Drug discovery for DNA break repair system by screening from TCM database and molecular
dynamics approach
Tung-Ti Changa1, Mao-Feng Sunb1, Kuan-Chung Chenc, Yung-Hao Wongc, Shun-Chieh Yangc, Hsin-Yi Chenc, Fuu-Jen Tsaide, Mark Fisherf, Chun-Lin Leeg, Wen-Chang Fanggand Calvin Yu-Chian Chencdfh*
a
Department of Chinese Pediatrics, China Medical University Hospital, Taichung, Taiwan ROC;bDepartment of Acupuncture, China Medical University Hospital, Taichung, Taiwan ROC;cLaboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical University, Taichung 40402, Taiwan ROC;dDepartment of Bioinformatics, Asia University, Taichung 41354, Taiwan ROC;eDepartment of Medical Genetics, Pediatrics and Medical Research, College of Chinese Medicine, China Medical University, Taichung 40402, Taiwan ROC;fHarvard-MIT Division of Health Sciences and Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA;gDepartment of Business Administration, National Taipei University, Taipei, Taiwan ROC;hComputational and Systems
Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA (Received 19 November 2010; final version received 13 December 2010)
In search for novel anti-tumour agents targeting non-homologous end joining pathway, we conducted a virtual screening using traditional Chinese medicine (TCM) on Ku heterodimer which plays an important role on DNA break repair system. Docking results gave glycyrrhizic acid and macedonoside C as potential TCM candidates. Both glycyrrhizic acid and macedonoside C were docked onto the Ku heterodimer with high-predicted binding affinities as evidenced in their Dock Score. These two ligands also interact with key residues which have been previously shown to influence Ku DNA-binding affinity. Both compounds show consistent hydrogen bonding interactions with key residues throughout the 10- ns dynamics simulation run. Furthermore, glycyrrhizic acid and macedonoside C are able to form additional hydrogen bond interactions with positively charged Ku heterodimer surface. Such interactions strengthen the binding interaction of the top two TCM compounds with Ku heterodimer. Glycyrrhizic acid and macedonoside C are products of licorice (Glycyrrhiza glabra), which is a commonly used herb in TCM formulations. Overall, we report glycyrrhizic acid and macedonoside C as potential anti-tumour agents.
Keywords: traditional Chinese medicine; docking; DNA break repair system; molecular dynamics; anti-tumour
1. Introduction
Non-homologous end joining (NHEJ) is the key mechanism responsible for repairing DNA double-strand breaks, the most lethal damage to genetic materials in a cell. This repairing mechanism is active in all stages of cell cycle, in contrast to homologous joining which is inactive in cell division [1]. In NHEJ, the repairing process starts with the binding of Ku protein (or also known as Ku86), to DNA end, preventing further damages to DNA sequence. More importantly, Ku protein functions as a molecular scaffold for other recruited NHEJ proteins, including DNA-dependent protein kinase catalytic subunit, Artemis, XRCC4 and Ligand IV, to bind (reviewed in [2]).
The Ku protein is a heterodimer made up of Ku70 (70 kDa) and Ku80 (80 kDa) subunits. These two subunits form a unique ring-like structure that encircles DNA with high affinity but without apparent sequence specificity [3,4]. The Ku heterodimer protein crystal also reveals that the protein does not interact with any DNA bases [3]. Furthermore, in addition to be a crucial member in NHEJ,
Ku proteins have also been found to function at telomere maintenance [5,6].
In the past, several studies were conducted to relate Ku protein with radiosensitivity. In mice model, deletion of Ku70 or Ku80 in a deficient level of p53 condition shows hypersensitivity to ionising radiation [7]. In head and neck cancer cell lines, the most radioresistant cell line has been associated with an increase expression of Ku70 [8]. On the basis of these studies, it can be deduced that disruption in Ku DNA-binding affinity could lead to hypersensitivity for ionising radiation. Moreover, it is possible that a therapeutic agent targeting Ku protein in cancer cells could enhance radiation responsive in radiation therapy.
We present a study focusing on designing novel anti-tumour agents that target NHEJ mechanism. More specifi-cally, our focus is on hindering binding and translocating of Ku heterodimer to DNA ends using molecules identified from the traditional Chinese medicine (TCM). It is not new that potential anti-tumour compounds, such as gallic acid, curcumin and quercetin, could be determined from TCM through various biochemical assays [9 – 11]. We, however,
ISSN 0892-7022 print/ISSN 1029-0435 online
q2011 Taylor & Francis
DOI: 10.1080/08927022.2010.548388 http://www.informaworld.com
*Corresponding author. Email: [email protected]; [email protected] Vol. 37, No. 9, August 2011, 804–811
used an in silico approach and used the world’s largest TCM database (TCM Database@Taiwan, http://tcm.cmu. edu.tw) for virtual screening. Molecular dynamic simu-lations were also used to investigate interactions found between Ku heterodimer and TCM molecules. Both virtual screening by molecular docking and molecular dynamic simulation were commonly applied in designing drugs targeting protein such as influenza haemagglutinin, phosphodiesterase five, epidermal growth factor receptor HER2, HIV enzymes and many more [12 – 22]. We are confident in using these two methods for finding potential Ku heterodimer inhibitors.
2. Materials and methods 2.1 Protein preparation
The crystal structure for Ku heterodimer was downloaded from Protein Data Bank (http://www.pdb.org), PDB ID: 1JEY [3]. Crystal water molecules, non-bonded heteroa-toms and synthetic DNA construct were removed from the protein crystal. The missing hydrogen atoms were added. The CHARMm force field was applied to the Ku heterodimer prior to any molecular simulation step. The binding site for docking of TCM molecules was set to the DNA-binding interface. In a study, Chen et al. [23] demonstrated that acetylation at Lys282, Lys338, Lys539 and Lys542 of Ku70 can suppress the DNA end binding activity. Mutagenesis study done by the same group also showed similar results. Hence, we specifically set the binding site covering Lys282 and Lys338 in an attempt to achieve the same effect. As for Lys539 and Lys542, both residues lie in COOH-terminal linker region which was not solved in 1JEY crystal.
2.2 Docking
A total of 20,000 natural products were downloaded from the TCM Database@Taiwan (http://tcm.cmu.edu.tw/) and were ionised, according to the physiological setting, using Discovery Studio 2.5. These compounds were also filtered using ADMET module in Discovery 2.5 to remove any potentially toxic compounds. TCM compounds that passed the ADMET screen were docked into the Ku heterodimer binding site using LigandFit module of Discovery Studio 2.5 [24]. In LigandFit, ligand conformations were generated by Monte Carlo simulation. The conformations were placed into the binding site using a shape-based matching method. The protein was held rigid throughout docking whereas the ligands were allowed to orientate inside the binding pocket for sampling different binding poses. Candidate ligand poses were energetically mini-mised using Smart Minimizer. Dock Score (DS) was used as the primary scoring function for ranking ligands poses. LigScore (LigScore1 and LigScore2) and piecewise linear potential (-PLP and -PLP2) were also calculated for
predicting binding affinities, but they were not used in ranking the TCM compounds. DS was used to calculate ligand – receptor interaction energy and ligand internal energy [24]. LigScore was used to evaluate receptor – ligand binding affinities by computing softened van der Waal descriptor and polar surface area descriptors [25]. The two versions of LigScore differ in that LigScore2 introduces desolvation penalty. PLP predicts ligand binding affinity by calculating hydrogen bond interactions between hydrogen bond donor, hydrogen bond acceptor and non-polar atom types [26]. The PLP2 function has an additional scaling factor that is based on the angle formed between receptor and ligand atoms.
2.3 Molecular dynamics simulation
The selected protein ligand complexes were taken for molecular dynamics simulation. The molecular dynamics simulation was conducted using the simulation module of Discovery Studio 2.5. Each complex was solvated in a water box using periodic boundary condition. The initial minimisation process consisted of two cycles of minimis-ation using 2000 steps of Steepest Descend and 2000 steps of Conjugate Gradient. The heating step was performed for 20 ps with a time step of 1 fs. The protein – ligand complex was heated from 50 to 310 K without constraint. The equilibration step was run for 100 ps at 310 K. The production run was conducted for 10 ns in NVT (constant temperature dynamics using Berendsen weak coupling method) condition at 310 K. The SHAKE algorithm was applied to constraint binding to hydrogen atoms. Root mean square deviation (RMSD) was calculated for both protein – ligand complexes and ligands. Total energy of the protein – ligand systems was also calculated.
3. Results
3.1 Docking of TCM compounds generates a list of potential candidates
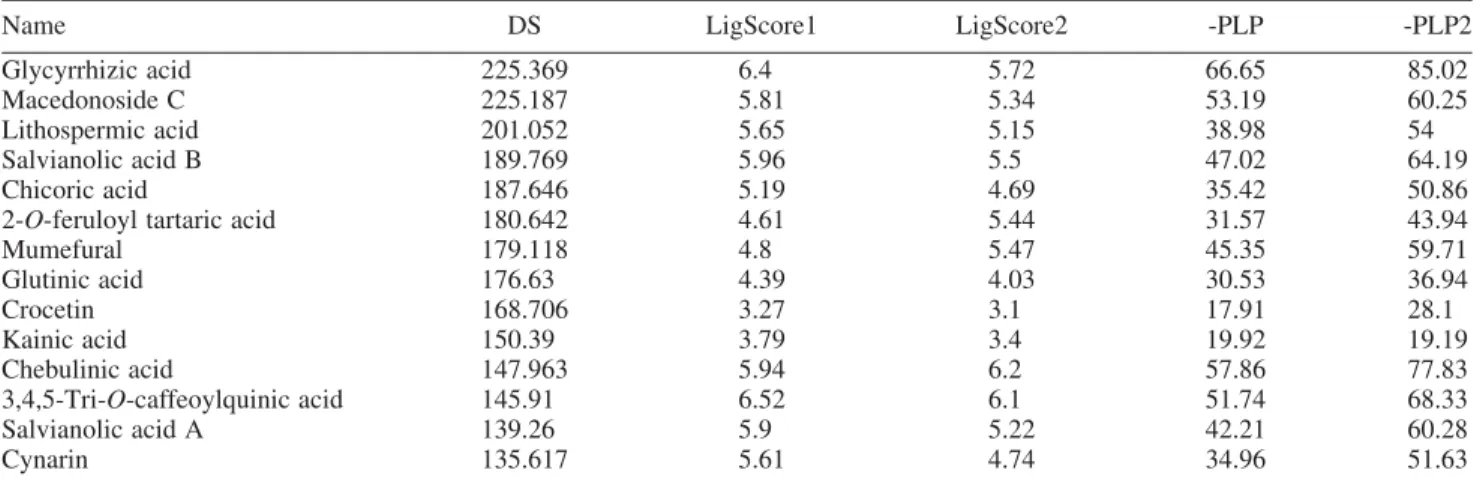
To screen for TCM compounds that can inhibit Ku heterodimer function, we performed molecular docking. Scoring function, DS, was used as the primary criterion for ranking ligands and their associated poses. We used DS because this scoring function results have been previously demonstrated for having positive correlation with com-pound bioactivities [24]. The docking result, ranked in descending order according to DS, is shown in Table 1. Glycyrrhizic acid, macedonoside C and lithospermic acid all have extremely well-docking results. Although not used in ranking the molecules, the top three TCM molecules all show very reasonable LigScore1, LigScore2, -PLP and -PLP2 values. Most significantly, glycyrrhizic acid has leading -PLP and -PLP2 values suggesting that hydrogen bond interactions are critical for binding of glycyrrhizic acid with Ku heterodimer. The -PLP for lithospermic
acid is significantly lower than for glycyrrhizic acid and macedonoside C. Thus, we did not conduct further analysis on lithospermic acid.
In addition to the high predicted binding affinity, there are other characteristics that make glycyrrhizic acid and macedonoside C as very potential Ku inhibitors. Both compounds carry negative charges in physiological pH. This characteristic favours the binding with Ku hetero-dimer as the DNA-binding surface has a positively charged lining. Furthermore, as shown in the protein synthetic DNA crystal (1JEY), the Ku ring for DNA is narrower than the
actual diameter of DNA, and protein residues are in close proximity with DNA [3]. In particular, Arg403 of Ku70 and Arg400 are found in the DNA minor groove with less than 6 A˚ away from DNA. Hence, the binding of the TCM molecules to Ku protein inhibits the protein – DNA interaction.
Both glycyrrhizic acid and macedonoside C are extracts of licorice (Glycyrrhiza glabra). Glycyrrhizic acid was determined with a broad spectrum of antiviral activities [27,28] and was also tested for anti-tumour activity [29]. However, this is the first study that investigates the effects of Table 1. TCM molecules docking result.
Name DS LigScore1 LigScore2 -PLP -PLP2
Glycyrrhizic acid 225.369 6.4 5.72 66.65 85.02
Macedonoside C 225.187 5.81 5.34 53.19 60.25
Lithospermic acid 201.052 5.65 5.15 38.98 54
Salvianolic acid B 189.769 5.96 5.5 47.02 64.19
Chicoric acid 187.646 5.19 4.69 35.42 50.86
2-O-feruloyl tartaric acid 180.642 4.61 5.44 31.57 43.94
Mumefural 179.118 4.8 5.47 45.35 59.71 Glutinic acid 176.63 4.39 4.03 30.53 36.94 Crocetin 168.706 3.27 3.1 17.91 28.1 Kainic acid 150.39 3.79 3.4 19.92 19.19 Chebulinic acid 147.963 5.94 6.2 57.86 77.83 3,4,5-Tri-O-caffeoylquinic acid 145.91 6.52 6.1 51.74 68.33 Salvianolic acid A 139.26 5.9 5.22 42.21 60.28 Cynarin 135.617 5.61 4.74 34.96 51.63
Note: DS, Dock Score; PLP, piecewise linear potential.
glycyrrhizic acid and macedonoside C on Ku protein of DNA repair pathway.
3.2 Glycyrrhizic acid and macedonoside C bind to key area on Ku heterodimer and form extensive hydrogen bond network with binding site residues
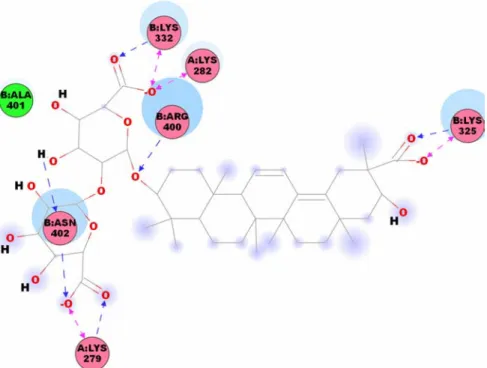
We have specified the binding site to regions covering Lys282 and Lys338 of Ku70 that were demonstrated to influence DNA-binding affinity. As illustrated in Figure 1, glycyrrhizic acid has hydrogen bond interaction with Arg318, Glu335 and Lys338 of Ku70 (Chain A), and Lys291 and Arg486 of Ku80 (Chain B). Macedonoside C has a slight different docking location from glycyrrhizic acid, but also has hydrogen bond interactions with Lys279 and Lys282 of Ku70 (Chain A), and Lys325, Lys332, Arg400 and Arg402 of Ku80 (Chain B; Figure 2). On the basis of the docking result, both TCM products have interactions with key residue, Lys282 or Lys338.
3.3 Molecular dynamic simulations show persistent hydrogen bonding interactions between top two TCM molecules and Ku heterodimer
Molecular docking provides a static view of protein – ligand interaction but does not provide any information of protein – ligand interaction in motion. Thus, we performed molecular dynamics simulation on Ku heterodimer in complex with glycyrrhizic acid or macedonoside C. The RMSD trajectories for the whole protein – ligand
complexes and for the ligands are shown in Figure 3. Significant difference is seen for whole molecule RMSD, but the top two compounds still share similar ligand RMSD trend. However, a close examination of the glycyrrhizic-and macedonoside C-bound Ku heterodimers reveals that there is no significant structural change at the binding Figure 2. Structure and docking pose of macedonoside C (colour online).
0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0 0 1 2 3 4 5 6 7 8 9 10 0.2 0.4 0.6 0.8 1.0 1.2 1.4 Glycyrrhizic acid Macedonoside C Whole molecule RMSD Time (ns) Ligand RMSD
Figure 3. Whole molecule RMSD of Ku – ligand complexes and ligand RMSD for TCM candidates.
region. Thus, binding of macedonoside C to Ku protein should not be the reason behind a smaller deviation from the starting structure and a lower total energy state in comparison with glycyrrhizic-bound Ku complex (Figure 4). Both the RMSD graph and the energy trajectory are ways to show that the protein – ligand complex reaches equilibrium during the simulation run. On the basis of this, the two TCM ligands all reach equilibrium during the simulation.
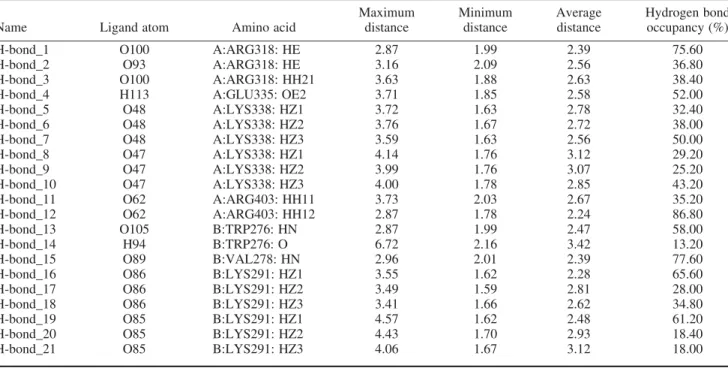
Analysis of glycyrrhizic acid hydrogen bond inter-action during the 10 ns of molecular dynamics simulation shows that all of the interactions observed from docking still persist during the molecular dynamics simulation
(Table 2). Additional interactions with Arg403 and Val278 are also observed. It is also apparent that there is at least one interaction between key residue Lys338 and glycyrrhizic acid during any given time of simulation run. Similarly, macedonoside C has hydrogen bond interactions with most of the residues observed from docking (Table 3). The exception is for the interaction with Lys282, which is not detected in the simulation run.
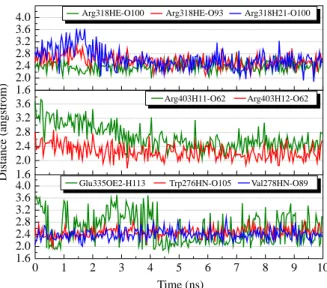
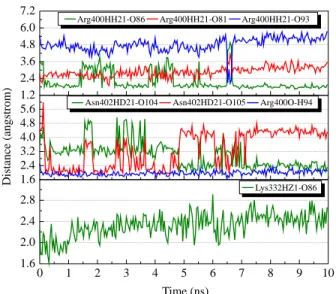
We selected hydrogen bond interactions between glycyrrhizic acid and Arg318, Arg403, Glu335, Trp276 and Val278 for further investigation. The separating distances between the ligand and the residues are shown in Figure 5. The ligand interactions with Arg318, Trp276 and Val278 are the most stable during the interaction run, with averaging distance around 2.5 A˚ . As illustrated in Figure 5, most of the interactions have similar trend that gradually becomes more stable after 5 ns of simulation run. This is most significant for the interaction of glycyrrhizic acid with Arg318 (Figure 5). For the interaction with Glu335, although there is a drop in interacting distance at the beginning of the simulation (Figure 6(a) and (b)), there is still no stable interaction, even to the end of simulation (see Supplementary Video 1). For macedonoside C, except for Lys279 and Lys325 that both have at least one hydrogen bond interaction with the ligand at any time of simulation, we selected all other interactions for further analysis. During the simulation run, some of the ligand interactions with Arg400 and Asn402 are among the most stable during the simulation run (Figure 7). For interaction between macedonoside C and Arg400, the most significant disruption occurs around 6.5 ns. As illustrated in Figure 8,
Table 2. Hydrogen bond frequency of glycyrrhizic acid during molecular dynamics simulation.
Name Ligand atom Amino acid
Maximum distance Minimum distance Average distance Hydrogen bond occupancy (%)
H-bond_1 O100 A:ARG318: HE 2.87 1.99 2.39 75.60
H-bond_2 O93 A:ARG318: HE 3.16 2.09 2.56 36.80
H-bond_3 O100 A:ARG318: HH21 3.63 1.88 2.63 38.40
H-bond_4 H113 A:GLU335: OE2 3.71 1.85 2.58 52.00
H-bond_5 O48 A:LYS338: HZ1 3.72 1.63 2.78 32.40
H-bond_6 O48 A:LYS338: HZ2 3.76 1.67 2.72 38.00
H-bond_7 O48 A:LYS338: HZ3 3.59 1.63 2.56 50.00
H-bond_8 O47 A:LYS338: HZ1 4.14 1.76 3.12 29.20
H-bond_9 O47 A:LYS338: HZ2 3.99 1.76 3.07 25.20
H-bond_10 O47 A:LYS338: HZ3 4.00 1.78 2.85 43.20
H-bond_11 O62 A:ARG403: HH11 3.73 2.03 2.67 35.20
H-bond_12 O62 A:ARG403: HH12 2.87 1.78 2.24 86.80
H-bond_13 O105 B:TRP276: HN 2.87 1.99 2.47 58.00
H-bond_14 H94 B:TRP276: O 6.72 2.16 3.42 13.20
H-bond_15 O89 B:VAL278: HN 2.96 2.01 2.39 77.60
H-bond_16 O86 B:LYS291: HZ1 3.55 1.62 2.28 65.60
H-bond_17 O86 B:LYS291: HZ2 3.49 1.59 2.81 28.00
H-bond_18 O86 B:LYS291: HZ3 3.41 1.66 2.62 34.80
H-bond_19 O85 B:LYS291: HZ1 4.57 1.62 2.48 61.20
H-bond_20 O85 B:LYS291: HZ2 4.43 1.70 2.93 18.40
H-bond_21 O85 B:LYS291: HZ3 4.06 1.67 3.12 18.00
0 1 2 3 4 5 6 7 8 9 10 –53,200 –53,000 –52,800 –52,600 –52,400 –52,200 –52,000 –51,800 –51,600 –51,400 –51,200 –51,000 –50,800 Glycyrrhizic acid Macedonoside C T otal ener gy (kcal/mol) Time (ns)
Figure 4. Energy trajectory of glycyrrhizic acid and macedonoside C.
Arg400 has a temporary shift in conformation. This change, however, is later reverted back to the confor-mation seen in Figure 8(a) and Supplementary Video 2. As for interaction with Lys282, although there is no hydrogen bonding observed during simulation run, macedonoside C is found above Lys282 that can still hinder Lys282 from interacting with DNA (Figure 8 and Supplementary Video 2).
Overall, several stable hydrogen bond interactions between the two top ranking TCM molecules and the binding site residues are observed. Glycyrrhizic acid forms stable interactions with Lys338, whereas macedonoside C forms interactions with residues close to Lys282 and effectively shelter Lys282. With stable and continuous hydrogen bond networks that could lock glycyrrhizic acid and macedonoside C to Ku heterodimer, we suggest that
Figure 6. Snapshots of glycyrrhizic acid in Ku binding site at (a) 2.50 ns and (b) 5.92 ns (colour online).
1.6 2.0 2.4 2.8 3.2 3.6 4.0 1.6 2.0 2.4 2.8 3.2 3.6 0 1 2 3 4 5 6 7 8 9 10 1.6 2.0 2.4 2.8 3.2 3.6 4.0
Arg318HE-O100 Arg318HE-O93 Arg318H21-O100
Distance (angstrom)
Arg403H11-O62 Arg403H12-O62
Glu335OE2-H113 Trp276HN-O105 Val278HN-O89
Time (ns)
Figure 5. Hydrogen bond distances of glycyrrhizic acid (colour online).
Table 3. Hydrogen bond frequency of macedonoside C during molecular dynamics simulation.
Name Ligand atom Amino acid
Maximum distance Minimum distance Average distance Hydrogen bond occupancy (%)
H-bond_1 O105 A:LYS279: HZ1 3.57 1.67 2.31 67.20
H-bond_2 O105 A:LYS279: HZ2 3.56 1.66 2.48 52.80
H-bond_3 O105 A:LYS279: HZ3 3.53 1.62 2.48 48.40
H-bond_4 O48 B:LYS325: HZ1 3.41 1.70 2.69 26.40
H-bond_5 O48 B:LYS325: HZ2 3.41 1.68 2.48 48.00
H-bond_6 O48 B:LYS325: HZ3 3.51 1.71 2.35 63.20
H-bond_7 O49 B:LYS325: HZ1 3.14 1.66 2.31 58.00
H-bond_8 O49 B:LYS325: HZ2 3.23 1.61 2.25 67.20
H-bond_9 O49 B:LYS325: HZ3 3.18 1.73 2.39 61.60
H-bond_10 O86 B:LYS332: HZ1 2.91 1.62 2.33 77.20
H-bond_11 H94 B:ARG400: O 2.49 1.70 1.97 100.00
H-bond_12 O86 B:ARG400: HH21 4.97 1.64 2.18 80.00
H-bond_13 O81 B:ARG400: HH21 4.13 1.89 2.88 12.80
H-bond_14 O93 B:ARG400: HH21 5.73 2.06 4.81 1.20
H-bond_15 O104 B:ASN402: HD21 5.14 1.84 3.02 19.20
these two TCM molecules can mimic the effect of acetylation and be potential anti-tumour agents.
4. Conclusion
We report two TCM compounds, glycyrrhizic acid and macedonoside C, as potential anti-tumour agents targeting Ku heterodimer, a key protein in DNA repair pathway. There are several important key features that make glycyrrhizic acid and macedonoside C promising Ku inhibitors. First, in physiological pH, both compounds are negatively charged and can have favourable interaction with positively charged Ku heterodimer DNA-binding interface. Both compounds interact with key residues that have been experimentally shown to influence DNA-binding affinity. Lastly, glycyrrhizic acid and macedonoside C have high predicted affinity for Ku heterodimer, as evidenced in their DS and form stable interaction with key binding site residues during the molecular dynamics simulation run. We, therefore, present these two compounds as potential anti-tumour agents.
Acknowledgements
This research was supported by grants from the National Science Council of Taiwan (NSC 99-2221-E-039-013), China Medical University (CMU98-TCM, CMU99-TCM, CMU99-S-02) and Asia University (CMU98-ASIA-09). This study was also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH99-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH99-TD-C-111-005). We are grateful to the National Center of High-performance Computing for the computer time and facilities.
Note
1. Equal contribution.
References
[1] M.H. Yun and K. Hiom, CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle, Appl. Opt. 459 (2009), pp. 460 – 463.
[2] B.L. Mahaney, K. Meek, and S.P. Lees-Miller, Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining, Biochem. J. 417 (2009), pp. 639 – 650.
[3] J.R. Walker, R.A. Corpina, and J. Goldberg, Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair, Nature 412 (2001), pp. 607 – 614.
[4] A. Rivera-Calzada, L. Spagnolo, L.H. Pearl, and O. Llorca, Structural model of full-length human Ku70 – Ku80 heterodimer and its recognition of DNA and DNA-PKcs, EMBO Rep. 8 (2007), pp. 56 – 62
[5] H.L. Hsu, D. Gilley, E.H. Blackburn, and D.J. Chen, Ku is associated with the telomere in mammals, Proc. Natl Acad. Sci. USA 96 (1999), pp. 12454 – 12458.
[6] G.B. Celli, E.L. Denchi, and T. de Lange, Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination, Nat. Cell Biol. 8 (2006), pp. 885 – 890. [7] D.S. Lim, H. Vogel, D.M. Willerford, A.T. Sands, K.A. Platt, and P. Hasty, Analysis of ku80-mutant mice and cells with deficient levels of p53, Mol. Cell. Biol. 20 (2000), pp. 3772 – 3780.
1.2 2.4 3.6 4.8 6.0 7.2 1.6 2.4 3.2 4.0 4.8 5.6 0 1 2 3 4 5 6 7 8 9 10 1.6 2.0 2.4 2.8 Distance (angstrom)
Arg400HH21-O86 Arg400HH21-O81 Arg400HH21-O93
Asn402HD21-O104 Asn402HD21-O105 Arg400O-H94
Time (ns)
Lys332HZ1-O86
Figure 7. Hydrogen bond distances of macedonoside C (colour online).
Figure 8. Snapshots of macedonoside C in Ku binding site at (a) 6.04 ns and (b) 6.56 ns (colour online).
[8] H.W. Chang, S.Y. Kim, S.L. Yi, S.H. Son, Y. Song do, S.Y. Moon, J.H. Kim, E.K. Choi, S.D. Ahn, S.S. Shin, K.K. Lee, and S.W. Lee, Expression of Ku80 correlates with sensitivities to radiation in cancer cell lines of the head and neck, Oral Oncol. 42 (2006), pp. 979 – 986.
[9] C.C. Su, J.S. Yang, C.C. Lu, J.H. Chiang, C.L. Wu, J.J. Lin, K.C. Lai, T.C. Hsia, H.F. Lu, M.J. Fan, and J.G. Chung, Curcumin inhibits human lung large cell carcinoma cancer tumour growth in a murine xenograft model, Phytother. Res. 24 (2010), pp. 189 – 192. [10] C. Lo, T.Y. Lai, J.H. Yang, J.S. Yang, Y.S. Ma, S.W. Weng, Y.Y.
Chen, J.G. Lin, and J.G. Chung, Gallic acid induces apoptosis in A375.S2 human melanoma cells through caspasedependent and -independent pathways, Int. J. Oncol. 37 (2010), pp. 377 – 385. [11] S.H. Wu, L.W. Hang, J.S. Yang, H.Y. Chen, H.Y. Lin, J.H. Chiang,
C.C. Lu, J.L. Yang, T.Y. Lai, Y.C. Ko, and J.G. Chung, Curcumin induces apoptosis in human non-small cell lung cancer NCI-H460 cells through ER stress and caspase cascade- and mitochondria-dependent pathways, Anticancer Res. 30 (2010), pp. 2125 – 2133. [12] C.Y. Chen, Virtual screening and drug design for PDE-5 receptor
from traditional Chinese medicine database, J. Biomol. Struct. Dyn. 27 (2010), pp. 627 – 640.
[13] C.Y. Chen, Y.H. Chang, D.T. Bau, H.J. Huang, F.J. Tsai, C.H. Tsai, and C.Y.C. Chen, Ligand-based dual target drug design for H1N1: Swine flu – a preliminary first study, J. Biomol. Struct. Dyn. 27 (2009), pp. 171 – 178.
[14] H.J. Huang, K.J. Lee, H.W. Yu, C.Y. Chen, C.H. Hsu, H.Y. Chen, F.J. Tsai, and C.Y.C. Chen, Structure-based and ligand-based drug design for HER 2 receptor, J. Biomol. Struct. Dyn. 28 (2010), pp. 23 – 37.
[15] H.J. Huang, K.J. Lee, H.W. Yu, H.Y. Chen, F.J. Tsai, and C.Y. Chen, A novel strategy for designing the selective PPAR agonist by the ‘sum of activity’ model, J. Biomol. Struct. Dyn. 28 (2010), pp. 187 – 200.
[16] T.T. Chang, H.J. Huang, K.J. Lee, H.W. Yu, H.Y. Chen, F.J. Tsai, M.F. Sun, and C.Y. Chen, Key features for designing phosphodi-esterase-5 inhibitors, J. Biomol. Struct. Dyn. 28 (2010), pp. 309 – 321.
[17] C.D. Yoo, S.C. Kim, and S.H. Lee, Molecular dynamics simulation study of probe diffusion in liquid n-alkanes, Mol. Simul. 35 (2009), pp. 241 – 247.
[18] D.X. Li, G.L. Chen, B.L. Liu, and Y.S. Liu, Molecular simulation of b-cyclodextrin inclusion complex with 2-phenylethyl alcohol, Mol. Simul. 35 (2009), pp. 199 – 204.
[19] F. Luan, H.T. Liu, Y. Gao, and X.Y. Zhang, QSPR model to predict the thermal stabilities of second-order nonlinear optical (NLO) chromophore molecules, Mol. Simul. 35 (2009), pp. 248 – 257. [20] J.H. Jing, G.Z. Liang, H. Mei, S.Y. Xiao, Z.N. Xia, and Z.L. Li,
Quantitative structure – mobility relationship studies of dipeptides in capillary zone electrophoresis using three-dimensional holographic vector of atomic interaction field, Mol. Simul. 35 (2009), pp. 263 – 269.
[21] A.M. Al-Mekhnaqi, M.S. Mayeed, and G.M. Newaz, Prediction of protein conformation in water and on surfaces by Monte Carlo simulations using united-atom method, Mol. Simul. 35 (2009), pp. 292 – 300.
[22] M.L. Mihajlovic and P.M. Mitrasinovic, Applications of the ArgusLab4/AScore protocol in the structure-based binding affinity prediction of various inhibitors of group-1 and group-2 influenza virus neuraminidases (NAs), Mol. Simul. 35 (2009), pp. 311 – 324. [23] C.S. Chen, Y.C. Wang, H.C. Yang, P.H. Huang, S.K. Kulp, C.C. Yang, Y.S. Lu, S. Matsuyama, C.Y. Chen, and C.S. Chen, Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation, Cancer Res. 67 (2007), pp. 5318 – 5327.
[24] C.M. Venkatachalam, X. Jiang, T. Oldfield, and M. Waldman, LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites, J. Mol. Graph. Model. 21 (2003), pp. 289 – 307.
[25] A. Krammer, P.D. Kirchhoff, X. Jiang, C.M. Venkatachalam, and M. Waldman, LigScore: A novel scoring function for predicting binding affinities, J. Mol. Graph. Model. 23 (2005), pp. 395 – 407. [26] D.K. Gehlhaar, G.M. Verkhivker, P.A. Rejto, C.J. Sherman, D.B.
Fogel, L.J. Fogel, and S.T. Freer, Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: Conformationally flexible docking by evolutionary programming, Chem. Biol. 2 (1995), pp. 317 – 324.
[27] R. Pompei, O. Flore, M.A. Marccialis, A. Pani, and B. Loddo, Glycyrrhizic acid inhibits virus growth and inactivates virus particles, Nature 281 (1979), pp. 689 – 690.
[28] J.C. Lin, Mechanism of action of glycyrrhizic acid in inhibition of Epstein – Barr virus replication in vitro, Antiviral Res. 59 (2003), pp. 41 – 47.
[29] J.G. Chung, H.L. Chang, W.C. Lin, H.H. Wang, C.C. Yeh, C.F. Hung, and Y.C. Li, Inhibition of N-acetyltransferase activity and DNA-2-aminofluorene adducts by glycyrrhizic acid in human colon tumour cells, Food Chem. Toxicol. 38 (2000), pp. 163 – 172.