Design, Synthesis, and Preclinical Evaluation of New 5,6-(or
6,7-)Disubstituted-2-(fluorophenyl)quinolin-4-one Derivatives as Potent
Antitumor Agents
Li-Chen Chou,1 Meng-Tung Tsai,1 Mei-Hua Hsu,1 Sheng-Hung Wang,2 Tzong-Der Way,3 Chi-Hung Huang,4 Hui-Yi Lin,1 Keduo Qian,5 Yizhou Dong, 5 Kuo-Hsiung Lee,5 Li-Jiau
Huang,1,* and Sheng-Chu Kuo1,*
1Graduate Institute of Pharmaceutical Chemistry, China Medical University, Taichung,
Taiwan; 2Institute of Cellular and Organismic Biology, Academia Sinica, 128 Academia
Road, Section 2, Nankang, Taipei 115, Taiwan; 3School of Biological Science and Technology, China Medical University, Taichung, Taiwan; 4Taiwan Advance Biopharm,
Inc., 12F, No. 25, Lane 169, Kangning Stl, Xizhi City, Taipei 221, Taiwan; 5Natural Products Research Laboratories, Eshelman School of Pharmacy, University of North
Carolina, Chapel Hill, North Carolina 27599-7568
*To whom correspondence should be addressed: S.C.K. Phone: +886-4-22030760; Fax: +886-4-22030760; E-mail: [email protected]. L.J.H. Phone:
+886-4-22053366-5609; E-mail: [email protected].
Abbreviations: 2-PQ, 2-phenylquinolin-4-one; SAR, structure-activity relationships; CHM-1, 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one; CHM-1–P-Na,
2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one monosodium phosphate; SPP, safety pharmacology profiling; NCI, National Cancer Institute; GI50, 50 % growth
inhibition; MG-MID, mean growth midpoint; TGI, total growth inhibition; 3-HP, 2-[(3-hydroxy-2-pyridinyl)methylene]hydrazinecarbothioamide; IGF-1R, insulin-like
growth factor-1 receptor; PDB, protein data bank; UGT1A1, UDP-glucuronosyltransferase 1A1;IV, intravenous, po, oral
Abstract
Our previous exploration of 2-phenylquinolin-4-ones (2-PQ) has led to an anticancer drug candidate CHM-1–P-Na. In order to develop additional new drug candidates, novel 2-PQ derivatives were designed, synthesized, and evaluated for cytotoxic activity. Most
analogs, including 1b, 2a-2b, 3a-3b, 4a-4b, and 5a-5b, exhibited significant inhibitory activity (IC50 values of 0.03-8.2 μM) against all tested tumor cell lines. As one of the
most potent analog, 2-(3-fluorophenyl)-5-hydroxy-6-methoxyquinolin-4-one (3b), selectively inhibited 14 out of 60 cancer cell lines in NCI evaluation. Preliminary
mechanism of action study suggested that 3b had a significant effect on the tyrosine autophosphorylation of IGF-1R. Safety pharmacology profiling of 3b showed no
significant effect on normal biological functions of most enzymes tested. Furthermore, sodium 2-(3-fluorophenyl)-6-methoxy-4-oxo-1,4-dihydroquinolin-5-yl phosphate (15),
the monophosphate of 3b, exceeded the activity of doxorubicin and was comparable to CHM-1–P-Na in a Hep3B xenograft nude mice model. In summary, 15 is a promising
clinical candidate, and is currently under preclinical study.
Introduction
During the last two decades, we have synthesized a series of substituted
2-phenylquinolin-4-ones (2-PQs)1–4 (Figure 1), and identified them as a new class of anticancer agents. In a recent in vivo evaluation of a series of 2-PQs with potent
cytotoxicity, excellent antitumor activity was identified with 2-(2-fluorphenyl)-6,7-methylenedioxyquinolin-4-one (CHM-1)4 (Figure 1) and the
sodium salt of its phosphate derivative (CHM-1–P-Na)4,5 (Figure 1). Furthermore, both acute toxicity and safety pharmacology profiling (SPP) studies supported that
CHM-1–P-Na is extremely safe to use. Based on the above findings, 2-PQs derivatives are a class of promising anticancer agents, which prompted us to design and develop
additional new promising analogs as potential clinical trials candidates.
As mentioned above, CHM-1–P-Na exhibited excellent antitumor activity, through
both oral and IV administration. Its unique structure possesses three key functional groups. The first key moiety is a phosphate group located on the 4-position of its
quinoline ring. As stated in our previous report,4 a pharmacokinetic study of CHM-1–P-Na confirmed its rapid bio-conversion into its active parent molecule CHM-1
following IV or po administration. It was also found that CHM-1–P-Na could be dephosphorylated by alkaline phosphatase into CHM-1 on the extracellular space of
SKOV-3 tumor. Because alkaline phosphatase is known to be over-expressed on the extracellular space of specific tumor cells, such as ovarian and hepatic carcinoma cells,
the introduction of a phosphate group appears to be a reasonable strategy for target delivery. Secondly, a methylenedioxy moiety bridges the 5- and 6-positions of the
quinoline ring. This moiety could form an orthoquinone upon metabolism, and subsequently be metabolized into more cytotoxic metabolites in hypoxic cells.4,6 Because
severe hypoxia is a common situation in locally advanced solid tumors, the incorporation of a methylenedioxy moiety becomes a meaningful antitumor approach. Thirdly, a
fluorine atom is located on the 2-phenyl group. In certain medicines, the unordinary nature of fluorine has been reported to impart a variety of properties, including enhanced
potency, improved duration of action, and attenuation of biliary clearance.7
Meanwhile, our prior SAR1–3 has established that the presence of a group with a lone
pair of electrons (for instance, OCH3, NRR, Cl, F) at both the 6-position of the quinoline
ring and 3’-position of the 2-phenyl group enhanced the cytotoxicity of 2-PQs. With this
finding in mind, we have designed compounds 1–5 (Figure 2) and their related phosphates as our target compounds based on the following attributes: (1) the presence of
a O-R group at the quinoline 6-position, (2) the presence of a fluorine atom on the 2-phenyl group, and (3) ability to be metabolized into an orthoquinone in vivo.
Among the target compounds (1–5) synthesized and evaluated, analog 3b appeared to be the most potent cytotoxic agent. Further studies, including safety pharmacology
profiling, mechanism of action studies, and molecular modeling of 3b, as well as in vivo antitumor evaluation on its monophosphate 15, were performed.
Chemistry
The synthetic route to 5,6,7,2’,3’,4’-substituted 2-phenylquinolin-4-ones (1, 2, 6) is illustrated in Scheme 1A. First, 3,4,5-substituted 1-amino-2-acetylbenzenes 7a~c were
reacted separately with 2,3,4-substituted benzoyl chlorides 8a~c to yield the corresponding amides 9a~i, which were subsequently cyclized in t-BuOH, in the
presence of t-BuOK, to afford the target compounds (1, 2, 6). Starting materials 6-amino-2,3-dimethoxyacetophenone 7a8, 6-amino-2,3-methylenedioxyacetophenone 7b9
and 4-benzyloxy-3-methoxy-6-nitroacetophenone 1010 were prepared according to previously reported methods. As shown in Scheme 1B, nitro 10 was reduced by reaction
with SnCl2 to give 6-amino-3-methoxy-4-benzyloxyacetophenone 7c.
As shown in Scheme 2, selective demethylation of compounds 1a~c with BCl3
provided the corresponding 2-(fluorophenyl)-5-hydroxy-6-methoxyquinolin-4-ones 3a~c, whose structures were confirmed by NOSY spectra. Catalytic hydrogenation of
compounds 2a~c and 7-benzyloxy-2-(fluorophenyl)-6-methoxyquinolin-4-ones 6a~c
yielded 2-(fluorophenyl)-5,6-dihydroxyquinolin-4-ones 4a~c and
2-(fluorophenyl)-7-hydroxy-6-methoxyquinolin-4-ones 5a~c, respectively.
The phosphorylation of 2-(3-fluorophenyl)-5-hydroxy-6-methoxyquinoline-4-one 3b is
shown in Scheme 3. Compound 3b was first reacted with tetrabenzylpyrophosphate 11 in the presence of NaH to yield 2-(3-fluorophenyl)-6-methoxyquinoline-4,5-diyl
bis(dibenzyl phosphate) 12 which was treated with MeOH at room temperature provided monophosphate 13. Its structure was confirmed by the chemical shift of the proton on the
3-position (δ 6.27) in the 1H-NMR spectra. Monophosphoric acid 14 was obtained by catalytic hydrogenation of 13. Finally, 14 was converted into water soluble sodium salt
15.
Results and Discussion
Growth inhibitory activity of 1–5 against human cancer cell lines
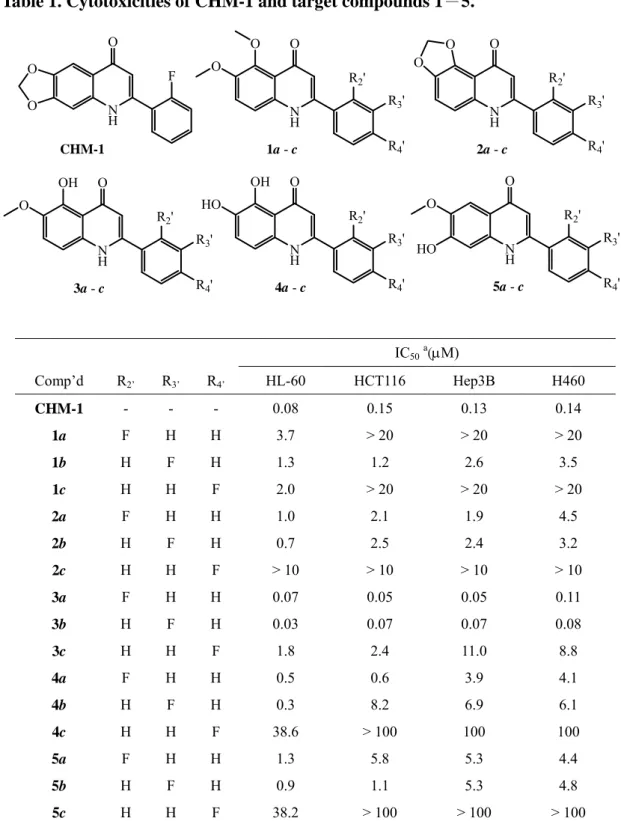
The 5,6-(6,7-)disubstituted 2-(fluorophenyl)quinolin-4-ones 1–5 and CHM-1 were screened against HL-60, HCT-116, Hep3B, H-460 human tumor cell lines and the results
are summarized in Table 1. Among the 5,6-dimethoxy derivatives 1a~c, the 3′-fluoro derivative 1b exhibited the greatest cytotoxicity, although it was less potent than the
positive control CHM-1. Meanwhile, both 2a and 2b, with a 5,6-methylenedioxy entity on the quinoline ring, demonstrated comparable activity to 1b, although again they were
less potent than CHM-1. While all three 5-hydroxy-6-methoxy derivatives 3a~c showed significant cytotoxicity, compounds 3a and 3b, with the fluorine in the 2′ and 3′ positions
respectively, were better than 3c, with the fluorine in the 4′ position. Changing the quinolone substitution pattern again from 5-hydroxy-6-methoxy (3a~c) to 5,6-dihydroxy
(4a~c) or 7-hydroxy-6-methoxy (5a~c) led to decreased potency. However, similar rank orders of potency were found among all derivatives regarding the position of the fluoro
substituent. Generally, 4′-fluorophenyl derivatives (1c–5c) were much less potent than the 2′- (1a–5a) and 3′-fluorophenyl (1b–5b) derivatives. In fact, only 1c and 3c showed
significant activity against some tested cancer cell line. Overall, compounds 3a and 3b are considered the most promising anticancer agents, as they exhibited potent broad
cytotoxic activity against all four cancer cell lines tested.
Compound 3b was submitted for evaluation of activity against NCI’s 60 human tumor
cell line panel (Figure 3). The results demonstrated that 3b was a potent antitumor agent against a broad range of human tumor cell lines [mean log GI50 (MG-MID) value of
-7.35]. As to total growth inhibition (TGI), 3b also displayed potent inhibitory activity (TGI<0.1 M) against the following 14 cancer cell lines: HL-60, SR, NCI-H522, Colo
205, HCC-2998, HT-29, SF-539, SNB-75, MDA-MB-435, IGROV1, OVCAR-3, NCI/ADR-RES, RXF-393, and DU-145.
Unique COMPARE Fingerprint of 3b
The mean graph pattern (fingerprint) of 3b was analyzed by a pattern-recognition
computer program (COMPARE), which contains a database covering similar types of fingerprints from over 300 known anticancer agents with various mechanisms of action.
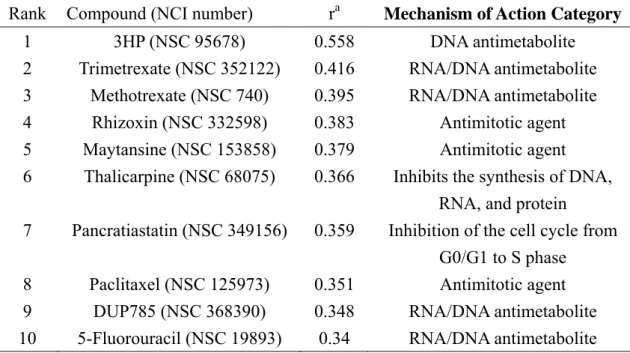
The results of COMPARE analysis in Table 2 suggested that the mechanism of action of
3b matched best with 2-[(3-hydroxy-2-pyridinyl)methylene]hydrazinecarbothioamide (3-HP). However, the low correlation coefficient (r<0.558) implied that its mechanism of action was different from those of the anticancer agents covered in the COMPARE
database.
Mechanism of Action of 3b
In our previous work,4 we reported a multiple-targeting action mechanism for CHM-1. More recently,11 we found that CHM-1 also affected the insulin-like growth factor-1
receptor (IGF-1R) which is gaining recognition as an attractive anti-cancer treatment target.12 Because IGF-1R is frequently over-expressed in a number of human tumors and
is considered an important anticancer target,13,14 we investigated the effect of 3b on IGF-1R expression in Hep3B cells, by examining receptor phosphorylation by the
IGF-1R tyrosine kinase, in the presence or absence of 3b. As shown in Figure 4, the cells were treated with different doses of 3b for 12 hours with DMSO as a control. Western
blot analysis of IGF-1R phosphorylation with the antibody against PY1135/1136-IGF-1R showed that the concentration of phosphor-IGF-1R decreased gradually with increasing concentration of 3b. The expression of IGF-1R, as evaluated by antibody to its -subunit,
served as loading control. The preliminary result indicated that 3b had a significant effect
on the tyrosine autophosphorylation of IGF-1R.
To delineate the interaction between 3b and IGF-1R kinase, structural models of
protein data bank (PDB) entries 1K3A15 and 3D9416 were selected for molecular modeling study. PDB 1K3A is a complex structure of human IGF-1R kinase, which
illustrates the ATP and Tyr-peptide substrate binding sites. On the other hand, PDB entry 3D94 illustrates the binding mode of the ATP-competitive inhibitor PQIP, which contains
a 2-phenylquinolinyl segment that can be superimposed on compound 3b due to structural similarity (Figures 5A and 5B).
During molecular modeling, the structural features of IGF-1R kinase with different ligand-bound states were first compared through molecular 3D superimposition. As
shown in Figure 6, the conformation of the activation loop changed dramatically between ATP-bound (model 1K3A; yellow color) and PQIP-bound (model 3D94; magenta color)
states. The autophosphorylation of Tyr-1131, Tyr-1135 and Tyr-1136 on the activation loop is also inhibited by PQIP.
The binding pocket of PQIP was then used for predicting the interaction mode of compound 3b against IGF-1R kinase. Molecular docking study showed that 3b interacted
with IGF-1R directly at the binding site of the 2-phenylquinolinyl segment of PQIP (Figure 7). The results indicate that the comparable binding is due to structural similarity.
Based on the molecular modeling study described above, the structure of PQIP is consists of two segments. One is an ATP-superimposable scaffold and the other is a
3b-superimposable scaffold (Figure 8). Furthermore, the 3b-binding site can be identified as a potential allosteric binding site, which is unique and different from the ATP-binding
site. Thus, 3b might inhibit autophosphorylation of the activation loop by changing its conformation, without significantly interfering with ATP-binding. Therefore, this model
could explain why the autophosphorylation of IGF-1R was inhibited by compound 3b. The exact action mechanism of 3b will be explored further.
Safety Pharmacology Profiling (SPP) of 3b
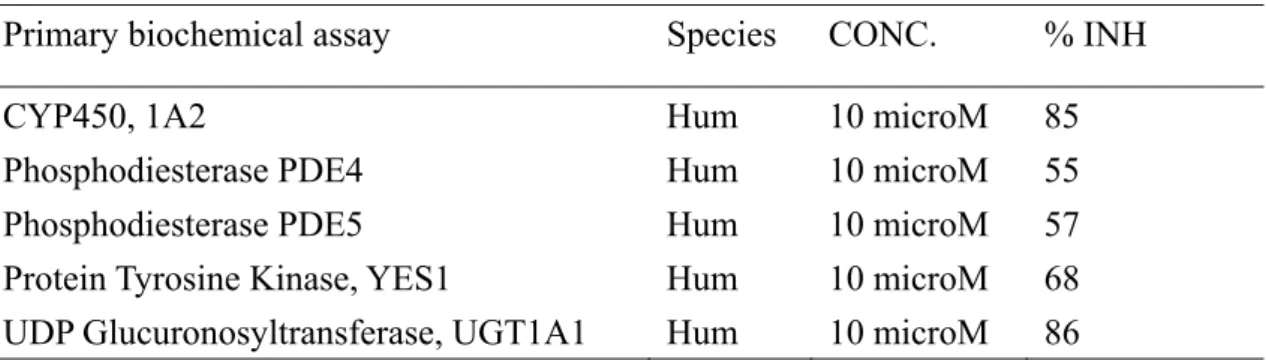
To explore its general pharmacological activities, 3b was tested in 158 types of enzyme spectrum screening assays (Table 3). At 10 M, 3b inhibited the activity of only the
activity was most profound against UDP-glucuronosyltransferase 1A1 (UGT1A1). Interestingly, a recent study by E. Selga identified UGT1A1 as an important gene code in
methotrexate resistance.17 The author speculated that UGT1A1 may be associated with the drug resistance of certain cancer cell lines. The above results indicated that the SPP of
3b differed from that of CHM-1.
In vivo antitumor activity of 15
The water soluble monophosphate of 3b (15) was evaluated in the Hep3B xenograft nude mice model by oral route (po) at doses of 7.5, 15, 30 mg/kg/day. As shown in
Figures 9A–C, 15 induced dose- and time-dependent inhibition of Hep3B tumor growth. Significant tumor growth suppression, at an extent exceeding that observed after 10
mg/kg/day oral dosing of doxorubicin, was detected after 7.5 mg/kg/day oral dosing of 15. Nearly complete tumor suppression was observed after 30 mg/kg/day oral dosing. During
the course of antitumor evaluation, no significant body weight changes were detected in either the tested or the control mice. Similarly, the dose- and time-dependent antitumor test results by IV administration, summarized in Figures 9D–F, resembled those
obtained by oral administration, and generally indicated slightly better antitumor activity.
Overall, the antitumor activity of 15 administrated po or IV is comparable with that of CHM-1–P-Na.
Conclusion
Novel 2-PQ analogs (1–5) were designed, synthesized, and evaluated for antitumor
activity. Preliminary SAR of the new analogs was established. As one of our most
promising target compounds, 3b was submitted for evaluation of activity against it NCI’s 60 human tumor cell line panel.18, 19 The NCI results confirmed that compound 3b had
potent inhibitory activity against 14 cancer cell lines. The preliminary result of a mechanistic study indicated that 3b had a significant effect on the tyrosine
autophosphorylation of IGF-1R, which is an attractive target for anti-cancer therapy. The SPP results from enzyme assays suggested that 3b has a clear-cut pharmacological effect
on tumor-associated UGT1A1. Furthermore, a second target compound, 15, the monophosphate derivative of 3b, demonstrated excellent antitumor activity in nude mice
bearing Hep3B xenograft, when administrated via po and IV routes. Excitingly, the antitumor activity of 15 exceeded that of doxorubicin, the most common drug used in current practice, and 15 was comparable to CHM-1–P-Na. In summary, 15 is a
Experimental Section
Chemistry. General Experimental Procedures. All reagents and solvents were obtained commercially and used without further purification. Reactions were monitored by thin-layer chromatography (TLC), using Merck plates with fluorescent indicator (TLC
Silica gel 60 F254). The following adsorbent was used for column chromatography: silica
gel 60 (Merck, particle size 0.063-0.200 mm). Melting points were determined on a
Yanaco MP-500D melting point apparatus and are uncorrected. IR spectra were recorded on Shimadzu IRPrestige-21 spectrophotometers as KBr pellets. NMR spectra were
obtained on a Bruker Avance DPX-200 FT-NMR spectrometer in CDCl3 or DMSO. The
following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartet; dd, double
doublet and m, multiplet. EI-MS spectra were measured with an HP 5995 GC–MS instrument. ESI-MS spectra were measured with a Finnigan LCQ ion-trap mass
spectrometer (TSQ Quantum, Thermo Finnigan Corporation, San Jose, CA). Elemental
analyses (C, H, and N) were performed on a Perkin-Elmer 2400 Series II CHNS/O
analyzer, and the results were within ±0.4% of the calculated values. The purity of tested compounds was ≥ 95 % as determined by HPLC conducted on a Shimadzu LC-10AT
apparatus equipped with a Shimadzu SPD-M10AVP diode-array detector and a Shimadzu SIL-10A auto-injector.
N-(2-Acetyl-3,4-dimethoxyphenyl)-2-fluorobenzamide (9a). To a solution of
2-fluorobenzoyl chloride (8a, 0.48 g, 2.46 mmol) in 40 mL of dry toluene were added
triethylamine (0.5 mL) and compound 7a8 (0.70 g, 4.43 mmol). The mixture was stirred at 55-60 oC for 30 min, and then poured into crushed ice, and extracted with EtOAc. The
organic layer was washed with brine, dried over MgSO4 and evaporated. The crude
product was purified by column chromatography (silica gel, EtOAc/n-hexane) to give 9a (0.5 g, 1.58 mmol) as a yellow solid. Yield: 64.1 %; mp 106108 °C; MS (EI, 70 eV):
m/z 317 (M+); 1H-NMR (DMSO-d6, 200 MHz): δ 2.45 (s, 3H), 3.76 (s, 3H), 3.81 (s, 3H),
7.14 (d, J = 2.6 Hz, 2H), 7.247.34 (m, 2H), 7.527.63 (m, 2H), 10.07 (s, 1H); 13C
NMR (DMSO-d6, 50 MHz): 31.91, 56.52, 61.42, 114.52, 116.70, 121.43, 124.16,
125.06, 126.81, 130.52, 131.50, 133.35 (d, J = 8.0 Hz), 145.98, 150.47, 159.61 (d, J = 247.5 Hz), 163.19, 201.38; Anal. (C17H16FNO4) C, H, N.
N-(2-Acetyl-3,4-dimethoxyphenyl)-3-fluorobenzamide (9b) was obtained from 7a
and 3-fluorobenzoyl chloride (8b). Yellow solid; Yield: 65.0 %; mp 9899 °C; MS (EI,
70 eV): m/z 317 (M+); 1H-NMR (DMSO-d6, 200 MHz): δ 2.43 (s, 3H), 3.76 (s, 3H), 3.81
(s, 3H), 7.037.15 (m, 2H), 7.397.71 (m, 4H), 10.18 (s, 1H); 13C NMR (DMSO-d 6, 50
MHz): 31.74, 56.45, 61.38, 114.30, 114.73 (d, J = 23 Hz), 119.04 (d, J = 21 Hz), 121.98, 124.13, 126.91, 131.12 (d, J = 7.5 Hz), 132.27, 136.89 (d, J = 6.5 Hz), 145.90,
150.65, 162.40 (d, J = 243 Hz), 164.67, 201.22; Anal. (C17H16FNO4) C, H, N.
N-(2-Acetyl-3,4-dimethoxyphenyl)-4-fluorobenzamide (9c) was obtained from 7a
and 4-fluorobenzoyl chloride (8c). Yellow solid; Yield: 64.7 %; mp 146147 °C; MS
(EI, 70 eV): m/z 317 (M+); 1H-NMR (DMSO-d6, 200 MHz): δ 2.43 (s, 3H), 3.76 (s, 3H),
3.81 (s, 3H), 7.037.14 (m, 2H), 7.267.35 (m, 2H), 7.887.95 (m, 2H), 10.14 (s, 1H);
13C NMR (DMSO-d
6, 50 MHz): 31.76, 56.48, 61.38, 114.33, 115.83 (d, J = 22 Hz),
121.91, 127.17, 130.65 (d, J = 9.0 Hz), 131.06, 132.21, 145.90, 150.53, 164.57 (d, J = 247 Hz), 164.94, 201.25; Anal. (C17H16FNO4) C, H, N.
N-(2-Acetyl-3,4-methylenedioxyphenyl)-2-fluorobenzamide (9d) was obtained from
7b9 and 8a. Yellow solid; Yield: 90.0 %; mp 165166 °C; MS (EI, 70 eV): m/z 301 (M+); 1H-NMR (DMSO-d 6, 200 MHz): δ 2.53 (s, 3H), 6.13 (s, 2H), 7.12 (d, J = 8.6 Hz, 1H), 7.287.38 (m, 2H), 7.567.61 (m, 1H), 7.727.82 (m, 1H), 7.85 (d, J = 8.8 Hz, 1H), 11.50 (s, 1H); 13C NMR (DMSO-d6, 50 MHz): 32.53, 102.57, 111.92, 112.52, 114.74, 116.94 (d, J = 22.5 Hz), 123.55 (d, J = 12.5 Hz), 125.41, 130.98, 131.91, 134.04 (d, J = 8.5 Hz), 144.46, 149.00, 157.18, 162.22, 199.61; Anal. (C16H12FNO4) C, H, N.
N-(2-Acetyl-3,4-methylenedioxyphenyl)-3-fluorobenzamide (9e) was obtained from
7b and 8b. Yellow solid; Yield: 95.0 %; mp 170171 °C; MS (EI, 70 eV): m/z 301 (M+); 1H-NMR (DMSO-d
7.43 (t, J = 8.6 Hz, 1H), 7.527.68 (m, 2H), 7.72 (d, J = 8.6 Hz, 2H), 11.56 (s, 1H); 13C
NMR (DMSO-d6, 50 MHz): 32.48, 102.61, 112.48, 112.62, 114.48 (d, J = 23 Hz),
114.89, 119.30 (d, J = 21.5 Hz), 123.56, 131.52 (d, J = 8.0 Hz), 131.96, 137.32, 144.61, 148.88, 162.61 (d, J = 243.5 Hz), 163.97, 199.88; Anal. (C16H12FNO4) C, H, N.
N-(2-Acetyl-3,4-methylenedioxyphenyl)-4-fluorobenzamide (9f) was obtained from
7b and 8c. Yellow solid; Yield: 84.0 %; mp 185186 °C; MS (EI, 70 eV): m/z 301 (M+); 1H-NMR (DMSO-d 6, 200 MHz): δ 2.51 (s, 3H), 6.13 (s, 2H), 7.12 (d, J = 8.6 Hz, 1H), 7.207.40 (m, 2H), 7.77 (d, J = 8.6 Hz, 1H), 7.897.97 (m, 2H), 11.58 (s, 1H); 13C NMR (DMSO-d6, 50 MHz): 32.55, 102.55, 111.20, 112.61, 114.54, 116.26 (d, J = 22 Hz), 130.22 (d, J = 7.0 Hz), 131.47, 132.41, 144.36, 148.97, 162.22, 164.67 (d, J = 248 Hz), 200.04; Anal. (C16H12FNO4) C, H, N. 2-Amino-4-benzyloxy-5-methoxyacetophenone (7c). To a solution of 1010
(1.0 g, 3.32 mmol) in anhydrous EtOH (100 mL) was added SnCl2·2H2O (3.7 g,
16.4 mmol). The mixture was refluxed for 2 h and then cooled to 25 °C, and
poured in 5% NaHCO3 solution. The precipitate was collected and washed with
H2O and then extracted with EtOAc. The extract was washed with H2O, dried
over MgSO4 and evaporated. The crude product was purified by column
72.2 %; mp 135137 °C; 1H-NMR (DMSO-d
6, 200 MHz): δ 2.39 (s, 3H), 3.66 (s, 3H),
5.03 (s, 2H), 6.38 (s,1H), 7.05 (s, 2H), 7.10 (s, 1H), 7.307.50 (m, 5H); 13C NMR
(DMSO-d6, 50 MHz): 28.21, 56.94, 69.87, 100.05, 109.70, 115.40, 128.34, 128.49,
128.93, 136.83, 139.45, 148.74, 154.64, 198.06; Anal. (C16H17NO3) C, H, N.
N-(2-Acetyl-5-benzyloxy-4-methoxyphenyl)-2-fluorobenzamide (9g) was obtained
from 7c and 8a. Yellow solid; Yield: 89.0 %; mp 142143 °C; MS (EI, 70 eV): m/z 393
(M+); 1H-NMR (DMSO-d6, 200 MHz): δ 2.60 (s, 3H), 3.82 (s, 3H), 5.16 (s, 2H), 7.107.50 (m, 8H), 7.567.67 (m, 1H), 7.807.89 (m, 1H), 8.57 (s, 1H), 12.45 (d, J = 4.0 Hz,1H); 13C NMR (DMSO-d6, 50 MHz): 29.08, 56.42, 70.41, 105.31, 115.28, 116.05, 117.08 (d, J = 22 Hz), 123.27 (d, J = 12.5 Hz), 125.60, 128.58, 128.95, 131.20, 134.45 (d, J = 8.5 Hz), 135.77, 136.50, 144.46, 152.98, 157.26, 162.35, 201.78; Anal. (C23H20FNO4) C, H, N.
N-(2-Acetyl-5-benzyloxy-4-methoxyphenyl)-3-fluorobenzamide (9h) was obtained
from 7c and 8b. Yellow solid; Yield: 86.6 %; mp 162163 °C; MS (EI, 70 eV): m/z 393
(M+); 1H-NMR (DMSO-d6, 200 MHz): δ 2.62 (s, 3H), 3.81 (s, 3H), 5.15 (s, 2H),
7.267.52 (m, 7H), 7.547.78 (m, 3H), 8.51 (s, 1H), 12.70 (s, 1H); 13C NMR (DMSO-d 6,
50 MHz): 29.10, 56.42, 70.40, 104.74, 114.45, 115.29, 115.83, 119.59 (d, J = 21.5 Hz),
144.43, 153.26, 162.71 (d, J = 244 Hz), 163.91, 202.48; Anal. (C23H20FNO4) C, H, N.
N-(2-Acetyl-5-benzyloxy-4-methoxyphenyl)-4-fluorobenzamide (9i) was obtained
from 7c and 8c. Yellow solid; yield: 67.1 %; mp 168169 °C; MS (EI, 70 eV): m/z 393
(M+); 1H-NMR (DMSO-d6, 200 MHz): δ 2.63 (s, 3H), 3.81 (s, 3H), 5.15 (s, 2H), 7.27.5 (m, 7H), 7.98.1 (m, 3H), 8.54 (s, 1H), 12.69 (s, 1H); 13C NMR (DMSO-d 6, 50 MHz): 29.11, 56.47, 70.41, 104.73, 115.39, 115.83, 116.27, 116.72, 128.53, 128.94, 130.17 (d, J = 9.0 Hz), 132.54 (d, J = 9.5 Hz), 136.48, 136.58, 144.33, 153.33, 164.24, 166.82, 202.48; Anal. (C23H20FNO4) C, H, N.
2-(2-Fluorophenyl)-5,6-dimethoxyquinolin-4-one (1a). To a suspension of 9a (0.50 g, 1.58 mmol) in t-butyl alcohol (30 mL) was added potassium t-butoxide (1.0 g, 8.93
mmol). The mixture was refluxed under argon for 20 h and evaporated. The residue was treated with a 10% NH4Cl solution (30 mL). The solid precipitate was collected and
washed with n-hexane and Me2CO. The crude product was recrystallized from MeOH to
afford 1a as yellow needles (0.27 g, 0.9 mmol). Yield: 57.1 %; mp 215217 °C; MS (EI,
70 eV): m/z 299 (M+); IR (KBr): 1628 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ
3.72 (s, 3H), 3.81 (s, 3H), 6.06 (s, 1H), 7.37.6 (m, 5H), 7.607.71 (m, 1H); Anal.
(C17H14FNO3) C, H, N. HPLC purity analysis. Colu mn: Thermo ODS HYPERSIL,
30/70. Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 1.999 min. Flow
rate: 1.0 mL/min. Purity: 98.35 %.
2-(3-Fluorophenyl)-5,6-dimethoxyquinolin-4-one (1b) was obtained from 9b. Yellow needles; yield: 53.1 %; mp 190192 °C; MS (EI, 70 eV): m/z 299 (M+); IR
(KBr): 1599 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.73 (s, 3H), 3.81 (s, 3H),
6.35 (s, 1H), 7.287.40 (m, 1H), 7.467.60 (m, 3H), 7.647.76 (m, 2H); Anal.
(C17H14FNO3) C, H, N. HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150
× 4.6 mm, 5 . Mobile phase: 0.01 M sodium hydrogen carbonate/acetonitril = 30/70.
Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 2.067 min. Flow rate: 1.0 mL/min. Purity: 98.54 %.
2-(4-Fluorophenyl)-5,6-dimethoxyquinolin-4-one (1c) was obtained from 9c. White needles; yield: 54.6 %; mp 227229 °C; MS (EI, 70 eV): m/z 299 (M+); IR (KBr): 1607
(C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.72 (s, 3H), 3.80 (s, 3H), 6.26 (s, 1H),
7.317.40 (m, 2H), 7.447.54 (m, 2H), 7.837.90 (m, 2H); Anal. (C17H14FNO3) C, H,
N. HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 .
Mobile phase: 0.01 M sodium hydrogen carbonate/acetonitril = 30/70. Detection
wavelength: PDA Ch1 254nm at 4 nm. Retention time: 2.054 min. Flow rate: 1.0 mL/min. Purity: 98.86 %.
2-(2-Fluorophenyl)-5,6-methylenedioxyquinolin-4-one (2a) was obtained from 9d. Yellow solid; yield: 47.6 %; mp 282283 °C; MS (EI, 70 eV): m/z 283 (M+); IR (KBr):
1605 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 5.92 (s, 1H), 6.11 (s, 2H), 7.09 (d,
J = 8.8 Hz, 1H), 7.277.38 (m, 3H), 7.557.70 (m, 2H), 11.71 (s, 1H); Anal.
(C16H10FNO3) C, H, N. HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150
× 4.6 mm, 5 . Mobile phase: methanol/0.05 % TFA in water = 75/25. Detection
wavelength: PDA Ch1 254nm at 4 nm. Retention time: 2.377 min. Flow rate: 1.0 mL/min. Purity: 99.16 %.
2-(3-Fluorophenyl)-5,6-methylenedioxyquinolin-4-one (2b) was obtained from 9e. White solid; yield: 44.9 %; mp 286288 °C; MS (EI, 70 eV): m/z 283 (M+); IR (KBr):
1609 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 6.11 (s, 2H), 6.19 (s, 1H),
7.197.36 (m, 3H), 7.557.67 (m, 3H), 11.71 (s, 1H); Anal. (C16H10FNO3) C, H, N.
HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 . Mobile
phase: methanol/0.05 % TFA in water = 75/25. Detection wavelength: PDA Ch1 254nm
at 4 nm. Retention time: 2.516 min. Flow rate: 1.0 mL/min. Purity: 98.63 %.
2-(4-Fluorophenyl)-5,6-methylenedioxyquinolin-4-one (2c) was obtained from 9f. White solid; yield: 45.9 %; mp 286288 °C; MS (EI, 70 eV): m/z 283 (M+); IR (KBr):
7.327.41 (m, 2H), 7.787.85 (m, 2H), 11.46 (s, 1H); Anal. (C16H10FNO3) C, H, N.
HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 . Mobile
phase: methanol/0.05 % TFA in water = 75/25. Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 2.500 min. Flow rate: 1.0 mL/min. Purity: 97.93 %.
7-Benzyloxy-2-(2-fluorophenyl)-6-methoxyquinolin-4-one (6a) was obtained from
9g. White solid; yield: 60.5 %; mp 132134 °C; MS (EI, 70 eV): m/z 375 (M+); 1H-NMR (DMSO-d 6, 200 MHz): δ 3.82 (s, 3H), 5.16 (s, 2H), 6.21 (s, 1H), 7.207.80 (m, 11H); 13C NMR (DMSO-d6, 50 MHz): 56.02, 70.40, 101.86, 104.14, 108.80, 116.77 (d, J = 21.5 Hz), 118.86, 123.30 (d, J = 13 Hz), 125.43, 128.50, 128.97, 131.24, 132.56 (d, J = 8.0 Hz), 136.58, 137.08, 144.73, 147.73, 152.52, 159.64 (d, J = 247 Hz), 174.57; Anal. (C23H18FNO3) C, H, N.
7-Benzyloxy-2-(3-fluorophenyl)-6-methoxyquinolin-4-one (6b) was obtained from
9h. White solid; yield: 64.3 %; mp 154155 °C; MS (EI, 70 eV): m/z 375 (M+); 1H-NMR (DMSO-d 6, 200 MHz): δ 3.83 (s, 3H), 5.17 (s, 2H), 6.56 (s, 1H), 7.307.50 (m, 8H), 7.557.60 (m, 1H), 7.607.80 (m, 2H); 13C NMR (DMSO-d 6, 50 MHz): 56.07, 70.45, 102.27, 103.72, 106.03, 114.71 (d, J = 23.5 Hz), 117.56 (d, J = 20.5 Hz), 118.44, 123.95, 128.56, 128.99, 131.50, 136.49, 137.41, 148.02, 148.44, 152.72, 165.13, 173.61; Anal. (C23H18FNO3) C, H, N.
7-Benzyloxy-2-(4-fluorophenyl)-6-methoxyquinolin-4-one (6c) was obtained from
9i. White solid; yield: 64.4 %; mp 248249 °C; MS (EI, 70 eV): m/z 375 (M+); 1H-NMR (DMSO-d 6, 200 MHz): δ 3.80 (s, 3H), 5.13 (s, 2H), 6.26 (s, 1H), 7.207.60 (m, 9H), 7.808.00 (m, 2H); 13C NMR (DMSO-d 6, 50 MHz): 55.96, 70.36, 101.41, 104.51, 106.61, 116.30 (d, J = 21.5 Hz), 119.27, 128.56, 128.99, 130.05 (d, J = 8.0 Hz), 136.60, 147.39, 148.06, 152.19, 163.63 (d, J = 246.5 Hz), 176.10; Anal. (C23H18FNO3) C, H, N.
2-(2-Fluorophenyl)-5-hydroxy-6-methoxyquinolin-4-one (3a). To a solution of 1a (0.2 g, 0.67 mmol) in CH2Cl2 (3 mL) was added 5 mL of BCl3 solution (1 M
in CH2Cl2) dropwise at 0 ± 1 °C. The mixture was stirred at 25 ± 1 °C for 2 h and
then poured into crushed ice, and extracted with EtOAc. The organic layer was washed with H2O, dried over MgSO4 and evaporated. The crude was purified by
column chromatography (SiO2, CHCl3: MeOH = 15:1) and recrystallized from
MeOH to give 3a. Yellow solid; yield: 24.1 %; mp 268270 °C; MS (EI, 70 eV): m/z
285 (M+); IR (KBr): 1604.77 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.78 (s,
3H), 6.11 (s, 1H), 7.01 (d, J = 7.4 Hz,1H), 7.367.48 (m, 3H), 7.547.72 (m, 2H), 12.25
(s, 1H), 14.54 (s, 1H); 13C NMR (DMSO-d
6, 50 MHz): 55.80, 106.22, 106.43, 112.88,
8.6 Hz), 135.09, 141.02, 146.27, 149.29, 158.92 (d, J = 247.7 Hz), 181.97; Anal. (C16H12FNO3) C, H, N. HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150
× 4.6 mm, 5 . Mobile phase: 0.01 M sodium hydrogen carbonate/acetone = 30/70.
Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 1.954 min. Flow rate:
1.0 mL/min. Purity: 96.04 %.
2-(3-Fluorophenyl)-5-hydroxy-6-methoxyquinolin-4-one (3b) was obtained from 1b and BCl3. Yellow solid; yield: 26.7%; mp 274276 °C; MS (EI, 70 eV): m/z
285 (M+); IR (KBr): 1606.70 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.77 (s,
3H), 6.33 (s, 1H), 7.11 (d, J = 8.8 Hz,1H), 7.337.48 (m, 2H), 7.517.76 (m, 3H), 12.09
(s, 1H), 14.56 (s, 1H); 13C NMR (DMSO-d6, 50 MHz): 57.19, 104.82, 106.97, 113.39,
115.09 (d, J = 23 Hz), 118.06 (d, J = 21 Hz), 121.07, 124.32, 131.64 (d, J = 9.0 Hz), 135.61, 136.16 (d, J = 8.0 Hz), 141.49, 149.64, 150.12, 162.64 (d, J = 242.5 Hz),
182.69; Anal. (C16H12FNO3) C, H, N. HPLC purity analysis. Column: Thermo ODS
HYPERSIL, 150 × 4.6 mm, 5 . Mobile phase: 0.01 M sodium hydrogen
carbonate/acetonitril = 30/70. Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 1.820 min. Flow rate: 1.0 mL/min. Purity: 99.13 %.
2-(4-Fluorophenyl)-5-hydroxy-6-methoxyquinolin-4-one (3c) was obtained from 1c and BCl3. Yellow solid; yield: 23.0%; mp 307309 °C; MS (EI, 70 eV): m/z
285 (M+); IR (KBr): 1610.56 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.76 (s,
3H), 6.25 (s, 1H), 7.08 (d, J = 9.0 Hz,1H), 7.347.43 (m, 3H), 7.827.89 (m, 2H), 12.01
(s, 1H), 14.60 (s, 1H); 13C NMR (DMSO-d6, 50 MHz): 57.19, 104.53, 106.84, 113.23,
116.47 (d, J = 22 Hz), 120.99, 130.55 (d, J = 9.0 Hz), 135.62, 141.45, 149.69, 150.64,
164.02 (d, J = 247 Hz), 182.59; Anal. (C16H12FNO3) C, H, N. HPLC purity analysis.
Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 . Mobile phase: 0.01 M sodium
hydrogen carbonate/acetonitril = 30/70. Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 1.822 min. Flow rate: 1.0 mL/min. Purity: 98.00 %.
2-(2-Fluorophenyl)-5,6-dihydroxyquinolin-4-one (4a). To a solution of 2a (0.1 g, 0.35 mmol) in anhydrous MeOH (30 mL) was hydrogenated in the
presence of 10% Pd/C (0.2 g) at 25 ± 2 °C for 40 h. The catalyst was filtered off and the filtrate was evaporated. The crude was purified by column
chromatography (SiO2, EtOAc: MeOH = 30:1) to give 4a. White solid; yield:
13.7%; mp 152154 °C; MS (EI, 70 eV): m/z 271 (M+); IR (KBr): 1622.13 (C=O) cm-1; 1H-NMR (DMSO-d
6, 200 MHz): δ 6.03 (s, 1H), 7.15 (d, J = 8.8 Hz,1H), 7.307.70 (m,
6H), 9.72 (s, 1H), 11.76 (s, 1H); 13C NMR (DMSO-d6, 50 MHz): 107.67, 108.57,
116.75 (d, J = 21.5 Hz), 120.54, 122.67, 123.36, 125.42, 126.70, 131.22, 132.49, 134.35, 144.30, 154.29, 159.43 (d, J = 248.5 Hz), 176.82; Anal. (C15H10FNO3) C, H, N.
HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 . Mobile
phase: methanol/0.05 % TFA in water = 75/25. Detection wavelength: PDA Ch1 254nm
at 4 nm. Retention time: 2.384 min. Flow rate: 1.0 mL/min. Purity: 96.50 %.
2-(3-Fluorophenyl)-5,6-dihydroxyquinolin-4-one (4b) was obtained from 2b. White solid; yield: 15.0%; mp 307308 °C; MS (EI, 70 eV): m/z 271 (M+); IR (KBr):
1608.63 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 6.25 (s, 1H), 7.15 (d, J = 8.8
Hz, 1H), 7.307.50 (m, 2H), 7.507.80 (m, 4H), 9.72 (s, 1H), 11.60 (s, 1H); 13C NMR
(DMSO-d6, 50 MHz): 106.38, 107.57, 114.65 (d, J = 23 Hz), 117.38 (d, J = 21.5 Hz),
120.91, 122.62, 123.91, 126.81, 131.52 (d, J = 8.5 Hz), 134.45, 137.17, 147.66, 154.36, 162.70 (d, J = 242 Hz), 176.82; Anal. (C15H10FNO3) C, H, N. HPLC purity analysis.
Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 . Mobile phase: methanol/0.05 %
TFA in water = 75/25. Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time:
2.485 min. Flow rate: 1.0 mL/min. Purity: 99.51 %.
2-(4-Fluorophenyl)-5,6-dihydroxyquinolin-4-one (4c) was obtained from 2c. White solid; yield: 13.9%; mp 332334 °C; MS (EI, 70 eV): m/z 271 (M+); IR (KBr):
1614.42 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 6.18 (s, 1H), 7.14 (dd, J = 9.0,
2.8 Hz, 1H), 7.337.42 (m, 3H), 7.59 (d, J = 8.8 Hz, 1H), 7.797.86 (m, 2H), 9.70 (s,
21.5 Hz), 120.71, 122.48, 126.73, 130.19 (d, J = 8.5 Hz), 131.38, 134.42, 148.24, 154.20, 163.70 (d, J = 247.5 Hz), 176.81; Anal. (C15H10FNO3) C, H, N. HPLC purity
analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 . Mobile phase:
methanol/0.05 % TFA in water = 75/25. Detection wavelength: PDA Ch1 254nm at 4 nm.
Retention time: 2.502 min. Flow rate: 1.0 mL/min. Purity: 99.40 %.
2-(2-Fluorophenyl)-7-hydroxy-6-methoxyquinolin-4-one (5a). Compound 6a (0.3 g, 0.80 mmol) was allowed to react in the same manner as described in the preparation of compound 4a to give 5a. White solid; yield: 61.3%; mp 277279 °C; MS (EI, 70 eV):
m/z 285 (M+); IR (KBr): 1622.13 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.82 (s,
3H), 6.04 (s, 1H), 7.01 (s, 1H), 7.327.50 (m, 3H), 7.507.67 (m, 2H), 10.22 (s, 1H),
11.68 (s, 1H); 13C NMR (DMSO-d6, 50 MHz): 55.52, 102.72, 105.37, 108.20, 116.28
(d, J = 22.5 Hz), 118.07, 122.94, 124.92, 130.75, 131.99 (d, J = 7.95 Hz), 136.45,
143.61, 146.58, 151.59, 158.98 (d, J = 246.9 Hz), 175.30; Anal. (C16H12FNO3) C, H,
N. HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 .
Mobile phase: methanol/0.05 % TFA in water = 75/25. Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 2.667 min. Flow rate: 1.0 mL/min. Purity: 99.39 %.
2-(3-Fluorophenyl)-7-hydroxy-6-methoxyquinolin-4-one (5b) was obtained from 6b. White solid; yield: 44.8%; mp 326328 °C; MS (EI, 70 eV): m/z 285 (M+); IR (KBr):
1606.70 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.81 (s, 3H), 6.24 (s, 1H), 7.12
(s, 1H), 7.277.42 (m, 2H), 7.477.70 (m, 3H), 10.20 (s, 1H), 11.44 (s, 1H); 13C NMR
(DMSO-d6, 50 MHz): 55.92, 103.35, 104.70, 106.67, 114.59 (d, J = 23 Hz), 117.26 (d,
J = 21 Hz), 118.85, 123.85, 131.47 (d, J = 8.0 Hz), 136.85, 137.16, 146.94, 147.33,
151.93, 162.69 (d, J = 242.5 Hz), 176.37; Anal. (C16H12FNO3) C, H, N. HPLC purity
analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 . Mobile phase:
methanol/0.05 % TFA in water = 75/25. Detection wavelength: PDA Ch1 254nm at 4 nm. Retention time: 2.754 min. Flow rate: 1.0 mL/min. Purity: 99.38 %.
2-(4-Fluorophenyl)-7-hydroxy-6-methoxyquinolin-4-one (5c) was obtained from 6c. White solid; yield: 42.5%; mp 352354 °C; MS (EI, 70 eV): m/z 285 (M+); IR (KBr):
1610.56 (C=O) cm-1; 1H-NMR (DMSO-d6, 200 MHz): δ 3.80 (s, 3H), 6.19 (s, 1H), 7.11
(s, 1H), 7.207.50 (m, 3H), 7.707.90 (m, 2H); 13C NMR (DMSO-d
6, 50 MHz): 55.89,
103.50, 104.55, 106.18, 116.30 (d, J = 21.5 Hz), 118.41, 130.03 (d, J = 8.5 Hz), 131.57, 137.22, 147.00, 148.01, 152.21, 163.58 (d, J = 246 Hz), 175.93; Anal. (C16H12FNO3)
C, H, N. HPLC purity analysis. Column: Thermo ODS HYPERSIL, 150 × 4.6 mm, 5 .
Mobile phase: methanol/0.05 % TFA in water = 75/25. Detection wavelength: PDA Ch1
254nm at 4 nm. Retention time: 2.736 min. Flow rate: 1.0 mL/min. Purity: 99.40 %.
stirred solution of 3b (0.12 g, 0.42 mmol) in dry THF (20 mL) was added NaH (96 mg, 4 mmol) at 0 ± 1 °C. After the mixture was stirred for 1 h, tetrabenzyl
pyrophosphate (11) (430 mg, 0.8 mmol) was added and stirring was continued for 25 min. The reaction mixture was filtered and washed with CH2Cl2. The filtrate
was concentrated under vacuum at a temperature below 30 °C. The residue was purified by column chromatography (SiO2, n-hexane: EtOAc) to give 12. Liquid;
yield: 95.0%; MS (EI, 70 eV): m/z 805 (M+); 1H-NMR (DMSO-d6, 200 MHz): δ 3.87 (s,
3H), 5.10 (s, 2H), 5.14 (s, 2H), 5.18 (s, 2H), 5.22 (s, 2H), 7.207.36 (m, 21H), 7.477.60 (m, 1H), 7.727.84 (m, 4H), 8.01 (d, J = 9.4 Hz, 1H); 13C NMR (DMSO-d 6, 50 MHz): 57.27, 69.63, 69.74, 70.12, 70.23, 110.20, 113.57, 114.03, 116.23, 116.92, 117.35, 119.48, 123.28, 128.10, 128.38, 128.70, 128.79, 128.85, 128.95, 131.35, 131.51, 135.79, 135.94, 136.32, 136.47, 140.41, 140.56, 145.39, 149.74, 149.82, 153.44, 153.57, 153.92, 160.71, 165.56; Anal. (C44H38FNO9P2) C, H, N.
Dibenzyl 2-(3-fluorophenyl)-6-methoxy-4-oxo-1,4-dihydroquinolin-5-yl phosphate
(13). A suspension of 12 (2.42 g, 3.0 mmol) in anhydrous MeOH (10 mL) was stirred at 25 °C for 24 h. The precipitates were collected and purified by column
chromatography (SiO2, n-hexane: EtOAc) to give 13. Yellow solid; yield: 80.0%;
mp 136138 °C; MS (ESI): m/z 544.5 (MH); 1H-NMR (DMSO-d
3.75 (s, 3H), 5.28 (s, 2H), 5.31 (s, 2H), 6.27 (s, 1H), 7.267.50 (m, 11H), 7.507.78 (m,
6H); 13C NMR (DMSO-d6, 50 MHz): 57.19, 69.32, 69.44, 108.51, 114.46, 114.93,
116.74, 117.38, 119.24, 123.92, 128.04, 128.51, 128.82, 131.49, 131.65, 136.74, 137.07, 137.23, 147.00, 160.29, 176.88; Anal. (C30H25FNO6P) C, H, N.
2-(3-Fluorophenyl)-6-methoxy-4-oxo-1,4-dihydroquinolin-5-yl dihydrogen phosphate (14). A suspension of 13 (250 mg, 0.46 mmol) in anhydrous MeOH (10 mL)
was hydrogenated in the presence of 10% Pd/C (125 mg) at 25 °C for 15 min. The catalyst and precipitate were collected and dissolved in 10% NaHCO3 solution
and then filtered. The filtrate was acidified with dil aq HCl and the precipitate was then collected and washed with acetone to give 14. Yellow solid; yield: 63.7%; mp 179181 °C; MS(ESI): m/z 366 (M + H)+; 1H-NMR (D
2O + NaOD, 200 MHz):
δ 3.76 (s, 3H), 6.53 (s, 1H), 7.05 (t, J = 8.4 Hz, 1H), 7.247.60 (m, 5H); Anal.
(C16H13FNO6P) C, H, N.
Sodium 2-(3-fluorophenyl)-6-methoxy-4-oxo-1,4-dihydroquinolin-5-yl phosphate
(15). To a stirred solution of NaHCO3 (0.34 g, 4.0 mmol) in H2O (20 mL) was added 14
(0.73 g, 2.0 mmol) at 0 ± 1 °C. After the addition was complete, the reaction mixture
was removed from the ice bath, stirred at 25 °C for 10 min and then filtered though Celite, after no dissolution from the solid was observed. The resulting filtrate (15
mL) was poured into acetone (60 mL), and kept in an ice bath for 1 h. The precipitate was collected and washed with ice-cooled acetone (10 mL×5). The
solid was dried under vacuum to give 15. Yellow solid; yield: 48.0%; mp >300 °C; MS(ESI): m/z 410 (M + H)+; 1H-NMR (D2O, 200 MHz): δ 3.72 (s, 3H), 6.54 (s,
3H), 6.99 (t, J = 7.8 Hz, 1H), 7.157.55 (m, 5H); Anal. (C16H11FNNa2O6P) C, H,
N.
Biological Assays
MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays.20, 21
L-60, HCT-116, Hep 3B, H460, Detroit 551 and HT29/FuR cells were treated with tested
compounds for the indicated periods. After treatment, cells were washed once with PBS and incubated with MTT (Sigma, St. Louis, MO, USA) for 2 h. The formazan precipitate
was dissolved in 150 μL of DMSO, and the absorbance was measured with an ELISA reader at 570 nm.
Mechanism of Action Study. Hep3B cells (5.3 × 107) were treated with 3b at different concentration or duration, and then placed in 1 ml of the lysis buffer (10% glycerol, 1% Triton X-100, 137 mM NaCl, 10 mM NaF, 1 mM EGTA, 5 mM EDTA, 1 mM sodium
pyrophosphate, 20 mM Tris-HCl, pH 7.9, 100 mM β-glycerophosphate, 1 mM sodium orthovanadate, 0.1% SDS, 10 µg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride, and
10 µg/ml leupeptin). The suspensions were centrifuged at 9000 g for 1 min in a model 3200 Eppendorf/Brinkman centrifuge, and the supernatant fraction was subsequently
centrifuged at 10,000 g for 60 min. protein content was determined against a standardized control, using the Bio-Rad protein assay kit (Bio-Rad Laboratories).
Molecular Modeling Study. Molecular flexible docking was performed by Dock 5.122. The Kollman partial charges were applied to protein models for force field calculation. Energy-optimized 3D coordinates and partial charges of small molecules were calculated
by Marvin 5.2.223, Balloon 0.624 and OpenBabel 2.2.325. There were 1000 orientations searched and 200 conformers generated per cycle identified in Dock program. The
docked conformers were then re-scored by HotLig26 to predicted protein-ligand binding modes. Protein structural superimposition was calculated and represented by Chimera
1.4.127.
Enzyme spectrum screen assay.28 MDS PharmaServices performed this testing under
In vivo antitumor activity assay. The Hep-3B tumor cell line was purchased from American Type Culture Collection (ATCC HB-8064, human ovarian carcinoma cells). The culture medium of DMEM, 90%; Fetal Bovine Serum, 10% and supplemented with
1% penicillin-streptomycin was used. The tumor cells were incubated in an atmosphere
containing 5% CO2 at 37 o
C.
Balb/c Nude mice used in this study were male, 4–6 weeks age, weighing 18–20g and provided by National Animal Center. All animals were housed in individually
ventilated cages racks (IVC Racks, 36 Mini Isolator system) under specific pathogen-free
(SPF) conditions throughout the experiment. Each cage (in cm, 26.7 length × 20.7 width × 14.0 height) was sterilized with autoclave and contained 8 mice. The animals were
maintained in a hygienic environment under controlled temperature (20–24 ºoC) and humidity (40%–70%) with a 12 hour light/dark cycle. The animals were given free
access to sterilized lab chow and sterilized distilled water ad libitum. All aspects of this
work, i.e., housing, experimentation and disposal of animals, were performed in general accordance with the Guide for the Care and Use of Laboratory Animals (National
Academy Press, Washington, D. C., 1996).
HB-8064) in male Balb/c Nude mice, compound 15 at doses 7.5, 15 and 30 mg/kg (IV or po, qd) was administered five days per week for four consecutive weeks and ceased at Day28.
The tumor size and body weight were monitored and recorded for 28 days. Human ovarian carcinoma cells (HEP-3B, ATCC HB-8064) with 2 × 106cells in 0.1 ml were
injected subcutaneously into the right flank of the mice. When the tumor growth reached >100 mm3 in volume (assumed as day 0), the tumor-bearing animals were assigned into
several groups (8 animals in each group) for study.
The body weight and tumor size were measured and recorded every 7 days during the
experiment periods of 28 days. Tumor volume (mm3) was estimated according to the formula of length × (width)2× 0.5 in mm3. Tumor growth inhibition was calculated as
T/C (treatment/control) by the following formula: T/C = (Tn – T0)/(Cn – C0) x 100% (T0:
Tumor volume of treated group in Day 0; Tn: Tumor volume of treated group in Day n;
C0: Tumor volume of control group in Day 0; Cn: Tumor volume of control group in Day
n).
Supporting Information Available: Literature references for enzyme spectrum screen assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments. The investigation was supported by research grants from the National Science Council of the Republic of China (NSC 96-2323-B-039-001; NSC
97-2323-B-039-001) awarded to S. C. Kuo, and the National Cancer Institute, NIH (CA17625-32) awarded to K. H. Lee.
References
1. Kuo, S.C.; Lee, H. Z.; Juang, J. P.; Lin,Y. T.; Wu,T. S.; Chang, J. J.; Lednicer, D.; Paull, K. D.; Lin, C. M. Synthesis and cytotoxicity of
1,6,7,8-substituted-2-(4’-substituted phenyl)-4-quinolones and related compounds: identification as antimitotic agents interacting with tubulin. J. Med. Chem. 1993, 36 (9), 11461156.
2. Li, L.; Wang, H. K.; Kuo, S. C.; Wu, T. S.; Lednicer, D.; Lin, C. M.; Hamel, E.; Lee, K.
H. Antitumor agents. 150. 2’,3’,4’,5’,5,6,7-Substituted 2-phenyl-4-quinolones and related compounds: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 1994, 37 (8), 11261135.
3. Li, L.; Wang, H. K.; Kuo, S. C.; Wu, T. S.; Mauger, A.; Lin, C. M.; Hamel, E.; Lee, K.
H. Antitumor agents. 155. Synthesis and biological evaluation of 3’,6,7-substituted
34003407.
4. Chou, L.C.; Chen, C. T.; Lee, J. C.; Way, T. D.; Huang, C. H.; Huang, S. M.; Teng, C.
M.; Yamori, T.; Wu, T. S.; Sun, C. M.; Chien, D. S.; Qian, K.; Morris-Natschke, S. L.; Hamel, E.; Lee, K. H.; Huang, L. J.; Kuo, S. C. Synthesis and preclinical evaluations
of 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one monosodium phosphate (CHM-1 - P-Na) as a potent antitumor agent. J. Med. Chem. 2010, 53 (4), 16161626.
5. Kuo, S. C.; Teng, C. M.; Lee, K. H.; Huang, L. J.; Chou, L. C.; Chang, C. S.; Sun, C.
M.; Wu, T. S.; Pan, S. L.; Way, T. D.; Lee, J. C.; Chung, J. G.; Yang, J. S.; Chen, C. T.; Huang, C. C.; Huang, S. M. Novel hydrophilic derivatives of 2-aryl-4-quinolones as
anticancer agents, WO 2008/070176 A1 (PCT), 2008. [Patent Applied in Taiwan (96146890)/USA (12/448088)/Australia (2007328034) /Canada (2670292)/China
(200780044796.9)/EU (07853279.3) /India (3052/EHENP/2009)/Japan (2009-540310)/Korea (10-2009-7014196)/New Zealand (577130)/Russian Fedeation
(2009124622)/South Africa (2009/03694)]
6. Chang, Y. H.; Hsu, M. H.; Wang, S. H.; Huang, L. J.; Qian, K.; Morris-Natschke, S. L.;
Hamel, E.; Kuo, S. C.; Lee, K. H. Design and synthesis of 2-(3-benzo[b]thienyl)-6,7-methylenedioxyquinolin-4-one analogs as potent antitumor
agents that inhibit tubulin assembly. J. Med. Chem. 2009, 52 (15), 48834891.
7. Hagmann, W. K. The many roles for fluorine in medicinal chemistry. J. Med. Chem.
2008, 51 (15), 43594369.
8. Press, J. B.; Bandurco, V. T.; Wong, E. M.; Hajos, Z. G.; Kanojia, R. M.; Mallory, R.
A.; Deegan, E. G.; McNally, J. J.; Roberts, J. R.; Cotter, M. L.; Graden, D. W.; Lloyd, J. R. Synthesis of 5,6-dimethoxyquinazolin-2(1H)-ones. J. Heterocyclic Chem. 1986,
23, 18211828.
9. Bandurco, V. T.; Schwender, C. F.; Bell, S. C.; Combs, D. W.; Kanojia, R. M.; Levine,
S. D.; Mulvey, D. M.; Appollina, M. A.; Reed, M. S.; Malloy, E. A.; Falotico, R.; Moore, J. B.; Tobia, A. J. J. Med. Chem. 1987, 30, 14211426.
10. Mizuta, Hironori.; Watanabe, S.; Sakurai, Y.; Nishiyama, K.; Furuta, T. Kobayashi, Y.; Iwamura, M. Design, Synthesis, Photochemical Properties and Cytotoxic Activities of
Water-Soluble Caged L-Leucyl-L-leucine Methyl Esters that Control Apoptosis of Immune Cells. Bioorg. Med. Chem. 2002, 10, 675683.
11. Unpublished data.
12. Riedemann, J.; Sohail, M.; Macaulay, V. M. Dual silencing of the EGF and type 1
IGF receptors suggests dominance of IGF signaling in human breast cancer cells.
13. Mayer, S. C.; Banker, A. L.; Boschelli, F.; Di, L.; Johnson, M.; Kenny, C. H.; Krishnamurthy, G.; Kutterer, K.; Moy, F.; Petusky, S.; Ravi, M.; Tkach, D.; Tsou, H.
R.; Xu, W. Lead identification to generate isoquinolinedione inhibitors of insulin-like growth factor receptor (IGF-1R) for potential use in cancer treatment. Bioorg. Med.
Chem. Lett. 2008, 18, 36413645.
14. Wood, E. R.; Shewchuk, L.; Hassel, A.; Nichols, J.; Truesdale, A. T.; Smith, D.;
Carter, H. L.; Weaver, K.; Barrett, G.; Leesnitzer, T.; Alvarez, E.; Bardera, A. I.; Alamillo, A.; Cantizani, J.; Martin, J.; Smith, G. K.; Jensen, D. E.; Xie, H.; Mook, R.;
Kumar, R.; Kuntz, K. Discovery of an inhibitor of insulin-like growth factor 1 receptor activation: Implications for cellular potency and selectivity over insulin receptor. Biochem. Pharmacol. 2009, 78(12), 14381447.
15. Favelyukis, S.; Till, J. H.; Hubbard, S. R.; Miller, W. T. Structure and autoregulation
of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001, 8(12), 10581163.
16. Wu, J.; Li, W.; Craddock, B. P.; Foreman, K. W.; Mulvihill, M. J.; Ji, Q. S.; Miller, W. T.; Hubbard, S. R. Small-molecule inhibition and activation-loop trans-phosphorylation of the IGF1 receptor. EMBO J. 2008, 27(14), 19851994.
Networking of differentially expressed genes in human cancer cells resistant to methotrexate. Genome Med. 2009, 1 (9), 83.
18. Paull, K.D.; Shoemaker, R. H.; Hodes, L.; Monks, A.; Scudiero, D. A.; Rubinstein, L.; Plowman, J.; Boyd, M. R. Display and analysis of patterns of differential activity of
drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81 (14), 10881092.
19. Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; Gray-Goodrich, M.; Campbell, H.; Mayo,
J.; Boyd, M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757766.
20. Chen, C. J.; Hsu, M. H.; Kuo, S. C.; Lai, Y. Y.; Chung, J. G.; Huang, L. J. (2E)-N,N-Dibutyl-3-(4-hydroxy-3-methoxyphenyl)acrylamide induces apoptosis and
cell cycle arrest in HL-60 cells. Anticancer Res. 2007, 27, 343−349.
21. Hsu, M. H.; Chen, C. J.; Kuo, S. C.; Chung, J. G.; Lai, Y. Y.; Teng, C. M.; Pan, S. L.;
Huang, L. J. 2-(3-Fluorophenyl)-6-methoxyl-4-oxo-1,4-dihydroquinoline- 3-carboxylic acid (YJC-1) induces mitotic phase arrest in A549 cells. Eur. J.
Pharmacol. 2007, 559, 14−20.
macromolecule-ligand interactions. J. Mol. Biol., 1982, 161, 269−288.
23. Marvin was used for drawing, displaying and characterizing chemical structures,
substructures and reactions, Marvin 5.2.2, 2009, ChemAxon (http://www.chemaxon.com)
24. Vainio, M. J.; Johnson, M. S. Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model., 2007, 47, 2462−2474.
25. Guha, R.; Howard, M. T.; Hutchison, G. R.; Murray-Rust, P.; Rzepa, H.; Steinbeck, C.; Wegner, J. K.; Willighagen, E. The blue obelisk - Interoperability in chemical
informatics. J. Chem. Inf. Model. 2006, 46(3): 991−998.
26. Wang, S.H., HotLig: A molecular surface-directed approach to scoring protein-ligand
interactions. Distributed by the author. Institute of Cellular and Organismic Biology, Academia Sinica, Taiwan. 2010.
27. Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. UCSF Chimera-a visualization system for exploratory research
and analysis. J. Comput. Chem., 2004, 25, 1605−1612. 28. References provided in Supporting Information.
Table 1. Cytotoxicities of CHM-1 and target compounds 1-5. N H O F CHM-1 O O N H O R2' O O R3' R4' 1a - c N H O R2' R3' R4' O O N H O R2' OH O R3' R4' N H O R2' R3' R4' OH HO N H O R2' O HO R3' R4' 2a - c 3a - c 4a - c 5a - c IC50a(M) Comp’d R2’ R3’ R4’ HL-60 HCT116 Hep3B H460 CHM-1 - - - 0.08 0.15 0.13 0.14 1a F H H 3.7 > 20 > 20 > 20 1b H F H 1.3 1.2 2.6 3.5 1c H H F 2.0 > 20 > 20 > 20 2a F H H 1.0 2.1 1.9 4.5 2b H F H 0.7 2.5 2.4 3.2 2c H H F > 10 > 10 > 10 > 10 3a F H H 0.07 0.05 0.05 0.11 3b H F H 0.03 0.07 0.07 0.08 3c H H F 1.8 2.4 11.0 8.8 4a F H H 0.5 0.6 3.9 4.1 4b H F H 0.3 8.2 6.9 6.1 4c H H F 38.6 > 100 100 100 5a F H H 1.3 5.8 5.3 4.4 5b H F H 0.9 1.1 5.3 4.8 5c H H F 38.2 > 100 > 100 > 100
Human tumor cells were treated with different concentrations of compounds for 48 h.
a Data are presented as IC
Table 2. Results of COMPARE correlation at GI50 level for compound 3b
Rank Compound (NCI number) ra Mechanism of Action Category
1 3HP (NSC 95678) 0.558 DNA antimetabolite
2 Trimetrexate (NSC 352122) 0.416 RNA/DNA antimetabolite
3 Methotrexate (NSC 740) 0.395 RNA/DNA antimetabolite
4 Rhizoxin (NSC 332598) 0.383 Antimitotic agent
5 Maytansine (NSC 153858) 0.379 Antimitotic agent
6 Thalicarpine (NSC 68075) 0.366 Inhibits the synthesis of DNA, RNA, and protein
7 Pancratiastatin (NSC 349156) 0.359 Inhibition of the cell cycle from G0/G1 to S phase
8 Paclitaxel (NSC 125973) 0.351 Antimitotic agent
9 DUP785 (NSC 368390) 0.348 RNA/DNA antimetabolite
10 5-Fluorouracil (NSC 19893) 0.34 RNA/DNA antimetabolite
Table 3. Inhibitory effects of 3b on enzyme spectrum screen assays for SPP.
Primary biochemical assay Species CONC. % INH
CYP450, 1A2 Hum 10 microM 85
Phosphodiesterase PDE4 Hum 10 microM 55
Phosphodiesterase PDE5 Hum 10 microM 57
Protein Tyrosine Kinase, YES1 Hum 10 microM 68
UDP Glucuronosyltransferase, UGT1A1 Hum 10 microM 86
※hum= human
※A standard error of the means in presented where results are based on multiple, independent determination.
Figure legends:
Figure 1. Structures of 2-PQs, CHM-1 and CHM-1–P-Na.
Figure 2. Structures of target compounds 1–5.
Figure 3. Differential activity patterns for compound 3b against 60 human cancer cell
lines. MG-MID: mean of log X values (X = GI50, TGI, and LC50). Delta: logarithm of
the difference between the MG-MID and the log X of the most sensitive cell line. Range: logarithm of the difference between the log X of the most resistant cell line and the log X of the most sensitive cell line.
Figure 4. Effect of 3b on basal receptor tyrosine phosphorylation (pTyr) in
Hep3B cells. Hep3B cells were treated with various concentrations of 3b at 37 ˚C for
12 h. Levels of phosphorylated IGF-1R and IGF-1R were analyzed by immunoblotting. Immunoblotting with -actin antibody demonstrated equivalent protein in each lane. Western blot data presented are representative of those obtained in at least three separate experiments.
Figure 5. (A) Chemical structure of compound 3b and PQIP. (B) Structural similarity
superimposable with 3b.
Figure 6. Structural superimposition of IGF1R model 1K3A and 3D94. The structural
features of IGF1R kinase were compared between ATP-bound (1K3A; yellow color) and PQIP-bound (3D94; magenta color) states. At PQIP-bound state, conformation of activation loop containing Tyr-1131, Tyr-1135 and Tyr-1136 changed dramatically, as compared with ATP-bound state.
Figure 7. Predicted binding mode of compound 3b with IGF1R kinase. Binding site of
3b is the same as that of 2-phenylquinolinyl segment of PQIP, which is beside the
ATP-binding site. The binding sites of ATP and peptide substrate are indicated as shaded regions according to model 1K3A.
Figure 8. Structure of PQIP can be identified as two segments, including ATP-superimposed scaffold and 3b-superimposed scaffold. Thus, 3b is probably an allosteric inhibitor, which might inhibit autophosphorylation of activation loop without interfering with ATP-binding.
(C) Mean body weight-time profiles in Hep3B xenograft nude mice (n = 11) following po dosing of doxorubicin at 10 mg/kg and 15 at 7.5, 15, and 30 mg/kg five days per week for four consecutive weeks.; (D) Mean tumor volume-time profiles (E) Mean tumor weight-time profiles (F) Mean body weight-time profiles in Hep3B xenograft nude mice (n = 11) following IV dosing of doxorubicin at 5 mg/kg and 15 at 7.5, 15, and 30 mg/kg five days per week for four consecutive weeks.
N H O R6 R7 R2' R3' 2-PQs N H O F CHM-1 O O N O F CHM-1-P-Na O O P O OH ONa Figure 1
c 0.125 0.25 0.5 1 (uM)
Compound 3b (12 hr)
p-IGF-1R
IGF-1R
β-actin
c 0.125 0.25 0.5 1 (uM)
Compound 3b (12 hr)
p-IGF-1R
IGF-1R
β-actin
Figure 4A B O O 10 O NO2 SnCl2 2H2O EtOH O O 7c O NH2 Scheme 1
N H O R2' O O R3' R4' 1a ~ c BCl3 CH2Cl2 N H O R2' OH O R3' R4' 3a ~ c N H O R2' R3' R4' O O H 2, Pd/C MeOH N H O R2' R3' R4' OH HO N H O R2' O O R3' R4' H2, Pd/C MeOH N H O R2' O HO R3' R4' 2a ~ c 6a ~ c 4a ~ c 5a ~ c
3a, 4a, 5a, R2' = F, R3' = H, R4' = H 3b, 4b, 5b, R2' = H, R3' = F, R4' = H 3c, 4c, 5c, R2' = H, R3' = H, R4' = F
![TraditionalMLCalgorithmsmainlytacklethebatchMLCproblem,wheretheinputdataarepresentedinabatch[24,28].Nevertheless,inmanyMLCapplicationssuchase-mailcategorization[22],multi-labelexamplesarriveasastream.Onlineanalysisistherefore dimensionreducermotivatedbyma](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)