounds. The similarities and the differences of these physical and chemical properties of the neutral and its posltlve iomc states are discussed. These theoretical findings were also compared with the theoretical and experimental results reported in the literature. The unique bonding properties of the hypermetalated compounds were specifically mvestlgated by electron density deformation maps.
1. Introduction
The hypermetalated compounds of alkali atoms and the molecular complexes formed between alkali atom and Lewis base molecule, such as the water or ammonia molecules, have attracted attention of both theoretical and experimental researchers in the past decade. On the theoretical side, the predictions of the moderate interaction between alkali atom and Lewis base molecule have been reported by quite a few groups [ l-9 1. The energetic stability and the nature of bonding of some hypermetalated compounds of lithium and sodium were also studied [ lo- 13 1. On the experimental side, clusters formed between the sodium atom and water clusters or ammonia clusters have been generated by pickup source and studied by photoionization mass spectrometer [ 14,15 1. Hyper- metallic oxide compounds of sodium, potassium and cesium have been investigated by the same technique
[ 16-l 81, More recently, some hypermetallic hy-
droxyl compounds of potassium were also been re- ported [ 19,201. These gas-phase studies gave us in- formation on the ionization potentials and sometimes bond energies of these compounds. However, none of these gas-phase studies have yielded further spec- troscopic information, such as the vibrational or ro- tational data of neutral species. Up to date the only vibrational information available was from the IR studies on the matrix-isolated alkali-atom-water/ ammonia complexes [ 2 l-24 1. Nevertheless, these vibrational data are still far from complete. In these experiments the major experimental difficulty is to generate a high enough concentration of these com- plexes either in the gas phase or being trapped in a low temperature matrix for good spectroscopic measurements.
Restricting ourselves to the potassium-containing species, there was only one report on the electrostatic calculation of KHZ0 complexes and an equilibrium bond dissociation energy of 5.0 kcal/mol was pre- 0301-0104/92/$05.00 0 1992 Elsevier Sctence Publishers B.V. All rights reserved.
336 Y.-W Hslao et al. /Neutral and ionic states of hypermetalatedpotassrum compounds dieted [ 61. No ab initio calculations were reported
on the bond energies and the vibrational frequencies of KHZ0 and KNH3. For the hypermetalated hy- droxyl compounds, the sodium species has been studied and the nature of the chemical bonding also has been elucidated theoretically [ 12 1. There were no reports on the corresponding potassium com- pounds. The simplest hypermetalated amide I&NH? was not studied theoretically either.
As for the potassium-containing ionic species, in experiments, the best-studied physical quantities are their enthalpies of complex formation. These include K+H20 [25,26],K+NH3 [26,27],andK:OH [28]. The other physical properties were not yet reported. On the theoretical side, there were only one Hartree- Fock calculation on K+H20 [ 291 and some electro- static calculations on both K+H*O and K+NHj
[ 26,301. In these studies the bonding energies and the vibrational frequencies were calculated at some- what lower levels of sophistication, Further studies on the physical pictures of these ions are needed.
In light of the recent experimental progress on the potassium-containing hypermetalated compounds and the potassium-atom-H,O/NH, molecular com- plexes, which have been carried out in the gas phase and also in the matrix-isolated form, we embarked upon the present theoretical study. The reported molecules are KI&O, K+H*O, KNH,, K+NH3, K,OH, K: OH, K2NH2, and K$NH*. This study serves as a theoretical check of the present available experimental results and also as a guide for further experimental studies.
2. Computational methods
All ab initio molecular orbital calculations were carried out with either restricted or unrestricted Har- tree-Fock theory using the GAUSSIAN 90 package of computer codes [ 3 I]. Electron correlation was calculated by Moller-Plesset perturbation theory up to the second order (MP2 ).
For the hydrogen atom, the contracted [ 2s] basis of Salez and Veillard [ 32 ] from the Huzinaga (6s) basis set [ 331 was used. For the nitrogen and oxygen atoms, the contracted [ 5s, 3p] basis sets of Gianolio et al. [ 341 from van Duijneveldt’s ( 13s 8p) basis sets [ 351 were used. A p-type polarization function
set was added to the H basis set and a d-type polari- zation function set was added to the N and 0 basis sets, respectively, in the same way as the standard 6- 3 11 G** [ 3 11. The potassium basis set was adapted from the Wachters’ contracted [8s, 4p] set of the
( 14s 9p) basis set [ 361. For the potassium atom, polarization functions were not added in the calcu- lations of geometries and vibrational frequencies. In the final calculations of the bond dissociation ener- gies and ionization energies, the (2d) set of polari- zation functions of Klein et al. [ 37 ] was also in- cluded in the potassium basis set.
With these basis sets, KOH, Hz0 and NH3 were first calculated up to the MP2 level. The equilibrium geometries of KOH agree with the best available the- oretical results to within 2.6% [38]. The electric di- pole moment agrees with each other to within 8%. These previous theoretical results have an estimated accuracy of 2% [ 381. The harmonic vibrational fre- quencies of HZ0 and NH, agree with the experimen- tal harmonic vibrational frequencies to within 2.5%
[ 39,401. The equilibrium geometries of NH3 and HZ0 agree with the experimental results to within 0.6%.
In the present report, KH20, KNH,, KIOH and K2NHZ and their ionic states are studied. The equi- librium geometries, the harmonic vibrational fre- quencies, the bond dissociation energies, and the adi- abatic and vertical ionization potentials were calculated. All the calculations were done up to MP2 level and the geometries were also optimized at MP2 level. for completeness, the bond dissociation ener- gies and the ionization potentials were then calcu- lated at these optimized geometries with the potas- sium basis set augmented with the d-type polarization functions. In the course of the calculations of KNH3 and K+NH3, the symmetries of the highest occupied doubly degenerate molecular orbitals, the e symme- try of C3,,, were undetermined by the available pro- gram. In these cases the symmetries were determined directly by the inspection of the molecular orbital maps. The energies of these molecular orbitals and the other physical quantities converge normally.
Y.-W. Hslao et al. /Neutral and lonlc states of hypermetalatedpotassrum compounds 337
3. Results and discussion
3.1. KH,O, KNH+ K + HzO, and K+ NH, molecular complexes
The physical and chemical properties of the potas- sium complexes are listed in the upper halves of ta- bles 1, 2, 3, 6, and 7 and the whole of tables 4 and 5. they are discussed in the following categories.
3.1.1. Structures and bonding
The upper part of table 2 lists the equilibrium ge- ometries of these potassium complexes obtained at MP2 level. Comparing the equilibrium geometries of free Hz0 and NH3 with those of the corresponding potassium molecular complexes and ions, one finds that these ligands only undergo minor changes dur- ing the formation of these potassium neutral or ionic complexes. This is a direct consequence of the com- paratively weak interactions between K or K+ with the water or ammonia molecules. In the case of H20, there is only a slight decrease of the HOH angle and a little increase of the OH distance in the formation of the potassium complexes. The ammonia molecule behaves similarly. The changes of the internuclear equilibrium distance between K and 0 or K and N as one goes from the neutral to the positive ionic state are minor too. This result also ensures that in the photoionization studies of these simple systems, the Table 1
Total energies (au) of potassium-containing compounds
adiabatic ionization potential would be close to the threshold ionization potential one usually measured in the experiments.
The LiH*O complex has been reported to be non- planar with a bending angle of 40.1’ between the Li- 0 and the Hz0 plane [ 8 1. The NaH20 complex also has a bending angle of 46.2”. The present study, with the K basis augmented with d-type polarization func- tions and calculated up to MP2 level, yields a planar structure for KH*O. In the paper of Broughton and Bagus [ 7 1, RbH20 has been assumed to be in the planar form. The optimized geometry of RbH20 has not been reported yet. For these alkali-atom-water molecule systems, the interaction potential along the bending angle is rather flat. A conclusive resolution of the correctness of this trend of the bending angles may eventually require some spectroscopic experi- ments in the gas phase. As for the ammonia com- plexes, a geometry of C3” was always obtained.
There have been quite a few theoretical discus- sions on the nature of the bonding between an alkali atom and a Hz0 or NH3 molecule [ l-91. The stabil- ities of these complexes have been attributed to sev- eral factors: the electron correlation effect in the complexes, the charge polarization on the metal atom, the charge transfer from the Lewis base to the metal atom and the electrostatic interaction of the unper- turbed molecular charge distributions. According to the result of the latest theoretical studies, the re-
Species State Symmetry Energy
HF MP2 K K+ Hz0 NH3 KHZ0 K+H20 KNH3 K+NH, K,OH K: OH KOH KzNHz K: NH2 KNH2 -599.1428761 - 598.9960480 -76.0563505 -56.2160117 -675.2061796 -675.0801899 -655.3657902 -655.2402027 - 1273.7838178 - 1273.6872233 -674.6157197 - 1253.9088100 - 1253.8103360 -654.7414354 - 599.2645937 -599.1177747 -76.2910626 - 56.4306848 -675.5641528 -675.4361878 -655.7041878 -655.5768208 _ 1274.27065 13 -1274.1719717 -674.9798304 - 1254.3787257 - 1254.2769801 -655.0872440
338
Table 2
Y.-W. Hslao et al. /Neutral and ionic states of hypermetalatedpotasslum compounds
Geometry index (results of MP2 )
Species Bond length (A) Interatomlc angle
KOorKN OH or NH Hz0 (Gv) 0.964 NH, (C,v) 1.015 KHz0 (Cl”) 2.770 0.966 K+H# (Czv) 2.725 0.966 KNHs (C,v) 2.971 1.018 K+NH, (C,,) 2.916 1.019 KOH (Cm,) 2.346 0.961 KzOH (Cl,) 2.524 0.966 K2’OH (C,, ) 2.562 0.967 KNH, (Czv) 2.535 1.023 K>NH> (L) 2.710 1.027 K:NHz (Cl,) 2.720 1.027
ported Mulliken populations of LiH*O, NaH20, and RbHzO indicate that the charge transfer from HZ0 to the metal atoms are O.O79e, O.O62e, and <O.OSe [ 7,9], respectively. The present study obtains a charge transfer of 0.025e for KHZ0 and 0.029e for KNH3. The reported theoretical dipole moment enhance- ments (in debye ), the electric dipole moment differ- ence between the complexes and the base molecules, are: 3.7 [6] or 3.0 [8] (LiH20), 3.3 (NaH,O) [6], 4.1 (LiNH3) [2], and 3.3 (NaNH3) [2]. In the present study the dipole moment enhancements are 1.0 and 1.4 for KHZ0 and KNH3, respectively. The theoretical equilibrium bonding energies (kcal/mol) of these complexes are 13.8 (LiH20) [ 81, 7.4
(NaH20) [9], 5 (RbH20) [7], 14.5 (LiNH3) [2], and 6.0 (NaNH3) [ 21. As shown in table 6, the pres- ent study yields 7.2 for KHZ0 and 7.5 for KNH3.
Some of the classifications of the above interaction forces have their origin in the classical electromag- netic theory. However, by ab initio molecular orbital methods, for instance, one can not unambiguously distinguish the charge-density shift due to the charge- transfer factor from the shift due to the dipole-in- duced-dipole factor. In this study we shall use the charge transfer obtained by Mulliken population analysis as a measure of the charge-transfer factor and the classical dipole-induced-dipole interaction be- tween the dipole moment of the Lewis base molecule and the alkali atom as a measure of the dipole-in- duced-dipole factor [ 93. Here the general trend of the
- L (HOH)= 104.2” L(HNH)=107.1” ~(KOH)=127.8” ~(KOH)=128.4” L(KNH)=112.8” ~(KNH)=114.1~ L (KOH)=lSO.O” ~(KOH)=121.9” L(KOK)=116.2’ ~(KOH)=108.1” ~(KOK)=143.8’= ~(KNH)=129.2” ~(KNK)=107.3” L(HNH)=101.5” L(KNK)=137.4” L(HNH)=IOI.I”
interaction strengths instead of their absolute values is emphasized.
Following the procedure of ref. [ 91, the dipole-in- duced-dipole interaction energy is [ 4 1 ]
E(r)=-/&~~(l+3cos2~)/2r6, (1)
in which ,~~a is the dipole moment of the Lewis base molecule, oM is the polarizability of the alkali atom, 13 is the angle between the dipole moment and the al- kali metal atom, and r is the distance between the al- kali atom and the center of the dipole. The dipole moments of Hz0 and NH3 are 1.85 and 1.47 D, re- spectively. The polarizabilities of the alkali atoms are 24.3, 23.6, 43.4 and 47.3 A3 for Li, Na, K, and Rb, respectively [ 42 1. Adapting the experimental geom- etries of Hz0 and NH3 and the theoretical bonding distances between the alkali atom and HZ0 or NH3, one obtains the dipole-induced-dipole interaction energies (kcal/mol): Li-H20 (- 14.8), Na-H,O (-4.3), K-H20 (-5.2), Rb-HZ0 (-4.3), Li-NH, (- 11.3), Na-NH, (-2.3), and K-NH3 (-2.7). The general trend of the above data suggests that the charge polarization of the alkali atom by a Lewis base molecule, electron correlation effect, and electro- static interactions are all playing important roles in the stabilization of the potassium complexes. The charge-transfer process, an important stability factor in the Li and Na complexes, becomes less important in the K and Rb cases. As for the ionic species, the major bonding force is electrostatic interaction be-
Y.-W Hsrao et al. /Neutral and lomc states of hypermetalatedpotawum compounds 339 tween the alkali ion and the Lewis base molecule.
3.1.2. Harmonic vibrational frequencies
The upper part of table 3 lists the normal mode harmonic vibrational frequencies of KI-120, KNH,, K+H?O and K+NH3. The vibrational frequency shifts of water and ammonia in both K and K+ complexes are tabulated in tables 4 and 5. The experimental har- monic vibrational frequencies of the isolated water and ammonia are also listed in tables 4 and 5. The only reported experimental vibrational spectra were those of matrix-isolated KHz0 and KNH3 measured by the IR spectroscopy [ 23,241. Up to date there have been no reports on the experimental vibrational fre- quencies of the ionic species yet. As indicated in ta- bles 4 and 5, the theoretical and experimental har- monic vibrational frequencies of NH3 and Hz0 agree with each other to within 2.3%. A direct comparison of the present harmonic vibrational frequencies of free-form molecular complexes with the observed vi- brational frequencies of the matrix-isolated com- plexes would not tell one the goodness of the agree- ments between these two sets of results. A comparison of the Hz0 and NH, vibrational frequency shifts in KHz0 or KNH3 between the theoretical and the ex- perimental results could offer better insight. Except for the bending mode of Hz0 in which the experi- ment shows a - 5 cm- ’ shift (but the theory predicts a 14 cm- ’ shift), the results agree very well. The rea- sons for the disagreement, although comparatively small in the absolute magnitude, may come from the theoretical inaccuracy in correctly predicting the minimum position of the bending angle between the plane of H20 and the K-O bond, because of the flat- ness of the potential along this coordinate, or it may just come from the change of the equilibrium geom- etry of the complex due to the presence of the rare- Table 3
Harmonic vibrational frequencies (MP2 ) (cm- ’ )
gas matrix in the matrix experiments. Further works, especially some experiments of the gas-phase vibra- tional analysis, are needed to clarify these disagreements.
Generally.speaking, the tables show that, in the case of HzO, there is an increase in the frequency of the bending mode and a decrease in the frequencies of both symmetric and antisymmetric stretching modes in the formation of both the neutral and ionic potas- sium complexes. For the NH3 molecule, however, the frequency shifts are somewhat more complicated. For the KNH3 complexes, there is an increase in the a, ( v2) mode, and a decrease for the other normal modes. For the K+NH3 ion, both the a, ( v2) and e( v,) normal modes show blue frequency shifts and the rest two modes have red frequency shifts. The vibrational frequencies of K+H,O have been calculated before through simplified Hartree-Fock potential by Kis- tenmacher et al. [ 29 1. The six vibrational frequen-
Table 4
Harmonic vibrational frequency shifts of K-H,0 and K+-Hz0 (cm-‘) Symmetry aI Hz0 HF 1732 MP2 1623 exp. ‘) 1648 KH,O HF 1751 MP2 1637 Av HP 19 MP2 14 exp. b, -5 K+H20 HF 1789 MP2 1682 Av HF 57 MP2 59 ‘) Ref. [39]. b, Ref. [24]. al(ul) 4131 3841 3832 4102 3798 -29 -43 -39 4104 3827 -27 - 14 b,(v,) 4238 3975 3943 4204 3928 -34 -47 4185 3929 -53 -46
=bO 170 (a,) 238 (bz) 265 (b,) 1637 (al) 3798 (a,) 3928 (b,) K+HZO 200 (a,) 375 (b,) 427 (bz) 1682 (a,) 3827 (a,) 3929 (b,) KNH3 157 (a,) 293 (e) 1156 (a,) 1659 (e) 3461 (a,) 3613 (e) K+NH, 189 (a,) 399 (e) 1262 (a,) 1673 (e) 3481 (a,) 3607 (e) K,OH 72 (a,) 228 (b,) 275 (a,) 336 (bz) 473 (b,) 3869 (a,) K: OH 87 (a,) 221 (a,) 336 (bl) 352 (b,) 582 (b,) 3843 (a,)
KzNHz 70 (a,) 252 (bz) 258 (aI ) 299 (b,) 362 (aI) 485 (bz) 1541 (a,) 3410 (a,) 3501 (b,) K: NH* 77 (a,) 206 (a,) 296 (b,) 313 (b,) 471 (a?) 554 (bz) 1543 (a,) 3412 (al) 3499 (b,)
340 Y.-W. Hslao et al. /Neutral and ~omc states oJhypermetalatedpota.wum compounds Table 5
Harmonic vibrational frequency shifts of K-NH3 and K+-NH9 (cm-‘)
Symmetry a,&) e(vJ aI (vi) ek)
NH3 HF 1083 1785 3686 3821 MP2 1043 1665 3502 3660 exp. ‘) 1022 1691 3506 3577 KNH3 HF 1202 1785 3660 3786 MP2 1156 1659 346 1 3613 AV HF 119 0 -26 -35 MP2 113 -6 -41 -47 exp. b, 90 -54 K+NH4 HF 1323 1795 3666 3770 MP2 1262 1673 3481 3607 AV HF 240 10 -20 -51 MP2 219 8 -21 -53 ‘) Ref. [40]. b, Ref. [23].
ties in their report were (in cm-‘): vr=3714.2, v2= 1631.8, u3= 176.1, v,=3769.0, uS=398.2 and ~~~444.3. For these ionic species Kebarle and co- workers also developed an electrostatic model for the calculations of the vibrational frequencies [ 26,301. Following their general procedure in determining the interaction potential between the potassium ion and a ligand, and assuming that the vibrational force con- stants and the geometry of the ligand remain un- changed in the formation of the complex, one can ob- tain the vibrational frequencies of K+H20 to be: u, = 3698, u2 - - 1612, v3=21 1, v,=3792, v,=490and v,=407 cm-’ and the vibrational frequencies of K+NH3 to be: ~~~3351, v2=1159, v,=2.16, ~~~3440, u5= 1547 and u,=263 cm-‘. Here the force constants of the H20 molecule are set at the funda- mental vibrational frequencies of vr = 3652, v2 = 1595, and ~~~3756 cm-’ and those of the NH, molecule aresetatv,=3337,vz=950,v,=3414,andv,=1628
cm-’ [40]. Additionally, in all these electrostatic calculation the intramolecular potential functions were assumed to be in the form of valence forces [ 43 1. Comparing these theoretical results with those of the present study, one finds that, for the K+H*O ion, the results of Kistenmacher et al. has a better agreement with the present values than those obtained by elec- trostatic model. Similarly, for the case of K+NH3, the agreement between the results of electrostatic model and the present values is at best only fair. If one looks further into the vibrational frequency shifts of H,O and NH3 in the formation of the ionic complexes, ex-
cept for the v2 mode of Hz0 and the v2 mode of NHX, the electrostatic model even predicts opposite shift directions for the rest of the vibrational modes, let along the shift magnitudes. In short, the electrostatic model, if considered at the present level of sophisti- cation, is not adequate in doing the vibrational nor- mal mode analysis.
3.1.3.
Bond dissociation energies and ionization
potentials
There are three commonly used measures for the bond strength of a molecule: equilibrium bond dis- sociation energy (D, ) , bond dissociation energy (D, ) and the enthalpy of bond dissociation (AHO,, Tzabsolute temperature). In the case that a molec- ular complex dissociates into an atomic species and a molecule, to a good approximation, these three quantities are related with each other through statis- tical mechanics by the following relations:
MO,=D,+;RT
with N= Avogadro number,
R =
gas constant, and Q”= fi [ l-exp( -hv,/kT)]-',
Y.-W. Hsiao et al. /Neutral and lomc states of hypermetalatedpotassium compounds 341
the vibrational partition function.
D,, and D,are re-
lated by
D,,=D,+ 1 i {Nhv,- C f tNhv,. products I reactants I
(3)
In thermodynamic measurements, one usually ob-
tained AHO, over a certain range of temperature. In
the experiments of photoionization, photodissocia-
tion, or spectroscopic measurements, the directly re-
lated energy quantity is D,,. However, for theoretical
studies, the first obtained energy quantity is
D,.In
this report all the data are adjusted to
D,, with thehelp of the above equations and the theoretical har-
monic vibrational frequencies. These results are listed
in the upper part of table 6. The agreement between
the theoretical values and the experimental results is
Table 6Theoretical and experimental dissociation energies (kcal/mol)
within the experimental uncertainties. The theoreti-
cal vertical and adiabatic ionization energies of these
complexes are listed in table 7. The agreement be-
tween the theoretical values and the experimental re-
sults is only fair. We shall come back to this point in
the end of this section.
3.2. K,OH, K2NH2, K: OH, and K2+ NH2
hypermetalatedpotassium compoundsThe physical and chemical properties of the bi-po-
tassium systems are listed in the lower parts of tables
1, 2, 3, 6 and 7. They are discussed in the following
categories:
3.2.1. Structures and bonding
Comparing the equilibrium geometries of the neu-
tral species and their positive ionic states, one finds
Reactions Theoretical Experimental
DOb’ D,(HF) D,(Hp) ‘) D,(MPZ) D (MP2*) ‘) e D,,(HF*) ‘) D,,(MP2*) a) KH20+K+H20 4.36 4.70 5.33 7.18 K+H,O+K+ +H,O 17.44 17.95 17.16 18.73 KNH3+K+NH3 4.33 4.71 5.59 7.48 K+NH,+K+ +NH, 17.66 18.23 17.80 19.55 K,OH+K+KOH 15.83 16.16 16.46 18.60 K:OH+K++KOH 47.35 46.77 46.67 47.07 KrNH2-+K+KNHl 15.37 15.25 16.87 19.22 K:NH2+K+ +KNHz 45.72 45.17 45.16 46.12 3.75 6.33 5.8” 16.48 17.30 18.4 d), 17.4 ‘) 3.59 6.47 7.6” 16.58 17.98 20.7 f), 18.5 ‘) 16.17 18.47 19.9 c’ 46.45 46.70 38.0 g’ 14.48 18.31 44.03 44.93 a) d-type polarization functions were augmented in the K basis set.
b, See text for the recalculations of some experimental data.
‘) Ref. [ 191. d, Ref. [25]. ‘) Ref. [26]. f, Ref. [27]. g, Ref. [28].
Table 7
Theoretical and experimental ionization energies (eV)
Reactions Vertical (MP2) Adiabatic (MP2) Adiabatic (MP2*) a) Experimental K+K+ 4.00 4.04 4.339 b’ K2+K2+ 3.56 3.67 4.0637 =) KHrO+K+HlO 3.48 3.48 3.54 3.92 d’ KNHS+K+NH, 3.47 3.47 3.51 3.87 *’ K,OH+K:OH 2.83 2.69 2.80 3.55 d’ K,NH,-+K: NH* 2.95 2.77 2.87 3.94 d,
‘) d-type polarization functions were augmented in the K basis set. b, Ref. [44]. ‘) Ref. [45]. d, Ref. [ 191.
342 Y.-FL Hslao et al. /Neutral and lomc states of hypermetalated potassium compounds
that the bonding distances between the center atom and the surrounding atoms only make small changes in going from the neutral species to ionic species. However, the angles between the alkali atoms undergo quite an appreciable change between these two states. These structure changes are just the geometrical manifestation of the nature of the chemical bonding in the molecules. According to the concept of hyper- metallic bonding proposed by Pople and co-workers, the extra valence electrons, after the completion of the filling of the molecular valance shell in accord- ance to the octet rule, go to the bonding between the metal atoms [ lo- 12 1. In this study the bi-potassium compounds have one extra valence electron for the hypermetallic bonding. Their positive ionic states become closed-shell systems. Apparently, the inter- action strength between the potassium atoms deter- mines their relative distance. Qualitatively speaking, the increase of the positive charge on the potassium atoms and the absence of the extra bonding electron between the potassium atoms as one goes from the neutral to the ionic state induce the large change of the potassium bonding angle.
One can look into the nature of the hypermetallic bonding in a more quantitative way. One good mea-
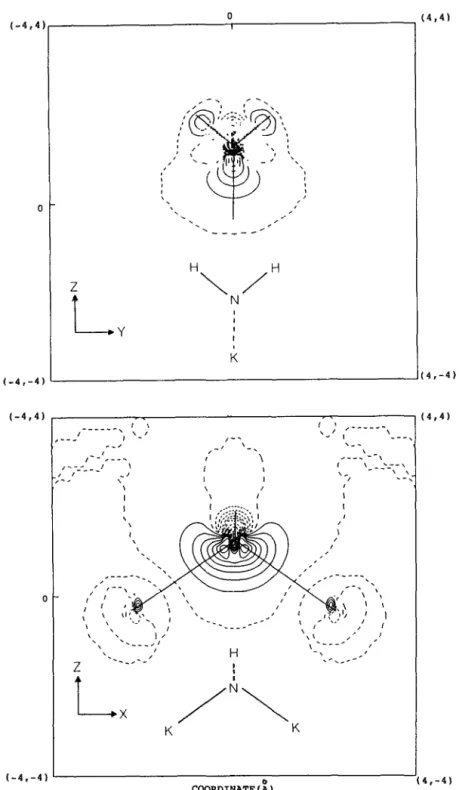
sure for the interaction is the overlap population be- tween potassium atoms. They are: 0.53 (K,), 0.35 (K,OH), 0.0 (K:OH), 0.30 (KZNH2), and 0.0 (K: NH2 ) at the MP2 level. These values suggest the large extent of the sharing of the extra valence elec- tron between the two potassium atoms in the neutral species. There is no such kind of electron-sharing in the ionic states. Another measure is the electron den- sity deformation in the formation of the molecule. Figs. 1 and 2 show the total electron density defor- mation maps of KzOH and K2NH2 on the xz and yz molecular planes, respectively, at the MP2 level. The deformation density is obtained by taking the differ- ence between the molecular density and the density of the sphericalized atoms [ 46 1. There are two gen- eral features emerging from figs. 1 and 2. One is that from the map of the xz molecular plane, there is a minor density deformation between the two potas- sium atoms. This indicates that a chemical bonding, although not a strong one, exists between these two potassium atoms. The second observation is that, comparing the density deformation around the oxy- gen atom or nitrogen atom of the present compounds
with those of Hz0 obtained by Mensching et al. [ 461, one finds that a semi-quantitative similarity in the deformation contours between these three molecular systems centering around the oxygen or nitrogen atom. To be more specific, if one disregards the mag- nitude of the net electron charges on the center atom, one finds that the electron density deformation of the two lone-paired electrons of HZ0 closely resemble the electron density deformation of the two potassium- oxygen bonds of K20H and also that of the two po- tassium-nitrogen bonds of K2NH2. The electron den- sity deformation of the two OH bonds of HZ0 ap- pears to be similar to the single OH bond of KzOH and, not unexpectedly, even is much closer to the two NH bonds of K2NH2. In the Hz0 and KPOH mole- cules, because of the different orientation of the hy- drogen atoms with respect to the oxygen atom, the density deformations around the hydrogen atoms be- tween these two systems show some minor difference in space. Nevertheless, the general features of the electron-density deformation around the oxygen atom are similar to each other. Figs. 1 and 2 suggest that the bonding between the potassium and oxygen or ni- trogen in KzOH or K2NH2 molecule has strong ionic character and is formed between two negatively- charged lone-paired electrons of the oxygen or nitro- gen atom and the positively-charged potassium at- oms. The OH bond of K20H and the NH bond of KzNHz have the same bonding character as the OH bond of HzO.
3.2.2. Harmonic vibrationalfrequencies
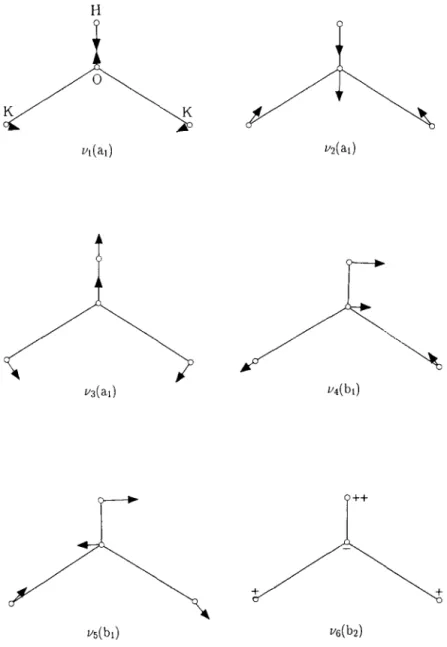
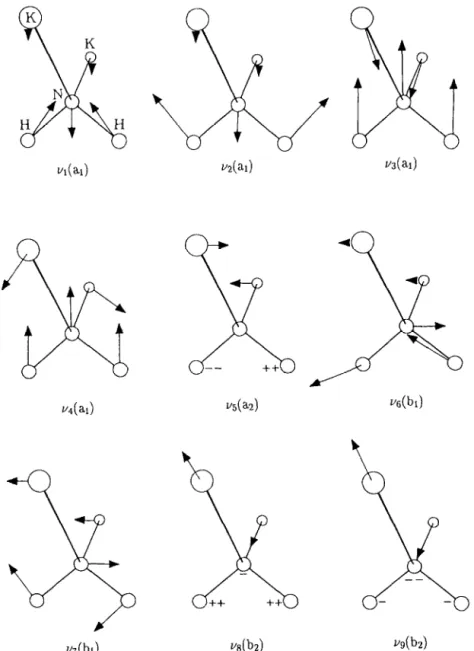
The lower part of table 3 lists the harmonic vibra- tional frequencies and the associated symmetries of the bi-potassium molecular systems. To the best of the authors’ knowledge, the experimental vibrational spectra of these molecular systems have not yet been reported.
Figs. 3 and 4 show the normal mode motions of KzOH and K2NH2, respectively. The corresponding ionic state has the identical normal mode motions. As one changes from the neutral species to their pos- itive ionic states, most of the normal modes do not change their vibrational frequencies much. For the KzOH and Kz OH systems, the b2( vg) vibrational mode almost preserves its vibrational frequency be- tween these two states. The bZ ( vg) mode is an um- brella motion between the H, K, K atoms and the 0
Y.-U’. Hsrao et al. /Neutral and ~otuc states ofhypermetalaredpotasswn compounds 343 _---_____/,-’ I’ 1-j - 11’ I-- , \’ /I I _I_’
_I’
If ’ I’ ’ \_- I 1 I.________---
‘1 i
L
X COORDINATE(i) (3,3) (3,-3) (3,3) (3,-3)Fg. 1. Electron density deformation map of KZOH along the yz and xz planes: long dashed line 0.00 e/A’, short dashed lme - 0.008n e/ ~3,sol~dline+0.008ne/~‘(n=1,2,3 ,... ).
344 Y.-W. Hsiao et al. /Neutral and iomc states of hypermetalatedpotassium compounds C I- I - )- t-4,-4 I-4,4) (4,4) (4,-4) (4,4) ;4,-4) COORDINATE(A)
Fig. 2. Electron density deformation map of KZNH2 along the yz and xz planes: long dashed line 0.00 e/A’, short dashed line -0.008n e/A3,solidline +0.008ne/A3(n=l,2, 3 ,... ).
Y.-W. Hslao et al. /Neutral and iomc ‘states of hypermetalated potassrum compounds 345
u&4
f-i-
-
-_+ +
A
e(h)
dbz)
Fig. 3. Vibratlonal normal modes of KzOH. The lengths and directions of the arrows indicate the relative vibrational magnitudes and their directions, respectively.
atom. Apparently, the ionization of the potassium electron does not alter the interaction force constant perpendicular to the C, axis of the molecule. The same phenomenon happens to the b, (v,) mode of the KzNHz and K: NH* pair. This suggests the electro- static nature of the interaction between the molecu- lar parts of K2 and OH in K,OH or the molecular parts of K2 and NH2 in Kz NH2. This observation is also consistent with the results of the electron density
deformation discussed in section 3.2.3. The largest vibrational frequency shifts are in the modes of b, (v,) and b, ( v5) for KzOH and a2 ( v5) for K2NH2. These modes are mainly involved in the stretching motion of KO in the K20H molecule or the twisting motion between NH2 and NK2 in the K2NH2 molecule. The increase of these normal mode frequencies as one goes from neutral to positive ionic state is a direct result of the increase of the interaction strength between
346 Y -W. Hs~ao et al. /Neutral and lomc states of hypermetalatedpotawum compounds
vl(al)
v2.(4
vh(ad
v3(ad v5(a2)44
4.
++
-
++
w(h)
Fig. 4. Vibratlonal normal modes of K2NH2. The lengths and dil their directions, respectively.
these bonds. The lowest vibrational frequency modes are the a, ( v3) mode of the K,OH/K$ OH pair and the a, ( v4) mode of the K,NH,/K$ NH2 pair. These are all bending motions for the K atom pair and are close to the vibrational frequency of the gaseous Kz, 92.0 cm-‘. This also implies the weak interaction of Kz with the rest part of the molecular complexes in these species.
4d
eOz)
-ections of the arrows mdicate the relatwe wbrational magnitudes and
3.2.3.
Bond dissociation energies and ionization
potentials
The lower halves of tables 6 and 7 show the bond dissociation energies and ionization potentials of the bi-potassium molecular systems. For the hydroxyl systems, the agreement of the dissociation energies between the theoretical results and the experimental values is very good. There are no reports on the ex-
Y-W. Hslao et al /Neutral and lonlcstates ofhypermetalatedpotassrum compounds 341 perimental measurements of the amide molecules yet.
The bond dissociation energies of the neutral species are always much lower than the corresponding ionic states. This reflects that the ion-electrical dipole in- teraction is much stronger than the hypermetallic bonding in these species. Note that the bonding en- ergy of the pure covalent bond of K2 is only 11.9 kcal/ mol. For the adiabatic ionization potentials, the agreement between the theoretical values and exper- imental data is poor. In an attempt to calculate the ionization potentials of some hypermetalated com- pounds of Na and Li, Wurthwei et al. also encoun- tered the same poor agreement between the results of theory and experiments [ 121. Further work is needed to clarify these situations.
4. Conclusions
The following conclusions can be drawn from the results of the present study of the neutral and ionic states of the potassium-containing hypermetalated compounds and molecular complexes.
(i ) Evidences suggest that electron correlation ef- fect, electrostatic interaction and charge polarization of the alkali atom are all play a part in stabilizing the KHZ0 and KNH3 complexes. The charge-transfer process is comparatively less important in these cases.
(ii) In the cases of KzOH and K2NH2, the two po- tassium atoms that are responsible for the so-called hypermetallic bonding are spatially oriented to the two lone-paired electrons of the center oxygen or ni- trogen atom. A weak bonding feature is developed between the two potassium atoms.
(iii) The majority of the geometrical and vibra- tional quantities of the neutral species is close to those of their ionic species studied in this report. Due to the different nature of the bonding as one changes from the neutral to ionic state, only those quantities directly related with this local change of the bonding undergo major changes.
Acknowledgement
This work is financially supported by the National Science Council, ROC. The authors are grateful for the help of Professor Yu Wang in the preparation of the electron density deformation maps.
References
[ 1 ] V.A. Nicely and J.L. Dye, J. Chem. Phys. 52 ( 1970) 4795. [2] M. Trenary, H.F. Schaefer III and P.A. Kollman, J. Am.
Chem. Sot. 99 ( 1977) 3885.
[ 31 M. Trenary. H.F. Schaefer III and P.A. Kollman, J. Chem. Phys. 68 (1978) 4047.
[4] L.A. Curtiss and D.J. Frurip, Chem. Phys. Letters 75 (1980) 69.
[5] J. Bentley and I. Carmichael, J. Phys. Chem. 85 (1981) 3821.
[6] J. Bentley. J. Am. Chem. Sot. 104 (1982) 2754.
[ 71 J.Q. Broughton and P.S. Bagus, J. Chem. Phys. 77 (1982) 3627.
[ 81 L.A. Curtiss and J.A. Pople, J. Chem. Phys. 82 (1985) 4230. [9] L.A. Curtiss, E. Kraka, J. Gauss and D. Cremer, J. Phys.
Chem. 91 (1987) 1080.
[ lo] P. van R. Schleyer, E.-U Wurthwein and J.A. Pople, J. Am. Chem. Sot. 104 (1982) 5839.
[ I I ] P. van R. Schleyer, E.-U. Wurthwein, E. Kaufmann, T. Clark and J.A. Pople, J. Am. Chem. Sot. 105 (1983) 5930. [ 121 E.-U. Wurthwein, P. van R. Schleyer and J.A. Pople. J. Am.
Chem Sot. 106 (1984) 6973.
131 A.E. Reed and F. Weinhold. J. Am. Chem. Sot. 107 ( 1985) 1919.
141 C.P. Schulz, R. Haugstatter, H.U. Tittes and I.V. Hertel, Phys. Rev. Letters 57 (1986) 1703.
151 C.P. Schulz, R. Haugstatter. H.U. Tittes and I.V. Hertel, Z. Physik D 10 ( 1988) 279.
161 C.H. Wu, H. Kudo and H.R. Ihle. J. Chem. Phys. 70 ( 1979) 1815.
171 P.D. Dao. K.I. Peterson and A.W. Castleman Jr., J. Chem. Phys. 80 (1984) 563.
18 ] H.G. Limberger and T.P. Martin, J. Chem. Phys. 90 ( 1989) 2979.
191 T.-C. Kuan. R.-C. Jiang and T.-M. Su, J. Chem. Phys. 92 (1990) 2553.
[ 201 R.-C. Jiang and T.-M. Su, Chem. Phys. Letters 18 1 ( 1991) 373.
[ 2 1 ] R.H. Hauge, P.F. Meier and J.L. Margrave, Ber. Bunsenges. Physik. Chem. 82 (1978) 102.
[ 221 L. Manceron, A. Loutelher and J.P. Perchard, Chem. Phys. 92 (1985) 75.
[23] S. Suzer and L. Andrews, J. Am. Chem. Sot. 109 (1987) 300
[ 241 S. Suzer and L. Andrews, Chem. Phys. Letters 140 ( 1987) 300.
[25] SK. Searles and P. Kebarle, Can. J. Chem. 47 (1969) 2619. [26] W.R. Davidson and P. Kebarle. J. Am. Chem. Sot. 98
(1976) 6125.6133.
[27] A.W. Castleman Jr., Chem. Phys. Letters 53 (1978) 560. [28] L.S. Kudm, M.F. Butman and K.S. Krasnov, Zh. Strukt.
Khim. 26 (1985) 65.
[ 291 H. Kstenmacher, H. Popkie and E. Clementi. J. Chem. Phys. 59 (1973) 5842.
348 Y.-W Hslao et al /Neutral and lomc states of hypermetalatedpotasscum compounds [ 3 I] M.J. Frisch, M. Head-Gordon, G. W. Trucks, J.B. Foresman,
H.B. Schlegel, K. Raghavachari, M.A. Robb, J.S. Binkley, C. Gonzalez, D.J. DeFrees, D.J. Fox, R.A. Whiteside, R. Seeger, C.F. Melius, J. Baker, R.L. Martin, L.R. Kahn, J.J.P. Stewart, S. Topiol and J.A. Pople. GAUSSIAN 90 (Gaussian Inc., Pittsburgh, PA, 1990).
[ 321 C. Salez and A. Veillard, Theoret. Chim. Acta 11 ( 1968) 441.
[33] S. Huzmaga, J. Chem. Phys. 42 (1965) 1293.
[ 341 L. Gianolio, R. Pavani and E. Clementi, Gazz. Chum. Ital. 108 (1978) 181.
[ 351 F.B. Van Duijneveldt. IBM Tech. Report RJ 945, December 1971.
[36] A.J.H. Wachters, J. Chem. Phys. 52 (1970) 1033. [ 371 M.L. Klein. J.D. Goddard and D.G. Boubds, J. Chem. Phys.
75 (1981) 3909.
[38] W.B. England, J. Chem. Phys. 68 (1978) 4896. [39] G. Strey, J. Mol. Spectry. 24 (1967) 87.
[40] J.L. Duncan and I.M. Mills, Spectrochim. Acta 20 (1964) 523.
[ 411 J.O. Hirschfelder, C.F. Curtiss, R.B. Bud, Molecular theory of gases and liquids (Wiley, New York, 1954).
[42] T.M. Miller and B. Bederson, Advan. At. Mol. Phys. 13 (1977) 1.
[43] G. Henberg, Molecular spectra and molecular structure, Vol. 2. Infrared and Raman spectra of polyatomic molecules (Van Nostrand, New York, 1945 ) .
[ 441 C.E. Moore, Atomic energy levels, Vol. 1, NSRD-NBS (US GPO, Washington, 197 I ).
[45] M. Broyer, J. Chevaleyre, G. Delacretaz, S. Martin and L. Woste, Chem. Phys. Letters 99 (1983) 206.
[46] L. Menschmg, W. Von Niessen, P. Valtazanos, K. Ruedenberg and W.H.E. Schwarz. J. Am. Chem. Sot. 111 (1989) 6933.