國立臺灣大學植物病理與微生物學系 博士論文
Department of Plant Pathology and Microbiology
National Taiwan University Doctoral Dissertation
海芋 potyvirus 之基因體序列分析、快速檢測及應用 RNAi 防治其感染之研究
Studies on genome sequence, rapid detection and RNAi-mediated control for calla lily potyviruses
胡文綺 Wen-Chi Hu
指導教授:張雅君 博士 Advisor: Ya-Chun Chang, Ph.D.
中華民國 98 年 7 月
July, 2009
目錄
中文摘要………i
Abstract ………iii
Introduction………1
Chapter I……….7
Abstract ………..9
Introduction………...11
Materials and methods....……….15
Results………..21
Discussion……….26
References………29
Tables………33
Figures………..35
Chapter II………..39
Abstract………41
Introduction ……….42
Materials and methods……….45
Results………..50
Discussion………55
References………59
Tables………64
Figures………..66
Chapter III………....71
Abstract………73
Introduction………..75
Materials and methods……….82
Results………..92
Discussion..……….102
References ………110
Tables………..119
Figures...121
Supplemental figures ………..135
中文摘要
海芋為天南星科,馬蹄蓮屬之多年生球根花卉。為廣受喜愛之觀賞花卉,因 此也成為國際間重要之經濟花卉作物。海芋原產非洲,引進台灣已超過十年,除 了組織栽培技術已開發成熟外,也培育出許多新品系。然而病毒病害防治仍為彩
色海芋培育之重點。目前被報導過可感染海芋之植物病毒已有9 屬 18 種。其中
長絲狀病毒之 Potyyvirus 是最早被報導,且已知可感染之病毒種類較多的一屬。
Potyvirus 為單鏈正意股 RNA 病毒,其基因體大小約 10 kb。而在台灣以 Dasheen mosaic virus (DsMV), Turnip mosaic virus (TuMV)以及 Zantedeschia mild mosaic virus (ZaMMV) 和 Zantedeschia mosaic virus (ZaMV)等四種 potyvirus 為感染海芋 之重要病毒。由於 Potyyvirus 屬病毒為目前已知數量最多之植物病毒,為能便於
檢測及了解其分子特性,因此首先發展能快速選殖及分析 potyvirus 全長度基因
體序列之策略。此方法應用 Potyvirus 屬之多組廣效性引子對進行 RT-PCR 並配 合terminal transferase 修飾病毒 5’端序列,以 RT-PCR 獲得病毒 5’端序列,經解 序及序列分析後可獲得病毒之正確全長度序列。廣效性引子對之設計則是利用目
前已知病毒之胺基酸序列進行多序列排倂分析,分別選取選取NIb、CI 及 HC-Pro
基因之高保守性區域設計出NIbF1、CIF2/CIR2 及 HCF4/HCR4 等引子。以此廣
效性引子對分別對七種不同之 potyvirus 進行 RT-PCR 測試,可獲得預期大小之
PCR 產物。利用此策略成功獲得兩種海芋重要病毒 ZaMMV 及 ZaMV 之 5'端及 全長序列。經序列分析後發現ZaMV 與 Konjac mosaic virus (KoMV)應為同種病
毒。另一方面,為能快速且同時檢測DsMV、TuMV、ZaMMV 以及 ZaMV 四種
病毒,利用病毒專一性引子對研發多引子對RT-PCR 檢測法,並加入植物之 nad5
mRNA 之專一性引子對,作為增幅植物樣品的內在對照。經由專一性及靈敏度
測試後,發現此方法可在一次反應中成功的偵測出不同病毒,且其靈敏度高於利
本論文亦希望研究利用基因沉寂(RNA silencing)機制引發植物對入侵病毒的抗性
作用。同樣以DsMV、TuMV、ZaMMV 以及 ZaMV 四種病毒為對象。分析其胺
基酸序列中高保守性區域,依照 TuMV 序列設計數種專一性引子對,藉由 PCR
將不同長度之HC-Pro, NIa 及 CP 基因片段擴增並選殖至 LITMUS 38i 載體,用以
產生病毒雙股RNA (dsRNA)。當以 dsRNA 與 TuMV RNA 共同接種於 Nicotiana benthamiana 植物,可得知較短之 dsRNA 片段對於病毒侵入之干擾效果較長片段
為差。為進一步在植物上進行分析,將兩段分別位在HC-Pro 及 NIa 基因,且感
染效果較佳之病毒片段,利用農桿菌轉型方式(Agrobacterium-mediated infiltration)
在植物上表現其雙股之hairpin RNA (hpRNA)。除了有效的干擾 TuMV 感染外,
似乎對於另一種potyvirus,Bean yellow mosaic virus (BYMV)也稍具干擾性。因
此將上述兩段含hpRNA 之載體送入菸草,並獲得轉基因菸草。對於 T0 及 T1 植
物初步分析可發現,80%以上的 hpRNA 轉基因菸草皆對 TuMV 感染具有抗性。
Abstract
Calla lily (Zantedeschia spp.), belonging to the family Araceae, are perennial bulbous flowers. Because calla lily is a favorite ornamental flower, it becomes an important economic flower crop worldwide. Calla lily original from Africa, and have been introduced into Taiwan more than 10 years. Tissue culture technique has been developed for calla lily propagation and many calla hybrids have been bred in Taiwan.
However, the viral disease control is important in calla lily cultivation. There are already 18 viruses have been reported which belonging to 9 genus. Potyirus is first reported and mainly calla lily-infecting virus in the field. Potyvirus is a positive sense stranded RNA virus with ~10 kb genome. Four viruseses are important in Taiwan, including Dasheen mosaic virus (DsMV), Turnip mosaic virus (TuMV), Zantedeschia
mosaic virus (ZaMV), and Zantedeschia mild mosaic virus (ZaMMV). For detection
and molecular characterization of potyviruses, the largest plant virus, full-length cloning and sequencing strategy was developed. The RT-PCR-based methods for detection and identification of virus are based on the use of degenerate primers for RT-PCR amplification, combined with modified 5’RACE by using terminal transferase to modify the 5’ end sequence of potyviruses genome. The complete sequence would be identified by cloning and sequencing. The degenerate primers
sequence alignment, theNIbF1, CIF2/CIR2, and HCF4/HCR4 were designed from potyviral NIb, CI and HC-Pro-coding regions. Expected PCR products were amplified by these primers from seven potyviruses. Complete genome sequences of ZaMMV and ZaMV, were successfully characterized. The sequence analysis reveals that ZaMV is the same with the Konjac mosaic virus (KoMV). On the other hand, in order to save time and simultaneous detection of DsMV, TuMV, ZaMV and ZaMMV in field, a multiplex RT-PCR assay was developed for these calla potyviruses. Specific primers for each virus were designed based on the sequences of 3’ terminal region of respective viruses. To prevent the false negative results, a primer pair specific to plant mitochondrial nad5 mRNA was used as an internal control of RT-PCR. After specific and sensitivity test, the multiplex RT-PCR can rapidly detect multiple targets in one single assay, and the detection sensitivity of multiplex RT-PCR was 25-625 times higher than that of I-ELISA depending on the virus. Furthermore, a control method that prevents the virus infection using the mechanism of RNA silencing was investigated. A dsRNA expression and screening system was used to obtain highly efficient interference fragments from the consensus regions of four calla lily-infecting potyviruses, DsMV, TuMV, ZaMMV and ZaMV. The viruses were chosen for multiple sequence alignment. Several TuMV specific primers were designed within the conserved regions of HC-Pro, NIa and CP genes. Different fragments were PCR
amplified and cloned into LITMUS 38i vector to produce viral dsRNA. Mechanical inoculation of different dsRNA transcripts with target virus on tobacco plants induced different levels of interference with virus infection. Two of the most effective fragments (located in HC-Pro and NIa genes) were further analyzed by using
Agrobacterium tumefaciens infiltration (agoinfiltration) as transient expression system
for hairpinRNA (hpRNA) expression. The successful interferences of TuMV infection were observed in the infiltrated leaves. Besides, Bean yellow mosaic virus (BYMV), another calla lily-infecting virus, was interfered by TuMV hpRNAs transiently expressed in plants. Several lines of transgenic Nicotiana benthamiana plants transformed with hpHC and hpNIa fragments were obtained. Over 80% of T0 and T1 transgenic plants revealed resistance response to TuMV infection.
Introduction
Brief preface of calla lily
Calla lily is the common name of the genus Zantedeschia spp. The genus
Zantedeschia belongs to the Aracreae family. There are seven species and two
subspecies of Zantedeschia: Z. aethiopica (L.) Spreng., Z. rehmannii Engl., Z.
jucunda Letty, Z. elliottiana (Watson) Engl., Z. pentlandii (Watson) Wittm., Z.
odorata P. L. Perry, Z. albomaculata, Z. albomaculata (Hook.) Baill. subsp. Valida
Letty (Kuehny, 2000). It is a tropical plant native in Africa. Now the crops are as outdoor garden plants, commercial cut flowers and flowering potted plants. New calla hybrids have been bred for ornamental purpose. The common disease of calla lily is bacterial rot caused by Erwinia carotovora subsp. carotovora and virus diseases (Chen et al., 2003; Kuehny, 2000).
Characterization of Potyvirus
Potyvirus is the one of largest plant virus groups, belonging to the family of
Potyvidiae, genus of Potyvirus. The family Potyviridae is classified into six genera
including: Ipomovirus, Potyvirus, Macluravirus, Rymovirus, Tritimovirus, and
Bymovirus based on vector transmission and genomic relatedness (Adams et al.,
2005).Members of the genus Potyvirus have particles at least 700 nm in length which
encapsidate a monopartite ssRNA genome 10 kb in length. The genome is characterized by a 5’ untranslated region (5’UTR) which is linked to a genome-linked protein (VPg), a single major open reading frame (ORF) and a 3’ UTR region terminating in a polyadenylated (polyA) tail (Ha et al., 2008). The genome organization is conserved in the genus and contains ten mature proteins: P1, helper component protease (HC-Pro), P3, 6k1, cylindrical inclusion protein (CI), 6k2, small nuclear inclusion protein (NIa; including the VPg and protease (NIa-Pro) domains), large nuclear inclusion protein (NIb) and coat protein (CP) (Adams et al., 2005).
The first protein of the genome, P1, is the least conserved among potyviruses. The function of P1 involved in amplification was supposed to its correlation with HC-Pro because it was able to enhance HC-Pro-mediated suppression of RNA silencing (Pruss
et al., 2004; Kasschau and Carrington, 1998).
The HC-Pro protein named after its dual features of helper component (HC) for aphid transmission and protease activity is an important viral product that is involved in many functions during the virus life cycle (Urcuqui-Inchima et al., 2001).
Mutagenesis studies and sequence alignments suggest that HC-Pro can be schematically divided into three regions. N-terminal region of HC-Pro is required for the aphid transmission. Three conserved motifs Ileu-Gly-Asn (IGN), Cys-(Cys/Ser)-Cys (CC/SC) (CCC) and Pro-Thr-Lys (PTK) have been found in
HC-Pro (Cronin et al., 1995). Many plant viruses have evolved counterstrategies to knock out post-transcriptional gene-silencing (PTGS) and encode PTGS suppressors.
In potyviruses, HC-Pro protein acts as a PTGS suppressor to interfere with plant defense through the suppression of PTGS mechanisms (Kasschau et al., 1998).
Among potyviruses, there is a relatively little similarity in P3 proteins compared to other proteins (Urcuqui-Inchima et al., 2001). P3 is postulated to participate in replication by direct interacting with NIb which is part of replication complex (Merits
et al., 1999). The cylindrical inclusion protein (CI) has been proved to have RNA
binding and RNA helicase activity (Kadare and Haenni, 1997). Genetic analysis indicated the CI protein is also involved in cell-to-cell passage of viral RNA-protein complexes (Carrington et al., 1998). On the other hand, like P3, CI is also regarded as a determinant of virulence to a plant resistance gene (Jenner et al., 2002). The potyviral 6K2 protein has been shown to be membrane associated (Restrepo-Hartwig and Carrington, 1994).
NIa is one of the nuclear inclusion proteins and is composed of the N-terminal viral genome-linked protein (VPg) domain and the C-terminal NIa-proteinase (Pro) domain (Schaad et al., 1997). NIa is a major proteinase for producing most functional viral proteins from polyprotein (Urcuqui-Inchima et al., 2001). NIb also a nuclear inclusion protein is the viral RNA-dependent RNA polymerase (RdRp) responsible
for genome replication of potyviruses, but how the interaction affects viral replication is unknown (Urcuqui-Inchima et al., 2001).
The coat protein (CP) is the only structure protein of potyvirus and is also involved in several functions including aphid transmission, viral movement, RNA amplification and encapsidation (Urcuqui-Inchima et al., 2001).
The full-length cloning and sequencing strategy of potyviruses
The genome of potyvirus is a single-stranded, positive sense RNA, with a 3’
polyA tail, similar to mRNA in plant cells. Therefore, RT-PCR is used for viral genome sequence identification. The complete genome of potyvirus has typically been obtained by constructing cDNA library or by primer walking using primers designed from the polyA sequence and degenerate primers designed on conserved genomic regions (Ha et al., 2008).
For the 5’ end sequence acquired, the strategies applied in 5’end mapping of mRNA are used in the RNA virus, including primer extension, cDNA library technique, and rapid amplification of cDNA ends (RACE), etc. The former two methods require relatively large amounts of RNA and often possess difficulty in identifying the 5' end of rare RNAs. Thus, RT-PCR with 5’RCAE is the common method for full-length genome sequencing of potyvirus. The 5’RACE was designed to
amplify the unknown 5’ end of a messenger RNA template using a defined internal site. Several alterations of this method were used for different purpose, as anchor RACE which introduced an oligonucleotide into the RNA or first-strand cDNA by RNA ligase; cRACE (circular first-strand cDNA-mediated RACE) were reported (Maruyama et al., 1995). Several commercial products were already developed.
PCR-based detection methods for plant RNA viruses
Several detection methods for plant RNA viruses were developed including protein-based enzyme-linked immunosorbent assay (ELISA) which is common usage method for many kinds of viruses. The other methods are PCR-based methods including reverse transcription-polymerase chain reaction (RT-PCR), immunocapture RT-PCR (IC-RT-PCR), multiplex RT-PCR and real-time PCR (Huang et al., 2005; Hu
et al., 2007; Huang et al., 2007). This detection method is based on targeting to the
viral nucleic acids. Most of these methods are designed for single virus detection in one reaction, except multiplex RT-PCR and TaqMan real-time RT-PCR (Lee and Chang, 2008). The multiplex RT-PCR technique has been applied to many plants for virus detection such as apple, (Menzel et al. 2002), citrus (Roy et al. 2005), olive (Bertolini et al. 2001), potato (Nie and Singh 2000; Du et al. 2006) and orchids (Lee and Chang 2006).
dsRNA triggered-PTGS in plant virus defense
Post-transcriptional gene silencing (PTGS) is a sequence-specific RNA degradation mechanism first discovered in transgenic plants (Napoli et al., 1990). It is closely related to RNA interference (RNAi) or RNA silencing in animals and other organisms, as well as it represents an ancient eukaryotic genetic phenomenon, which is also involved in the defense against viruses (Tenllado and Diaz-Ruiz, 2001;
Waterhouse et al., 2001). The pathway of PTGS is triggered by Dicer-like (DCL, the ribonuclease III-like enzymes) proteins in plants. These proteins play the role to cut dsRNAs from virus into siRNA (small interfering RNA) (Waterhouse and Fusaro, 2006). The dsRNA andhpRNA have been demonstrated that the double-stranded form RNAs can induce PTGS more efficiency than single-stranded RNA in plants (Smith et
al., 2000). In recent years, by applying dsRNA-derived from viral sequences to induce
RNA silencing and confer virus resistance in host plants was reported in various kinds of plant viruses (Tenllado et al., 2003; Vargas et al., 2008), such as the coat protein genes of different viruses (Wisniewski et al., 1990; Kalantidis et al., 2002; Vargas et
al., 2008) and viral suppressor, HC-Pro (Negri-Nicola et al., 2005), or viral replicase,
54-kDa protein of Pepper mild mottle virus (PMMoV) (Tenllado et al., 1995).
Chapter I
Molecular analysis of two potyviruses in Zantedeschia spp.
plants in Taiwan
Abstract
The full-length genomic sequences of two potyviruses, Zantedeschia mosaic virus (ZaMV) and Zantedeschia mild mosaic virus (ZaMMV) from Zantedeschia spp. in Taiwan were determined. One universal d(T) primer combined with a potyviral degenerate primer, NIbF1, which was designed from the consensus sequences in NIb gene, were used to amplify the 3’ portion (from polyA to 3’ portion of NIb gene) of potyvirus genome by RT-PCR. Two pairs of degenerate primers located in the CI (CIF2/CIR2) and HC-Pro (HCF4/HCR4) coding regions were designed according to the conserved sequence and could amplify specific fragments of potyviruses.
Virus-specific primers were designed according to the cloned sequences to amplify the intervening sequences. The 5’ terminal region was cloned by 5’RACE method that used terminal transferase to add extra oligomers to the end of cDNA and followed by PCR reaction. The complete sequence of ZaMV is 9852 nts and 3096 amino acids.
Sequence analysis of ZaMV showed that it has 90% nucleotide sequence identity to the Konjak mosaic virus F isolate (KoMV-F), but has 12 more AT-rich nucleotides compared with the 5’UTR sequence of KoMV. However, it indicated that ZaMV and KoMV is the same species of the genus Potyvirus. Moreover, the complete genome of ZaMMV is 9973 nts and 3176 amino acids. The sequence analysis reveals that ZaMMV is an individual member of Potyvirus group. This RT-PCR method along
with degerate primers was effective to obtain specific fragments from potyvirus member. Combined with the terminal transferase involved 5’ RACE method could easily acquire a full-length sequence of Potyvirus species. This strategy has widely appliction in detection and molecular sequencing of potyviruses.
Introduction
The genus Potyvirus belonging to the family Potyviridae contains over 200 species and more than 1,000 plant species were infected with different potyviruses (Adams et al., 2005). The genome of a potyvirus is positive-sense, single-stranded polyadenylated RNA molecule about 10 kb, covalently linked with a virus-encoded VPg protein at 5’ end. The genome contains a large unique open reading frame (ORF), which encodes single polyprotein. Pre-mature polyprotein is processed by three viral proteinases, P1, HC-Pro, and NIa, respectively, into 10 functional proteins: the first N-terminal protein (P1), the helper-component proteinase (HC-Pro), the P3 protein, the cytoplasmic inclusion protein (CI), the nuclear inclusion protein a (NIa) containing VPg and proteinase domains, the nuclear inclusion protein b (NIb) also the RNA-dependent RNA polymerase (RdRp), the coat protein (CP), and two small proteins 6K1 and 6K2 (Adams et al., 2005). According to the gene function and multiple sequence analysis, HC-Pro, CI, NIa, NIb, and CP genes showed the conserved characterization intra-genus. On the other hand, the 5’UTR, P1 protein, P3 protein, and 3’UTR region are species specific (Zheng, 2008).
Furthermore, due to the large genome size of potyvirus, complete genome sequence data is not easily acquired. Although Tobacco etch virus (TEV) was reported in 1921, the complete genome sequence was not published until 1986 (Allison et al.,
1986). Only six species of potyviruses have been completely cloned and sequenced between 1985 and 1992 including TEV, Tobacco vein mottling virus (TVMV) (Domier et al., 1986), Plum pox virus (PPV) (Lain et al., 1989; Maiss et al., 1989),
Potato virus Y (PVY) (Robaglia et al., 1989) and Pea seed-borne mosaic virus
(PSbMV) (Johansen et al., 1991), Turnip mosaic virus (Nicolas and Laliberté, 1992).
Although observation of new species of potyvirus constantly reported in resent years, the most complete genome sequences are the new isolates of known potyvirus species.
Because of the large RNA genome, reverse-transcription polymerase chain reaction (RT-PCR) method must be used in molecular cloning of potyvirus group. However, to amplify the long PCR fragments, purified viral RNA or large amount total RNA were required. Besides, it is difficult to obtain and handle whole genome or even long cDNA fragment until resent years. Another question is about the 5’UTR, the very diverge region of Potyvirus, and an AU-rich conserved ‘Poty box’ sequence is located at 5’UTR region of most Potyvirus species (Chen et al., 2001). However, the precisely sequence of the 5’-proximal end is still not easily to identify. This is the problem to acquire full-length genome sequence data of a new potyvirus.
Here we used degenerate primer RT-PCR method to amplify several species of potyviruses. According to the sequence alignments of three conserved gene regions,
and PHCR4, respectively were designed to amplify the cDNA fragments of different potyviruses. For identification the 5’ end sequence of potyvirus, a modified rapid amplification of 5' complementary DNA ends (5’RACE) method was developed. This method used terminal transferase for transferring additional deoxynucleotides into the 3’end of cDNA, and followed by PCR amplification which was amplified by specific viral primer and oligo primer such as dT or dC primers, instead of RCAE kit. Using this combined method, there were at last two complete sequences of new reported potyviruses, Zantedeschia mosaic virus (ZaMV) and Zantedeschia mild mosaic virus (ZaMMV), obtained and used for sequence analysis. The other new reported potyvirus, Basella rugose mosaic virus (BaRMV), was also completely sequenced by the same cloning strategy (Jhu, 2005; unpublished data).
Zantedeschia spp., commonly called calla lily, belongs to the family Araceae and
is originated from Africa. Several cultivars of Zantedeschia plants were introduced into Taiwan from New Zealand, the USA and the Netherlands about twenty years ago.
Since 1998, a potyvirus was found in calla lilies in Taichung area, the molecular evidence suggested a new species of potyvirus at that time, and thus called Zantedeschia mosaic virus (ZaMV). Several isolates were obtained during 1998 - 2000 in Taiwan, that were named ZaMV-BG, ZaMV-DB, ZaMV-SG, and ZaMV-Zan (Chen, 1998). ZaMV could cause mosaic symptom on Zantedeschia spp. and
Philodendron selloum but could not infect other indicator plants such as
Chenopodium amaranticolor (Chenopodiaceae), Nicotiana benthamiana
(Solanaceae), etc. The virus was also detected and reported in South Korea in 2002 (ZaMV-KR) (Kwon et al., 2002). In addition, another potyvirus caused mild mosaic symptom of tissue-cultured seedlings of calla lily was observed in 2000 also at Taichung area by Huang and designated as Zantedeschia mild mosaic virus (ZaMMV).
In addition to Dasheen mosaic virus (DsMV), the most common potyvirus in aroid plants, another potyvirus Turnip mosaic virus (TuMV), was also reported in Taiwan.
According to the virus detection on field survey in 2003-2004 (Huang et al., 2007) and 2005-2006 (data not shown), ZaMV and ZaMMV were widespread within mid-Taiwan. To study the molecular characterization of ZaMV and ZaMMV, the complete genomic sequences of ZaMV-SG isolate (from Zantedeschia spp. cv. Super Gold) and ZaMMV-Zun isolate (Huang and Chang, 2005) were obtained using the degenerate primer RT-PCR and modified 5’ RACE method described above. The genomes of ZaMV and ZaMMV are 9852 and 9973 nt long, respectively. Sequence analysis with other potyviruses indicated that ZaMV is the same species as Konjak
mosaic virus (KoMV) (Nishiguchi et al., 2006). However, ZaMMV is a new calla
lily-infecting potyvirus.
Materials and methods
Virus source and sample collection
The viruses used in this study include Basella rugose mosaic virus (BarMV),
Dasheen mosaic virus (DsMV), Potato virus Y (PVY), Turnip mosaic virus (TuMV),
Zucchini mosaic virus (ZYMV), Zantedeschia mild mosaic virus (ZaMMV) and
Zantedeschia mosaic virus (ZaMV). PVY and TuMV were maintained in Nicotiana
benthamiana plants. ZYMV was inoculated and maintained in Zucchini squash. In
addition, DsMV, ZaMMV and ZaMV were maintained in calla lily plants. Inoculated plants were grown in pots at 25oC under a 16/8-h photoperiod with 60% humidity.
Plant total RNA extraction
Total RNA was extracted from 100 mg of infected plant tissue using Plant Total RNA Miniprep System (Viogene, Sunnyvale, CA, USA) according to the manufacturer’s protocol. The leaf tissue was ground into powder with liquid nitrogen and mixed with 450 μl of RX or PRX extraction buffer, the lysate was filtrated using a Shearing Tube. And then the filtrate was mixed with 230 μl of absolute ethanol, transferred to a new Plant Total RNA Mini Column. After centrifugation to remove filtrate, the column was then washed once with WF Buffer and twice with WS Buffer.
Finally, plant total RNA was eluted with 50 μl of RNase-free ddH2O. The amount and
quality of total RNA was analyzed by 1% agarose gel electrophoresis. Plant total RNA was used directly for RT-PCR or stored at -20oC for further use.
Degenerate primers
Degenerate primers were designed based on amino acid sequence alignment of the conserved regions in NIb, CI, and HC-Pro genes of potyviruses. The NIb degenerate primer, NIbF1, corresponds to the ‘GNNSGQP’ motif. The CIF2 and CIR2 primers are corresponding to the ‘TRPLAEN’ and ‘IENGVDLT’ motifs within the CI genes.
HCF4 and HCR4 degenerate primers are located in the ‘PSLCDNQ’ and
‘KxFTKMVR’ motifs in the HC-Pro gene. The sequence and corresponding motifs of universal and degenerate primers used in the genome amplification of potyvirus groups are list in Table 1.
First-strand cDNA synthesis and viral genome cDNA cloning strategy
For first-strand cDNA synthesis, 8.5 μl of plant total RNA (0.4 μg/μl) and 2 μl of 10 μΜ dT-Bam primer (Hu and Chang, 2004) were mixed and denatured at 65oC for 10 minutes and then placed on ice for 5 minutes. For 25 μl reverse transcription (RT) reaction, 1.0 μl of AMV reverse transcriptase (10 U/μl) (Promega, Madison, WI, USA), 5 μl of AMV RT 5X reaction buffer (Promega, Madison, WI, USA), 1 μl of 10
mΜ dNTPs, 0.5 μl of rRNasin (40 U/μl) (Promega, Madison, WI, USA) and 7 μl of sterile distilled water were added to the previous mixture and incubated at 42 oC for 1 hour.
Double-stranded cDNA fragments were produced by polymerase-chain reaction (PCR) methods. Different strategies were used to clone viral genome from the cDNA templates. First, three degenerate primer pairs, PNIbF1/dT-Bam, PCIF2/PCIR2, and PHCF4/PHCR4, were used to amplify the conserved domains of the potyvirus NIb, CI and HC-Pro gene respectively. Second, specific primers were designed based on the nucleotide sequences of above fragments in order to amplify the intervening genomic fragments between them (Fig. 1A). PCR reactions were performed using 0.5 μl of DyNAzyme™ II DNA polymerase (2 U/μl, Finnzymes Inc., Espoo, Finland) in total 20 μl PCR mixture, containing 1 μl of 10 mM dNTPs, 1 μl each of 5 mM sense and antisense primers, 2 μl RT product, 2 μl of 10X DyNAzymeTM II DNA polymerase buffer and 12.5 μl sterile distilled water. The amplification protocol involved one cycle of 94oC for 5 min; 26~28 cycles of 94oC for 30 sec, 55oC for 35 sec; 72oC for 2 min; and a final 72oC extension for 7 min. The annealing temperature and extension time of individual PCR reaction depended on the primer pair used. PCR products were analyzed by 1% agarose gel electrophoresis, and expected amplification products were eluted by GFX™ PCR DNA and Gel Band Purification Kit (GE
Healthcare, Buckinghamshire, UK). The purified PCR fragments were ligated into the pGEM-T® Easy Vector (Promega, Madison, WI, USA) or pCR®II-TOPO® Vector (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol, and transformed into E. coli DH5α. After blue white selection and the colony PCR screening, plasmid DNAs were purified by Mini-M™ plasmid DNA Extraction System (Viogene, Sunnyvale, CA, USA) and checked by the restriction enzyme digestion to get correct clones for advanced sequencing.
5’ RACE method
Sequence of 5’-terminal of virus genome was determined using 5’ rapid amplification of cDNA ends (RACE) procedure. The 5’RACE method added extra nucleotides to the first-strand cDNA-end by terminal transferase. A virus specific primer was used to prime first-strand cDNA synthesis as described above except the dT-Bam primer was replaced by viral specific primers near the 5’ end. After RT reaction, the cDNA was precipitated with ethanol and resuspended in one third volume of distilled water. To add homopolymeric dC or dG tail to the cDNA 3’ end, 10 μl of condensed RT product, 0.5 μl Terminal Transferase (20 U/μl, NEB, Beverly, MA, USA), 5 μl of 10X NEB Buffer 4, 5 μl of 2.5 mM CoCl2, 1 μl of 10 mM dCTP (or dGTP) and 28.5 μl distilled water were mixed and incubated at 37oC for half hour.
The enzyme was then inactivated by incubating the reaction solution at 70oC for 20 minutes. dG-Not (to complement the dC oligo-mer), dC-T7 (complementary of dG oligomer) and virus-specific primers were used for PCR reaction. To obtain authentic 5’ terminal sequence, modified RACE was performed with dG-Not or dC-T7 primer and another specific primer closer to the 5’ end of virus genome than the primer used for first-strand cDNA synthesis.
Sequencing and sequence comparison
The cDNA clones were sequenced using universal primers or virus specific primers with ABI PRISM® BigDye™ terminator cycle sequencing ready reaction kit (Perkin-Elmer Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions. Automated sequencing was done on ABI PRISM™ 310 Genetic Analyzer (Perkin-Elmer Applied Biosystems, CA, USA). Each nucleotide of the viral genome was determined by sequencing at least two independent clones.
Sequence analyses and assembly were done using programs from the Wisconsin (GCG) package. The sequence comparisons of ZaMV and ZaMMV to known viral sequences using BLAST program at the National Centre for Biotechnology Information (NCBI) (http://www.ncbi.nih.gov/NCBI). The entire ORFs comparisons with other potyviruses were performed using program Align X in Vector NTI Suit 9.0
(Invitrogen) and ClustalX (Thompson et al., 1997). The alignment results were exported by GeneDoc program (Nicholas et al., 1997).
.
Results
Conserved degenerate primers designation
Amino acid sequences of potyviruses were aligned and used for degenerate primer design. The first primer set, HCF4/HCR4, corresponding to the ‘PSLCDNQ’ motif in the central region and ‘KxFTKMVR’ motif in C-terminal of HC-Pro gene were designed to amplify an approximately 0.6-kb product within HC-Pro coding region.
The second pair of degenerate primers, CIF2/CIR2, were designed based on the sequence of ‘TRPLAEN’ and ‘IENGVDLT’ motifs in mid-region of CI gene and were able to amplify about 0.6-kb fragment from potyvirus groups. For the 3’ terminal region cloning, the ‘GNNSGQP’ conserved motif of NIb (RNA dependent RNA polymerase) gene was selected for NIbF1 primer (Table 1).
Degenerate primer RT-PCR amplification assay
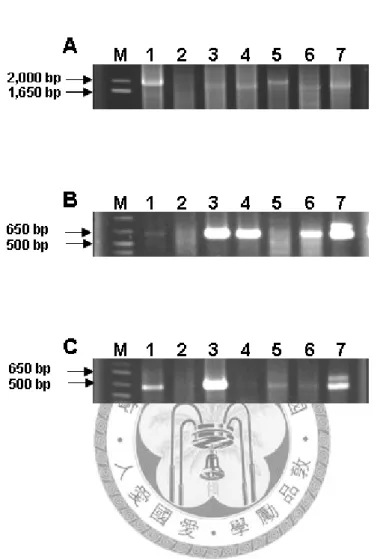
To evaluate the specificity of the degenerate primers, seven potyviruses species were tested by RT-PCR using dT-Bam/NIbF1, CIF2/CIR2 and HCF4/HCR4 primer pairs. In the RT reaction, dT-Bam primer reacted with the poly-A tail of viral RNA present in total RNAs of infected plants. A 1.6 ~ 1.8-kb cDNA fragments were then amplified by PCR using NIbF1 and dT-Bam primers from BarMV, DsMV, PYV, TuMV, ZaMMV, ZaMV, and ZYMV-infected plant samples (Fig. 2A). The
amplification efficiency varied in different virus-infected samples. It could be due to either the variation of the virus titer in infected plants or the non-optimized RT-PCR reaction, etc. The CIF2/CIR2 and HCF4/HCR4 primers amplified fragments of expected ~0.6-kb products from the same total RNA samples of seven potyviruses (Fig. 2B, and 2C). The results of RT-PCR suggested that degenerate primer-based PCR method is useful in amplification of several members of Potyvirus species. It indicated the application of these primers not only in cloning of specific fragments but also in potyvirus detection.
Amplification and sequencing of the complete genomes of ZaMMV and ZaMV
According to the degenerate primer and RT-PCR-based strategy, three expected size fragments which were amplified from HC-Pro, CI, and NIb regions of ZaMV and ZaMMV-infected samples were cloned into pGEM-T® Easy Vector. After colony PCR selection, three independent clones were sequenced. Sequencing data showed that the dT-Bam/NIbF1 amplified DNAs contained nucleotide sequences corresponding to the partial NIb gene, complete CP gene and 3’ UTR followed by a poly-A tail. Three 0.6-kb fragments amplified by CIF2 and CIR2 clones were proved to contain part of CI gene. And other three clones derived from HCF4 and HCR4 were confirmed to include the HC-Pro fragment. The subsequent RT-PCR using
virus-specific primers which were designed from previously sequenced fragments was to amplify and obtain the interval sequences (Fig. 1A).
The 5’-terminal sequence of viral genomic RNA including 5’UTR was resolved by using the 5’RACE method. Specific primer located at 5’-end of sequenced HC-Pro gene was used to initiate the synthesis of first-strand cDNA of 5’-terminal sequence of viral RNA. After dC (ZaMMV) or dG (ZaMV) tailing of cDNA was performed, PCR was carried out with dG-Not or dC-T7 and virus-specific primers. PCR products were analyzed by agarose gel and about 2 ~ 3-kb cDNA fragments were eluted from the agarose gel and cloned into vector as before. The 5’-terminal region consists of complete 5’UTR, P1 gene and part of HC-Pro gene. To confirm the sequence of the 5’-terminal region of the viral genome, 5’UTR was amplified again by PCR using dG-Not (dC-T7) and specific primer near the 5’ end of P1 gene with the same cDNA template tailing with oligo-nucleotids (data not shown).
The full-length of ZaMMV is 9973 nts excluding polyA tails, and encoded an open reading frame of 3176 amino acids. The genome of ZaMV is 9852 nts and encodes a putative polyprotein of 3096 amino acids. The 5’UTR of ZaMV and ZaMMV were A/T-rich with terminated in several A residues, typical of other potyviruses (Fig. 3A). The 5’UTRs also contained sequences similar to the potybox-like blocks: “TGAACAG” in ZaMMV and “TCAACAG” in ZaMV (Fig.3A)
(Chen et al., 2001; Ha et al., 2008).
Sequence comparisons
The entire ORF of ZaMV and ZaMV were compared with other potyviruses using BLAST program. The result revealed that ZaMMV is most similar to Soybean mosaic
virus (SMV) (68% amino acid sequence similarity of polyprotein). The P1 protein of
ZaMMV had 50% sequence similarity to Bean common mosaic virus (BCMV-NL1).
The amino acid sequence comparisons of polyprotein confirmed the phylogenetic analysis data of CP sequence, that ZaMMV belongs to the BCMV subgroup (Huang and Chang, 2005). Comparison of the putative amino acid sequence of HC-Pro among DsMV, TuMV, ZaMMV and ZaMV revealed the similarity is ranging from 60%
~77%, and HC-Pro gene of ZaMMV is closer to DsMV (77% similarity) than TuMV and ZaMV. Sequence comparisons also revealed several functional motifs of HC-Pro present in ZaMMV and ZaMV genomes such as PTK motif involved in aphid transmission, and CCC, the systemic movement functional motif (data not shown) (Cronin et al., 1995).
However, the sequence comparison of the full-length sequence of ZaMV and KoMV revealed high homology (96% amino acid sequence similarity, 90%
nucleotide sequence identity) (Table 2). It indicated that ZaMV is the same species as
KoMV. Interestingly, the 5’UTR sequence of KoMV is 152 nt but ZaMV is 167 nt.
The sequence identity between KoMV and ZaMV is 81% in 5’UTR region, which is lower than entire genome sequence. However the 3’UTR alignment shows 93%
sequence identity between them (data not shown). In addition, the complete sequence of ZaMV compared with KoMV and other three calla lily-infecting potyviruses including DsMV, TuMV, and ZaMMV indicated these viruses are distinct species, except KoMV and ZaMV (Table 2). Moreover, the 5’UTR comparison revealed 50%
sequence identity between DsMV and ZaMV (Table 2). The sequence alignments of DsMV and ZaMV showed there is a highly identical 40-bp sequence at 5’end of 5’UTR between these two viruses (Fig. 3B).
Discussion
In this work, several degenerate primers were designed for potyviruses. The GNNSGQP region of the NIb gene is highly consensus in members of Potyvirus group. In 1997, Gibbs and Mackenzie reported a primer that matched the GNNSGQ motif of the NIb and used the primer to amplify cDNA from potyviruses as well as other genera in the family Potyviridae (Gibbs and Mackenzie, 1997). A few years later, Chen et al. published a degenerate primer which was extended version of the GNNSGQP primer with a few more nucleotides in 2001 (Chen et al., 2001). In this study, the NIbF1 primer also corresponds to GNNSGQP motif and is successfully used for RT-PCR amplification of potyviruses.
The CIF2 and CIR2 degenerate primers located on the segment Ia and segment V within the CI gene (Kadare and Haenni, 1997) are different from the CIFor and CIRev degenerate primers which were designed in segment I and V of CI gene (Ha et al., 2008). Besides, the fragments amplified by CIFor/CIRev are ~100 bp longer than CIF2/CIR2. The degenerate primers of HC-Pro, HCF4/HCR4, designed to amplify the 3’ half region of the HC-Pro gene of potyvirus (Fig. 1A) are also different from the recently published degenerate primers HCFor and HCRev (Ha et al., 2008). Using these degenerate primers could easily amplify the central (CI) and near 5’ region (HC-Pro) of potyviral genome.
For complete genome sequencing of potyvirus, 5’RACE is a useful method. To improve the anchoring efficiency of the first strand cDNA, the terminal transferase was used for transferring the homopolymer into cDNA end instead of adaptor ligation or oligomer annealing of other RACE method. Because of the A residues often in the first 1 ~ 4 nt of 5’UTR region of potyviruses, dC or dG was chosen for modification the cDNA end. The advantages of this strategy include: 1) easily amplifying the genome fragments of unknown sequence of new potyvirus species and 2) efficiently obtaining the variable 5’ region of potyviruses. Besides the application to complete sequencing of potyvirus genome, the concept of this strategy is simple and suitable for other plant viruses as long as the conserved degenerate primers exist.
Using the degenerate primers in combination with genome-specific primers and modified 5’RACE. We characterized the complete genomes of two calla lily-infecting potyviruses. Through the sequence analysis, ZaMMV is proved a distinct species of potyviruses as previously reported (Huang and Chang, 2005). Although the sequence analysis indicated ZaMV should be an isolate or strain of KoMV, the biological characterization of these viruses is not similar. The major difference of these two viruses is the host range. KoMV F was isolated from konjak plants with mosaic symptoms but also showed a wide range of host plants including Aizoacea, Araceae,
Chenopodiacea and Solanacea plants (Nishiguchi et al., 2006). In contrast to KoMV,
ZaMV has a narrow host range only in Araceae plants (Chang et al., 2001; Kwon et
al., 2002). The other difference is the 5’UTR sequence between ZaMV and KoMV.
When compared the first 50 nt of 5’UTR region of ZaMV and KoMV with other eight potyviruses, all show AAA(A/T)TAAA at the first eight residues except KoMV (Fig.
3A). Because the biological features are so different in these two viruses, we have no information about the infectivity of the ZaMV isolated from Taiwan or Korea to konjak (Amorphophallus konjac K. Koch, family Aracea) plant and vice verse. This means more molecular and biological information are needed before we can understand the relationship of ZaMV and KoMV.
The full-length cloning and sequencing strategy developed in this study successfully detected numerous of potyviruses including a new potyvirus, BarMV (Huang and Chang, 2006) and several other viruses: DsMV, PVY, TuMV, and ZYMV. Due to the efficiency and convenience, using this method should promote the molecular characterization of potyvirus, the important group of plant RNA viruses.
References
Adams, M. J. Antoniw, J.F., and Fauquet, C. M. 2005. Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol. 150:
459–479.
Allison, R., Johnston, R.E., and Dougherty, W.G. 1986. The nucleotide sequence of the coding region of tobacco etch virus genomic RNA: Evidence for the synthesis of a single polyprotein. Virology 154: 9-20.
Chang, Y.-C., Chen, Y.-L., & Chung, F.-C. 2001. Mosaic disease of calla lily caused by a new potyvirus in Taiwan. Plant Dis. 85: 1289.
Chen, J. Chen, J. Chen, J., and Adams, M. J. 2001. Molecular characterization of an isolate of Dasheen mosaic virus from Zatedeschia aethiopica in China and comparisons in the genus Potyvirus. Arch Virol. 146: 1821-1829.
Cronin, S., J. Verchot, R. Haldeman-Cahill, M. C. Schaad, and J. C. Carrington. 1995.
Long-distance movement factor: a transport function of the potyvirus helper component proteinase. Plant Cell 7: 549-59.
Domier, K. M., Franklin, M., Shahabuddin, G. M., Hellmann, J. H., Overmeyer, S. T., Hiremath, M. F. E., Siaw, G. P. Lomonossoff, J. G., and Rhoads, R. E. 1986.
The nucleotide sequence of tobacco vein mottling virus RNA, Nucleic Acids Res.
14: 5417–5430.
Gibbs, A, J., Mackenzie, A. 1997. A primer pair for amplifying part of the genome of all potyvirids by RT-PCR. J Virol. Methods 63: 9–16.
Ha, C., Coombs, S., Revill, P. A., Harding, R. M., Vu, M., and Dale, J. L. 2008.
Design and application of two novel degenerate primer pairs for the detection and complete genomic characterization of potyviruses. Arch Virol. 153: 25-36.
Huang, C. H., and Chang, Y. C. 2006. Basella rugose mosaic virus, a new potyvirus infecting Basella rubra. Plant pathol. 55: 819.
Huang, C.-H., and Chang, Y.-C. 2005. Identification and molecular characterization of Zantedeschia mild mosaic virus, a new calla lily-infecting potyvirus. Arch Virol.
150: 1221-1230.
Hu, W.-C., and Chang, Y.-C. 2004. A new mosaic disease of Amazon lily in Taiwan.
Plant Pathol. 53: 240.
Jhu, H.-J. 2005. Molecular cloning and characterization of Basella rugose mosaic
virus and the isolate infecting Anredera cordifolia. Master thesis. National Taiwan
University. Taipei, Taiwan.
Johansen, E., Rasmussen, O. F., Heide, M., and Borkhardt, B. 1991. The complete nucleotide sequence of pea seed-borne mosaic virus RNA. J. Gen. Virol. 72:
2625-2632.
Kadare, G., and Haenni, A.-L. 1997. Virus-encoded RNA helicases. J. Virol. 71:
2583-2590.
Kwon, S. B., Ha, J. H., Yoon, J. Y., and Ryu, K. H. 2002. Zantedeschia mosaic virus causing leaf mosaic symptom in calla lily is a new potyvirus. Arch Virol. 147:
2281-2289.
Nicolas, O., and Lalibert, J.-F. 1992. The complete nucleotide sequence of turnip mosaic potyvirus RNA. J. Gen.l Virol. 73: 2785-2793
Nicholas, K. B., Nicholas, H. B., and Deerfield, D. W. 1997. GeneDoc: analysis and visualization of genetic variation. Embnew News. 4: 14.
Nishiguchi, M., Yamasaki, S., Lu, X.-Z., Shimoyama, A., Hanada, K., Sonoda, S., Shimono, M., Sakai, J., Mikoshiba, Y., and Fujisawa, I. 2006. Konjak mosaic virus: the complete nucleotide sequence of the genomic RNA and its comparison with other potyviruses. Arch Virol. 151: 1643–1650.
Rana, G. L., Vovlas, C., and Zettler, F. W. 1983. Manual transmission of dasheen mosaic virus from Richardia to nonaraceous host. Plant Dis. 67: 1121-1122.
Robaglia, C., Durand-Tardif, M., Tronchet, M., Boudazin, G., Astier- Manifacier, S.
and Casse-Delbart, F. 1989. Nucleotide sequence of potato virus Y (N strain) genomic RNA. J. Gen. Virol. 70: 935-947.
Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997.
The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876-82.
Wu, C.-I. 2004. Molecular characterization and antibody preparation of Turnip mosaic
virus isolate infecting calla lily. Master thesis. National Taiwan University. Taipei,
Taiwan.
Zheng, L., Wayper, P., Gibbs, A. J., Fourment, M., Rodoni, B. C., and Gibbs, M. J.
2008. Accumulating variation at conserved sites in Potyvirus genomes is driven by species discovery and affects degenerate primer design. Plos ONE 3(2): e1568.
Table 1. Universal and degenerate primers used in this study
a Nucleotide at degenerate positions are represented by a single letter code; R = A and G; Y = C and T; B = C, G and T; D
= A, G and T; N = A, C, G and T.
Potyvirus HC-Pro gene primer
Potyvirus HC-Pro gene primer
Potyvirus CI gene primer
Potyvirus CI gene primer
Potyvirus NIb gene primer
5’RACE PCR primer 5’RACE PCR primer 3’ first-strand primer
Usage
TAATACGACTCACTATAGGGCCCCCCCCCCC dC-T7
AGCTGGATCCTTTTTTTTTTTTTTTTTT dT-Bam
GATGCGGCCGCGGGGGGGGGGGGGGGGGG dG-Not
KxFTKMVR PSLCDNQ IENGVDLT TRPLAEN GNNSGQP
Conserved motif
CGCGCACCATCTTGGTRAARTCYTT PHCR4
CCTCCCTGCTGTGCGATAATCAGCT PHCF4
GTCCAGGGTCACGCCRTTYTCDAT PCIR2
ACCCGGCCCCTGGCNGARAAYGT PCIF2
GGBAAYAATAGTGGNCAACC PNIbF1
Sequence (5’- 3’)a Primer
Potyvirus HC-Pro gene primer
Potyvirus HC-Pro gene primer
Potyvirus CI gene primer
Potyvirus CI gene primer
Potyvirus NIb gene primer
5’RACE PCR primer 5’RACE PCR primer 3’ first-strand primer
Usage
TAATACGACTCACTATAGGGCCCCCCCCCCC dC-T7
AGCTGGATCCTTTTTTTTTTTTTTTTTT dT-Bam
GATGCGGCCGCGGGGGGGGGGGGGGGGGG dG-Not
KxFTKMVR PSLCDNQ IENGVDLT TRPLAEN GNNSGQP
Conserved motif
CGCGCACCATCTTGGTRAARTCYTT PHCR4
CCTCCCTGCTGTGCGATAATCAGCT PHCF4
GTCCAGGGTCACGCCRTTYTCDAT PCIR2
ACCCGGCCCCTGGCNGARAAYGT PCIF2
GGBAAYAATAGTGGNCAACC PNIbF1
Sequence (5’- 3’)a Primer
96%
59%
65%
62%
aab
25% 90%
25%
nta 24%
29% 81%
28%
5’UTR 50%
KoMV ZaMMV
TuMV DsMV
Table 2. The percentage of amino acid similarity and nucleotide identity of the whole genome and 5’UTR among ZaMV, KoMV and three calla lily-infecting potyviruses
a the complete nucleotides (nt) sequence identity of ZaMV
compared with DsMV (AJ298033), TuMV, ZaMMV (NC_011560), and KoMV (AB219545).
b the amino acid (aa) sequence similarity of the polyprotein.
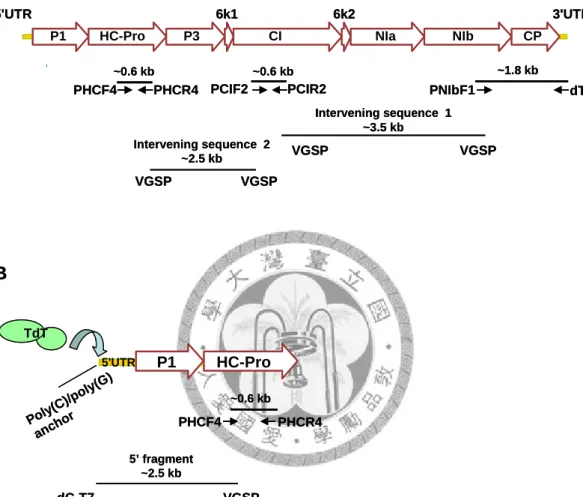
Fig 1. Stragtegy for cloning of the complete genome of potyvirus and the relative positions of the degenerate primers. (A) Genome organization of potyvirus and the cloning strategy of genomic fragments amplified by degenerate and viral gene specific primers (VGSP). (B) 5’RACE strategy by anchoring poly(C) or poly (G) into the 5’end by terminal transferase. The 5’
fragments were amplified by VGSP and the universal dC-T7 or dG-Not primer.
P1 HC-Pro P3
6k1 CI
6k2
NIa NIb CP
PNIbF1 PCIR2
PCIF2 PHCF4 PHCR4
3'UTR 5'UTR
dT-Bam
~0.6 kb ~0.6 kb ~1.8 kb
Intervening sequence 1
~3.5 kb Intervening sequence 2
~2.5 kb VGSP VGSP
VGSP VGSP
A
P1 HC-Pro P3
6k1 CI
6k2
NIa NIb CP
PNIbF1 PCIR2
PCIF2 PHCF4 PHCR4
3'UTR 5'UTR
dT-Bam
~0.6 kb ~0.6 kb ~1.8 kb
Intervening sequence 1
~3.5 kb Intervening sequence 2
~2.5 kb VGSP VGSP
VGSP VGSP
A
5’ fragment
~2.5 kb
VGSP dC-T7
dG-Not
P1 HC-Pro
5'UTR
PHCF4 PHCR4
~0.6 kb
B
Poly(C)/poly(G) anchor TdT
5’ fragment
~2.5 kb
VGSP dC-T7
dG-Not
P1 HC-Pro
5'UTR
PHCF4 PHCR4
~0.6 kb
B
Poly(C)/poly(G) anchor TdT
Fig 2. Amplification of potyvirus by reverse transcriptase-PCR from inoculated plant total RNA and analyzed by 1% agarose gel electrophoresis. RT-PCR products amplified by dT-Bam/PNIbF1 (A), PCIF2/PCIR2 (B) and PHCF4/PHCR4 (C) primers, respectively. M, 1 kb plus DNA ladder (Invitrogen), 1. BaRMV, 2. DsMV, 3. PVY, 4. TuMV, 5. ZaMMV, 6.
ZaMV, 7. ZYMV. Arrows to the left indicate size of markers.
Fig 3. The alignments of the 5’UTR sequence of four calla potyviruses and six potyviruses. (A) The proximal-50 nucleotides in the 5’UTR of six potyviruses: BCMV (U34972), PVA (Z21670), PPV (AJ234957), JGMV (Z26920), ZYMV (L29569), KoMV (AB219545), and four calla lily-infecting viruses: TuMV, DsMV, ZaMV and ZaMMV. the underlines indicated the
‘potybox-like blocks‘. (B) 5’UTR sequence alignments with DsMV, ZaMV and KoMV.
A A A
B B
Chapter II
Detection of four calla potyviruses by multiplex RT-PCR
using nad5 mRNA as an internal control
Abstract
Dasheen mosaic virus (DsMV), Turnip mosaic virus (TuMV), Konjac mosaic virus
(KoMV) and Zantedeschia mild mosaic virus (ZaMMV) are important potyviruses previously identified in calla lily plants in Taiwan. In order to save time and cost of virus detection, a multiplex RT-PCR assay was developed for these calla potyviruses.
Specific primers for each virus were designed based on the sequences of 3’ terminal region of respective viruses. To prevent the false negative results, a primer pair specific to plant mitochondrial nad5 mRNA was used to produce a 185-bp fragment as an internal control of RT-PCR. The specificities of primers were confirmed by means of simplex and multiplex PCR assays. Optimal primer concentration ratio was identified by multiplex PCR assay. Total RNAs purified from virus-infected plants were used directly or mixed in different combinations, and then tested by multiplex RT-PCR. The result indicated that the expected RT-PCR products could be specifically amplified and identified on the basis of their molecular sizes. The detection sensitivity of multiplex RT-PCR was 25-625 times higher than that of indirect-ELISA (I-ELISA) depending on the virus. When applied to field surveys, multiplex RT-PCR could detect more single as well as mixed infection samples than I-ELISA. Accordingly, our multiplex RT-PCR assay provides a simple, rapid and reliable method for multiple potyvirus detection in calla lily.
Introduction
Calla lily is the common name of the members of Zantedeschia spp. which belong to the family Araceae and are classified into seven species and two subspecies (Kuehny 2000). As tropical plants native to Africa, calla lilies are now grown as outdoor garden plants, cut flowers and flowering potted plants. Many cultivars of calla lily have been introduced into Taiwan from New Zealand, the Netherlands and the United States for more than 10 years. However, virus diseases are one of the major factors limiting calla lily production (Chen et al. 2003; Huang et al. 2007). Calla lily has been reported as the natural host of various plant viruses, mainly potyviruses (Huang et al.
2007). There are six potyviruses identified in this plant and five of them have been found in Taiwan, including Calla lily latent virus (CLLV) (Chen et al. 2004), Dasheen
mosaic virus (DsMV) (Chen et al. 1998), Turnip mosaic virus (TuMV) (Chen et al.
2003), Zantedeschia mosaic virus (ZaMV) (Chang et al. 2001), and Zantedeschia mild
mosaic virus (ZaMMV) (Huang and Chang 2005). Among them, DsMV is a
well-known potyvirus and reported in many kinds of aroid plants (Rana et al. 1983;
Zettler and Hartman 1987). TuMV is an important cruciferous plant virus with a broad host range. ZaMV which was recently classified as the isolate of Konjak mosaic
virus (KoMV) is probably the most prevalent virus infecting calla lily (Chang et al.
2001; Kwon et al. 2002; Nishiguchi et al. 2006; Huang et al. 2007). A newly
identified calla virus, ZaMMV, was found widely spread in the fields in Taiwan and probably in other countries (Huang et al. 2007). The major symptoms of calla lily induced by these four viruses are mosaic, yellow stripe, green island and mild mosaic (Zettler and Hartman 1987, Chang et al. 2001; Pham et al. 2002; Chen et al. 2003;
Huang and Chang 2005). CLLV alone does not produce symptoms in calla lilies and may not have a direct impact on the crop (Chen et al. 2004). For that reason we selected DsMV, TuMV, KoMV (ZaMV) and ZaMMV as the detection targets in our study. According to the field surveys in Taiwan, mixed infections by potyviruses are very common in calla lilies (Huang et al. 2007). Severe or mixed infection could cause leaf and flower distortion, stunting, growth reduction and yield loss (Hu et al.
2007).
Tissue culture is an important propagation method for calla lily in addition to tuber production in Taiwan. Viruses can be maintained within the plants during the growing season and storage stage due to systemic viral infection. Therefore, tissue culture together with reliable virus detection methods is essential for production of virus-free plantlets and tubers. Several detection methods for individual calla potyviruses were recently developed such as reverse transcription-polymerase chain reaction (RT-PCR), immunocapture RT-PCR (IC-RT-PCR), dot-blot hybridization, and enzyme-linked immunosorbent assay (ELISA) (Huang et al. 2005; Hu et al. 2007;
Huang et al. 2007). These methods target only single viruses in one reaction. To save time and cost of virus detection, multiplex RT-PCR is chosen because it can rapidly detect multiple targets in one single assay with a small amount of sample. This technique has been successfully applied to many plants for virus detection, including apple (Menzel et al. 2002), citrus (Roy et al. 2005), olive (Bertolini et al. 2001), potato (Nie and Singh 2000; Du et al. 2006), orchids (Lee and Chang 2006), and other crops.
In this study, we established a multiplex RT-PCR system for simultaneous detection of four calla potyviruses in field samples. To rule out false negative results, one primer pair specific to plant mitochondrial NADH dehydrogenase (nad5) gene was incorporated to amplify the product of plant nad5 mRNA as the internal control.
This is the first multiplex RT-PCR developed for aroid plants.
Material and methods
Virus isolates and plant materials
Four potyviruses, DsMV, TuMV, ZaMMV and KoMV (ZaMV), were previously isolated from calla lilies. Isolates of DsMV-ZAN (Chen et al. 1998), KoMV (ZaMV-ZAN) (Chang et al. 2001), ZaMMV-ZUN (Huang and Chang 2005) and TuMV-ZAN were separately maintained on tissue culture plantlets of cultivar ‘Black Magic’ or Philodendron selloum by mechanical inoculation (Huang and Chang 2005).
TuMV was also maintained in Nicotiana benthamiana. These plants were kept at 25oC with 16 h photoperiod in a greenhouse. For the disease survey, field-grown calla lily plants were randomly collected from the Taiwan Seed Improvement and Propagation Station (TSIPS) in Taichung County.
Plant total RNA extraction
Plant tissues collected from healthy, virus-inoculated and field grown plants were used to extract total RNA following the protocol of Plant Total RNA Extraction Miniprep System (Viogene, Sunnyvale, CA, USA). In brief, 0.1 g leaf tissue was ground into fine powder in liquid nitrogen and then transferred to a microfuge tube.
After being mixed with 450 μl PRX extraction buffer, the lysate was filtered using a Shearing Tube. The filtrate was mixed with 230 μl absolute ethanol, transferred to a
new Plant Total RNA Mini Column, and filtered by centrifugation. This column was then washed once with WF Buffer and twice with WS Buffer. Finally, plant total RNA was eluted with 50 μl RNase-free ddH2O. The quality of total RNA was analyzed by 1% agarose gel electrophoresis. Plant total RNA was used directly for RT-PCR or stored at -20oC for further use.
Primer design
Specific primer pairs of four calla potyviruses were designed based on the sequences of 3’ terminal region of each individual virus, with the help of the Primer Premier 5 program (Premier Biosoft Int., Palo Alto, CA, USA) to avoid primer dimer formation.
The names, targets and sequences of primers and the expected product sizes are shown in Table 1.
Viral and plant nad5 cDNA clones construction
Plant mitochondrial nad5 gene (mt) and four viral cDNA fragments were prepared from virus-infected plant total RNA by RT-PCR amplification. The first-strand cDNA was synthesized using dT-Bam (5’-AGCTGGATCC(T)18-3’) or mtR1 primers. PCR reactions were carried out using dT-Bam and PNIbF1 (5’-GGBAAYAATAGTGGNCAACC-3’) primers for potyviruses (Hsu et al. 2005), or
mtR1/mtF2 primers for plant mitochondrial nad5 gene gene. RT-PCR products were analyzed in 1% agarose gels and the desired cDNA fragments were purified by GFXTM PCR DNA and Gel Band Purification Kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The purified fragments were cloned into pGEM-T® Easy Vector (Promega, Madison, WI, USA). The correct cDNA clones were confirmed by sequencing and then used as templates for PCR experiments.
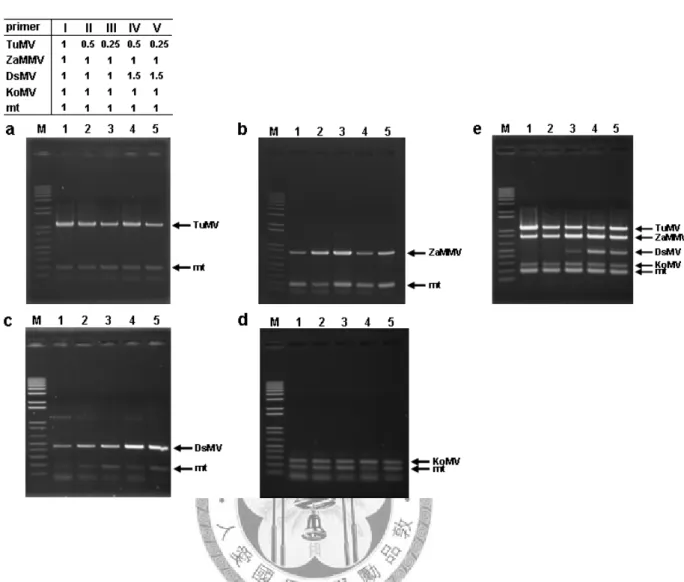
Multiplex PCR
Five pairs of reverse and forward primers (Table 1) were used in the multiplex PCR reaction. Different primer mixtures containing each primer 10x concentrated were prepared according to the experimental design. The final concentration of every primer was 0.125 μM in original multiplex PCR. To adjust the primer ratio, TuMV primer concentration was decreased to 0.5 and 0.25x and DsMV primer was increased to 1.5x of original concentration (0.125 μM). For multiplex PCR, the 20 μl reaction mix contained 2 μl template mixture (2 ng per cDNA clone), 2 μl 10x primer mixture, 2 μl 10x DyNAzymeTM II DNA polymerase buffer (Finnzymes, Inc., Espoo, Finland), 0.5 μl dNTPs (10 mM), 0.5 μl DyNAzymeTM II DNA polymerase (2 U μl-1, Finnzymes, Inc.) and 13 μl ddH2O. The amplification was carried out in GeneAmp® PCR system 2400 or 2700 (Perkin-Elmer Applied Biosystems, Foster City, CA, USA).
PCR program for DNA synthesis was 95oC for 5 min, followed by 30 cycles of 95oC for 35 s, 56oC for 35 s, 72oC for 1 min 30 s, and a final elongation step at 72oC for 7 min. Eight μl of PCR products were analyzed by 1.5% agarose gel electrophoresis in 1X TAE buffer (40 mM Tris-acetate, 1 mM EDTA). The DNA fragments were stained with ethidium bromide for 10 min and examined under UV illumination.
Multiplex RT-PCR
The multiplex RT-PCR protocol has separate RT and PCR steps. Total RNA was extracted as described above. For 25 μl RT reaction, 7 μl plant total RNA together with 2 μl dT-Bam primer (10 μM) and 1 μl mtR1 primer (5 μM) were heated at 65oC for 10 min, cooled at 4oC for 5 min and then 15 μl RT mixture [5 μl 5x first strand buffer (Promega, Madison, WI, USA), 1 μl dNTPs (10 mM), 0.5 μl rRNasin (40 U μl-1, Promega), 1 μl AMV reverse transcriptase (10 U μl-1, Promega) and 7.5 μl ddH2O] was added. After incubating RT reaction solution at 42oC for 60 min, the multiplex PCR reaction was performed as previously described except that 2 μl RT product was used as template.
Indirect enzyme-linked immunosorbent assay (I-ELISA)
The antisera to DsMV, TuMV, ZaMMV and KoMV were previously prepared in our
laboratory using recombinant capsid proteins as the antigens (Huang et al. 2005; Hu et al. 2007; Huang et al. 2007). I-ELISA was performed according to the protocol of Agdia Inc. (Elkhart, IN, USA) with some modification. One hundred mg of plant tissue was ground in 1 ml of indirect sample extraction buffer [ISE buffer, 15 mM Na2CO3, 35 mM NaHCO3, 2% polyvinylpyrrolidone (MW 40,000), pH 9.6]. One hundred μl of the extracts were coated to the 96-well ELISA plate and then assayed as previously described (Lee and Chang 2008). Each sample assayed in triplicate. A sample was regarded as positive if the A405 value exceeded twice the mean value of healthy controls.
Results
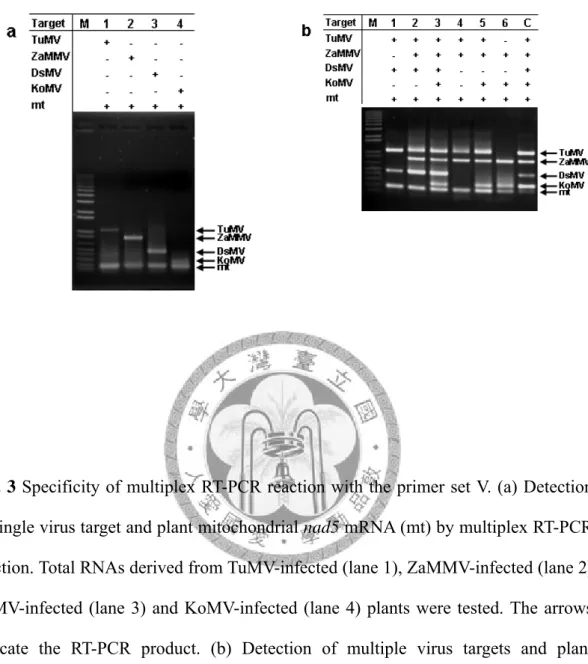
Specificity of detection primers tested by simplex and multiplex PCR
The performances of the designed primers were first tested using viral and plant nad5 cDNA clones as templates in simplex and multiplex PCR assays. The final concentration of each primer was 0.125 μM in these PCR reactions. Initially, the specificity of individual primer pairs was analyzed by PCR reaction with single cDNA template. The results of simplex PCR demonstrated that the expected PCR products were successfully generated by each single specific primer pair (Fig. 1a). When individual cDNA clones were tested by the multiplex PCR reaction, the expected fragments were specifically amplified by the primer mixture of TuMV, ZaMMV, DsMV, KoMV and mt control (Fig. 1b). Although a lower yield of the products of DsMV, KoMV and mt were obtained in multiplex PCR compared with simplex PCR, all tested primers were specific to their targets. To further test the specificity of these primers with five templates simultaneously, the same amount of each individual cDNA clone was combined and then used for the multiplex PCR assay. According to the gel analysis, the PCR products of TuMV, ZaMMV, KoMV and mt control were successfully amplified but the DsMV fragment was hardly visible (Fig. 1c) indicating that the multiplex PCR needed modification in order to detect multiple targets.