I
國立臺灣大學生命科學院微生物與生化學研究所 碩士論文
Graduate Institute of Microbiology and Biochemistry
College of Life Science
National Taiwan University Master Thesis
根毛農桿菌對黃豆之基因轉形研究
Agrobacterium rhizogenes-mediated
soybean gene transformation
郭致均 Chih-Chun Kuo
指導教授:李昆達 博士 Advisor: Kung-Ta Lee, Ph.D.
中華民國九十七年七月
July, 2008
II
國立臺灣大學碩士學位論文 口試委員會審定書
根毛農桿菌對黃豆之基因轉形研究
Agrobacterium rhizogenes-mediated
soybean gene transformation
本論文係郭致均君(R95B47104)在國立臺灣大學微生物 與生化學研究所完成之碩士學位論文,於民國九十七年七月九 日承下列考試委員審查通過及口試及格,特此證明
口試委員:
(簽名)
(指導教授)
系主任、所長
(簽名)
III
Abstract
Agrobacterium rhizogenes-mediated soybean multiple genes transformation was
investigated in this study. Soybean Tainan #2 was selected as the material for the highest germination rate of sterile plants and high number of tissue segments for root induction.
Two reporter genes were used for co-transformation study, gfp from pCAMBIA 1302 and gus from pCAMBIA 1201. Transformation efficiency of pCAMBIA 1201 and pCAMBIA 1302 was 5.5 x 105 and 8.25 x 105 for wild-type A. rhizogenes, and efficiency lowered to an average of 9.5 x 102 for transforming the second vector into pre-transformed A. rhizogenes. Co-transformation was carried out either by two binary vectors in one A. rhizogenes (2BV) or two binary vectors in separated A. rhizogenes and infecting soybean at the same time (2AR). Root induction rate from cotyledon was 48.0% for WT A. rhizogenes, 39.6% for 2BV, and 68.2% for 2AR, independently. We gained two hairy root clones after liquid culture, which both expressing GFP but not GUS. As a result, soybean hairy roots are successfully induced by A. rhizogenes transformants, and foreign gene is successfully transformed and expressed in hairy roots.
Soybean multiple genes co-transformation is not yet fully constructed and still needs much effort to get a further understanding and for further applications.
Keywords: A. rhizogenes, soybean, hairy roots, GFP, GUS
IV
中文摘要
本論文在進行根毛農桿菌對黃豆之多重基因共轉形之探討。黃豆臺南二號有 最高之無菌植株發芽率且可用於農桿菌感染之組織材料數高而被選作後續研究材 料。選用兩個廣泛使用之報導蛋白質進行黃豆之多基因共轉形:帶有綠色螢光蛋
白質 (GFP) 之質體 pCAMBIA 1302 及帶有 β-葡萄糖酸苷酶 (GUS) 之質體
pCAMBIA 1201。首先探討對野生型農桿菌進行單一質體轉形時, pCAMBIA 1201 轉形效率為5.5 x 105,pCAMBIA 1302 效率為 8.25 x 105;對已帶有一質體之農桿
菌轉形平均轉形效率則降低至9.5 x 102。黃豆多基因共轉形操作分為單一菌體同時
攜帶兩質體 (2BV) 或兩質體分別位於兩菌體來同時感染黃豆 (2AR)。黃豆子葉之
根狀組織誘導率在野生型農桿菌及2BV、2AR 組別分別為 48.0%、39.6%及 68.2%。
經過液態搖瓶培養後,得到的兩株毛狀根都會表現 GFP 而不表現 GUS。本研究 已初步建立黃豆之毛狀根誘導系統、將外源基因送入黃豆染色體並在毛狀根中成 功表現出異源蛋白質綠色螢光蛋白質。黃豆之多基因共轉形系統仍未完整建立,
後續仍需要許多努力以對其有完整之了解並得以應用。
關鍵字:根毛農桿菌、黃豆、毛狀根、綠色螢光蛋白質、β-葡萄糖酸苷酶
V
Abbreviations
A. rhizogenes Agrobacterium rhizogenes
A. tumefaciens Agrobacterium tumefaciens
CaMV 35S Cauliflower mosaic viral 35S dNTP Deoxyribonucleotide triphosphate
E. coli Escherichia coli
EDTA Ethylene diamine tetraacetic acid
HPLC High performance liquid chromatography LB Left Border
MS medium Murashige and Skoog basal medium
nos Nopaline synthase
PAGE Polyacrylamide gel electrophoresis PCR Polymerase chain reaction
RB Right Border
Ri Root inducing
SDS Sodium dodecyl sulfate T-DNA Transfer DNA
Ti Tumor-inducing
X-Gluc 5-bromo-4-chloro-3-indolyl glucuronide
VI
Index
Agrobacterium rhizogenes 根毛農桿菌
Agrobacterium tumerfaciens 根瘤農桿菌
callus 植物禦傷組織
CaMV 35S promoter 花椰菜嵌紋病毒35S 啟動子 constitutive promoter 持續性啟動子
electrophoresis 電穿孔法
elicitor 誘引劑
GFP 綠色螢光蛋白質
GUS β-葡萄糖酸苷酶
hairy roots 毛狀根
inducible promoter 誘導性啟動子
PCR 聚合酶連鎖反應
primer 引子
promoter 啟動子
reporter gene 報導基因
restriction enzyme 限制酶
Western blotting 西方墨漬法
VII
Contents
口試委員會審定書 ... II Abstract ... III 中文摘要 ... IV Abbreviations ... V Index ... VI Contents ... VII Contents of tables and figures ... X
Chapter 1 Introduction ... 1
1.1 Transgenic plant cell cultures ... 2
1.1.1 Plant cell cultures ... 2
1.1.2 Agrobacterium-mediated gene transformation ... 4
1.1.3 Hairy root cultures ... 7
1.2 Multiple genes co-expression ... 8
1.3 Reporter proteins ... 9
1.4 Soybean ... 10
1.5 Research aim ... 12
Chapter 2 Materials and Methods ... 13
2.1 Plasmid constructions ... 14
2.1.1 Binary vectors ... 14
2.1.2 Two reporter genes in one T-DNA region ... 15
2.1.3 Two T-DNA regions in one binary vector ... 17
2.2 Bacteria transformation methods ... 18
2.2.1 Heat shock method for E. coli transformation ... 18
2.2.2 Electroporation for A. rhizogenes transformation ... 19
VIII
2.2.3 Transformation efficiency of A. rhizogenes ... 20
2.2.4 PCR confirmation ... 21
2.3 Establishment of hairy root clones ... 22
2.3.1 Soybean sterile plants germination ... 22
2.3.2 Preparation of A. rhizogenes for induction of soybean hairy roots ... 23
2.3.3 Induction of soybean hairy roots ... 24
2.3.4 Sterilization of hairy roots ... 25
2.3.5 Liquid cultures of hairy roots ... 25
2.4 Confirmation of foreign DNA in root clones ... 26
2.4.1 Extraction of genomic DNA ... 26
2.4.2 Confirmation of foreign genes with PCR ... 27
2.5 Functional GFP and GUS confirmation ... 28
2.5.1 Direct observation of GFP ... 28
2.5.2 GUS activity assay ... 29
2.6 Protein quantification ... 29
2.6.1 Extraction of cytosol protein ... 29
2.6.2 Quantification of total protein ... 30
2.6.3 Western blotting for detection of GFP ... 30
Chapter 3 Results ... 33
3.1 A. rhizogenes transformation ... 34
3.2 Selection of soybean materials ... 34
3.3 Establishment and selection of transgenic root clones ... 35
3.4 Liquid cultures of transgenic root clones ... 36
3.5 Transgenic root clones gene confirmation ... 38
Chapter 4 Conclusions and Discussions ... 41
IX
4.1 A. rhizogenes transformation ... 42
4.2 Soybean and transgenic soybean roots ... 43
4.3 Perspectives ... 47
Tables and Figures ... 51
References ... 71
Appendix ... 79
A.1 Molecular cloning ... 80
A.2 Microbe culture ... 81
A.3 Plant culture ... 81
A.4 Protein purification and detection ... 82
X
Contents of tables and figures
Table 1 Proteins of medical relevance produced in plant cell cultures ... 52
Table 2 Recent reports on valuable metabolites produced by hairy roots ... 54
Table 3 A. rhizogenes transformation efficiency ... 55
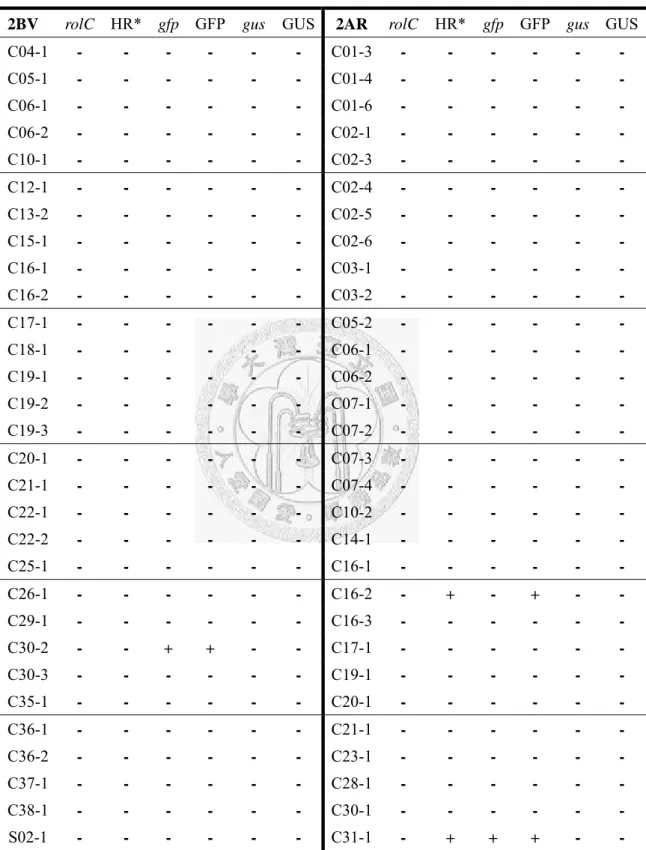
Table 4 Induction rate of soybean roots ... 56
Table 5 Gene confirmation of 2BV and 2AR root clones ... 57
Figure 1 Agrobacterium-mediated genetic transformation model. ... 58
Figure 2 Binary vectors pCAMBIA 1201 and pCAMBIA 1302 ... 59
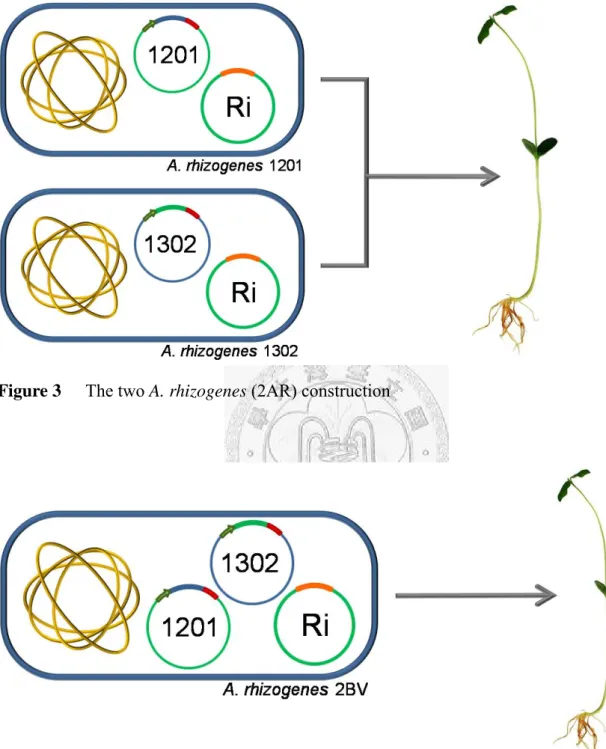
Figure 3 The two A. rhizogenes (2AR) construction ... 60
Figure 4 The two binary vector (2BV) construction ... 60
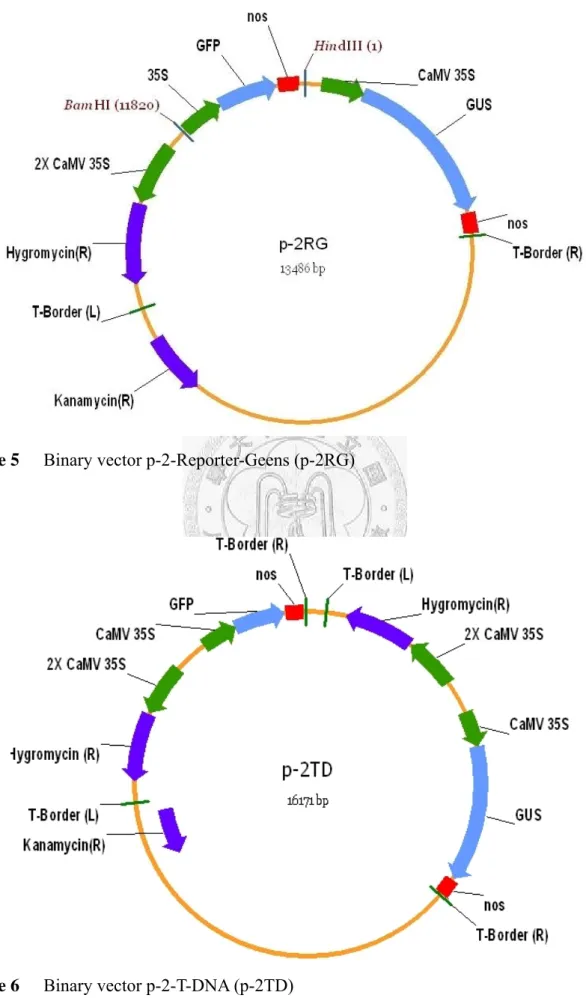
Figure 5 Binary vector p-2-Reporter-Geens (p-2RG) ... 61
Figure 6 Binary vector p-2-T-DNA (p-2TD) ... 61
Figure 7 Calibration curve of A. rhizogenes OD600 vs. colony number ... 62
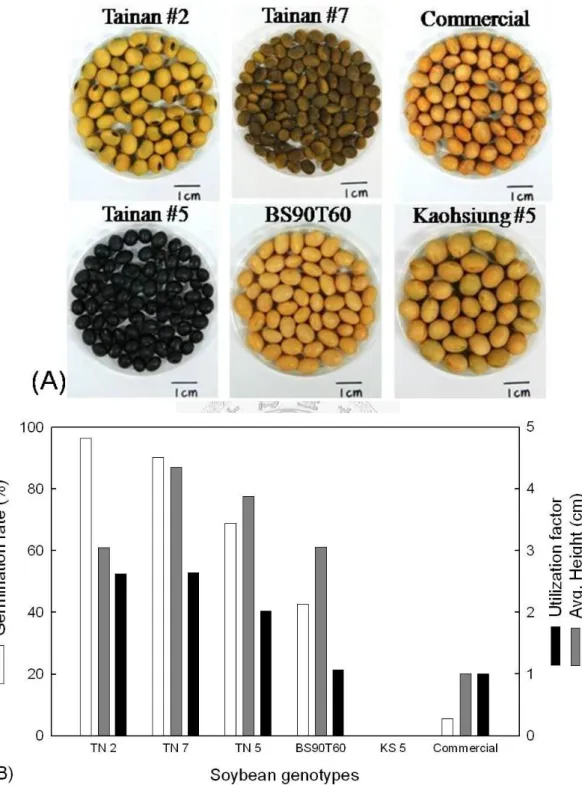
Figure 8 Soybean germination tests ... 63
Figure 9 Growth of wild-type root clones ... 64
Figure 10 Growth of 2BV root clones ... 65
Figure 11 Growth of 2AR root clones ... 66
Figure 12 Morphology of transgenic roots ... 67
Figure 13 PCR confirmation of transgenic hairy roots ... 68
Figure 14 GFP confirmation under fluorescence microscope ... 69
Figure 15 SDS-PAGE of protein extract of root clones. ... 70
1
Chapter 1
Introduction
2
1.1 Transgenic plant cell cultures
1.1.1 Plant cell cultures
Plants have been an important source of food for the past centuries, on top of that, plants are also sources of medical compounds. Many pharmaceuticals are derived from plant secondary metabolites, such as digitalis, L-DOPA, morphine, codeine, and reserpine. Anticancer drugs such as vincristine, vinblastine, and taxol are also derived from plants (Dicosmo and Misawa, 1995). The use of whole plants for the synthesis of recombinant proteins has received a great deal of attention because of advantages in economy, scalability and safety compared with traditional microbial and mammalian production systems. However, production systems that use whole plants have several disadvantages, including the needs of long periods and large areas for whole plant cultivation; the lack of several intrinsic benefits of cultured cells such as precise control over growth conditions, batch-to-batch product consistency, and the ability to produce recombinant proteins in compliance with good manufacturing practice (Hellwig et al., 2004).
The term “plant cell cultures” means the propagation of any plant-derived cell tissue in gently agitated liquid culture, and they can be classified as unorganized cultures (e.g. callus, suspension, and the protoplast culture) and organized cultures (e.g.
hairy roots or embryo cultures) depending on the tissue source and level of
3
differentiation (James and Lee, 2001). For the past three decades, significant effort was invested in producing high-value secondary metabolites such as shikonin (Yamamura et al., 2003), taxol (Hezari and Croteau, 1997; Hezari et al., 1997), and gesenoside (Palazon et al., 2003), via plant cell cultures.
In recent years, the number of recombinant proteins used for therapeutic applications has increased dramatically. These demands have driven the development of a variety of improvements in protein expression technology in a wide range of expression systems (Andersen and Krummen, 2002). Plant cells, like microbes, can be maintained in simple, synthetic media, but like animal cells they can synthesize complex multimeric proteins and glycoproteins. Plant cell cultures combine the merits of whole-plant systems with those of microbial and animal cell cultures, and already have an established track record for the production of valuable therapeutic secondary metabolites (Hellwig et al., 2004). The first recombinant protein produced in plant cells was reported nearly 30 years ago (Sijmons et al., 1990), since then, over 20 different recombinant proteins have been produced in plant cell cultures, including antibodies, enzymes, hormones, growth factors and cytokines (Table 1). Although most interest has been focused on microbial and animal cell cultures during the past 15 to 20 years, plants and plant cells are now considered as viable and competitive expression systems for
4
large-scale protein production (Doran, 2000), and >100 recombinant proteins have now been produced in a range of different species (Twyman et al., 2005).
Genetically engineered transgenic plant cell cultures have many advantages as sources of proteins compared with human or animal fluids/tissues, recombinant microbes, transfected animal cell lines, or transgenic animals. Among these are (a) production of raw material on an agricultural scale at low cost with the possibility of using the edible plant material directly; (b) reduced capitalization costs relative to fermentation methods; (c) rapid scale up of production; (d) correct eukaryotic assembly of multimeric proteins such as antibodies; and (e) increased safety, since plants do not serve as hosts for human pathogens, such as HIV, prions, hepatitis viruses, etc (Larrick et al., 2001).
1.1.2 Agrobacterium-mediated gene transformation
Plant transformation has become a core research tool in plant biology and a practical tool for cultivar improvement (Birch, 1997). There are two classes of plant transformation technologies: 'non-natural' or in vitro methods, and 'natural' methodologies. Among the in vitro technologies are microinjection, direct DNA uptake into protoplasts (with or without electroporation), and microprojectile (or particle) bombardment. 'Natural' technologies include the use of viral vectors (that will result in
5
transient but not stable transformation) and Agrobacterium tumefaciens T-DNA mediated transformation (Gelvin, 1998). The non-natural methods can normally express the transformed genes in as little as few hours post introduction, which are suitable methods for studying genetic regulatory networks such as promoter activity and RNA silencing (Hellens et al., 2005). These methods result in transient expressing and the foreign genes introduced are frequently unstable following propagation, leading to difficulties for gaining regenerated transgenic plants. For detailed examination of introduced genes expression pattern, stably transformed plants are needed. Hence, natural methods, typically Agrobacterium-mediated transformation, is required (Finnegan and McElroy, 1994).
Agrobacterium tumerfaciens is a kind of Gram-negative soil bacterium which is
also a plant pathogen. It genetically transforms its host by transferring a well-defined DNA segment from its tumor-inducing (Ti) plasmid to the host-cell genome. The integration and expression of T-DNA genes ipt (tmr) and iaaM (tms) result in the overproduction of auxin and cytokinin, leading to the tumor phenotype of crown gall disease (Binns, 2002).
Agrobacterium rhizogenes is closely related to Agrobacterium tumerfaciens in
terms of physiology and DNA homology, which also infect at plant wound sites and transfer T-DNA from the root-inducing (Ri) plasmid to plant genome. Integration and
6
expression of the root loci (rol) genes and the aux genes leads to the development of the hairy-root phenotype and synthesis of novel low-molecular-weight compounds called opines (Cho et al., 2000).
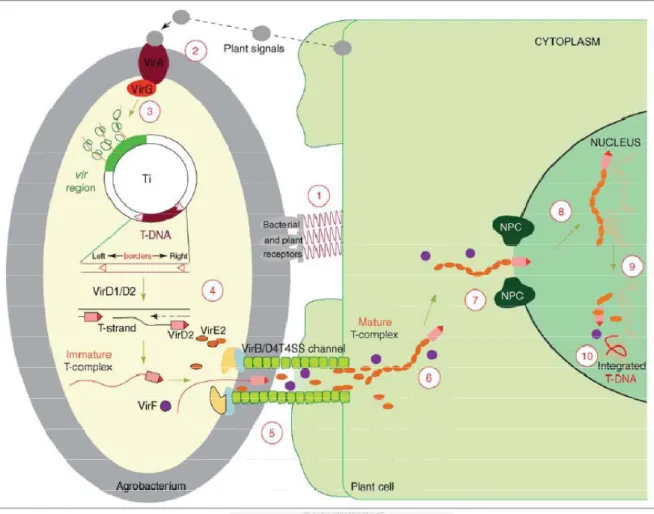
The vir region, located on the Agrobacterium Ti/Ri plasmid, encodes most of the bacterial virulence (Vir) proteins used by the bacterium to produce its T-DNA and to deliver it into the plant cell. The T-DNA segment is a specific element delimited by two 25-bp direct repeats called left border (LB) and right border (RB) on the Ti/Ri plasmid.
The genetic transformation process describes as follows. The transformation process begins with the bacterium–plant attachment (Figure 1; step 1) which is facilitated by chromosomal virulence protein A and B (ChvA, ChvB). Plant wound signals such as acetosyringon binds to the transmembrane VirA/VirG protein complex and triggers the expression of vir region on Ti/Ri plasmid (Figure 1; steps 2 and 3). A single-stranded (ss) T-DNA molecule (T-strand) (Figure 1; step 4) is then produced by the combined action of the bacterial VirC, VirD1, and VirD2 proteins. The ssT-DNA then exported into the host cell by a type IV secretion system formed by VirD4 and several proteins in the VirB family (Figure 1; step 5). The full length of the T-strand molecule is coated with numerous VirE2 molecules and nicked with VirD2 at both ends, these molecules confer to the T-DNA the structure and protection needed for its travel to the host-cell nucleus (Figure 1; step 6). It is mainly during the last steps of the transformation
7
process: transport through the cytoplasm (Figure 1; step 6), nuclear import (Figure 1;
step 7), intranuclear transport (Figure 1; step 8), T-DNA uncoating (Figure 1; step 9) and integration (Figure 1; step 10) (Tzfira and Citovsky, 2006).
A. rhizogenes not only is able to transfer the T-DNA on its Ri plasmid in cis, but
also able of transferring the T-DNA on a small, autonomous binary vectors in trans, thereby enabling the production of transgenic plants containing other foreign genes carried on a second plasmid. This method has been used to produce transgenic plants in several species (Christey, 1997).
1.1.3 Hairy root cultures
Hairy roots are formed from plants infected by A. rhizogenes at wound site.
Hairy-root cultures grow rapidly, show plagiotropic root growth and are highly branched on hormone-free medium (Cho et al., 2000).
Many plant secondary metabolites of interest are accumulated in roots, and they may not be occurred in undifferentiated cell. Hairy root cultures, in the differentiation degree of tissue, have been proposed as an alternative method of producing plant secondary metabolites. Not only because they have genetic and biochemical stability, the capability to synthesize secondary products at levels comparable to that of the original plants, but also the capability to synthesize for a longer period compared to
8
original plant roots (Chang et al., 2005). A number of plant species including many medicinal plants have been successfully transformed with A. rhizogenes (Sevón and Oksman-Caldentey, 2002). Valuable metabolites were produced by hairy roots of several plant species (Table 2).
1.2 Multiple genes co-expression
Over the past few years, there has been a growing realization that metabolic pathways must be studied in the context of the whole cell rather than at the single pathway level, and that even the simplest modifications can send ripples throughout the entire system. Attention has therefore shifted away from reductionist, single-gene engineering strategies and towards more complex approaches involving the simultaneous overexpression and/or suppression of multiple genes (Capell and Christou, 2004). Therefore, the most important value of multiple genes co-expression system might be the use in metabolic engineering.
Multiple genes co-expression system can also be used in multiple proteins production. Heterologous gene expression can lead to the production of nonfunctional target proteins, this is often due to the absence of cofactors or post-translational modifications required for function, stability or folding. Coexpression of multiple genes in such as the members of a stable multiprotein complex or a protein with a chaperone,
9
can in many cases alleviate these problems (Bross et al., 1993; Li et al., 1997; Rivas et al., 2005). Co-expression system can also be used in expression of two functional related proteins, such as protein Der p 1 and Der p 2 which can be used in immunotherapy procedures (Taketomi et al., 2006).
1.3 Reporter proteins
For the study of multiple genes co-expression in plant, efficient and easy-to-use reporter proteins are required. Commonly used reporter proteins in plant gene transformation usually involve fluorescent and luminescent proteins; examples include the green fluorescent protein (GFP) and the enzyme luciferase, which catalyzes a reaction with a luciferin to produce light. Another common reporter used in plant is β-glucuronidase (GUS) which can be easily assayed within fresh cells or tissues. Our lab had successfully expressed both GFP and GUS in tobacco hairy roots (Liu, 2004;
Chiang, 2006), the two reporter proteins were chosen for this study.
The green fluorescent protein (GFP) isolated from Aequorea victoria mgfp5 has been used as a reporter protein in many studies. It is a protein of 238 amino acid residues with a molecular weight of 27 kDa. GFP has a major absorbance wavelength at 395 nm and a minor one at 475 nm. The major emission wavelength is at 508 nm when excited at 395 nm (Heim and Tsien, 1996). The intrinsic fluorescence of the GFP allows
10
non-destructive, in-vivo imaging of events within cells (Boevink et al., 1999). The mature purified protein is highly stable; remain fluorescent up to 65°C, pH 11, 1%
sodium dodecyl sulphate (SDS) or 6M guanidinium chloride. GFP can also function as a protein tag since it tolerates both N- and C-terminal fusions to a variety of proteins and can be expressed in a broad range of host organisms (Cubitt et al., 1995).
The β-glucuronidase (GUS) is from Escherichia coli gusA, with a mutation at amino acid 358 form N to Q to avoid N-linked glycosylation. Furthermore, an intron was inserted inside the coding sequence to ensure that expression of glucuronidase activity is derived from eukaryotic cells, not from expression by residual Agrobacterium cells (Ohta et al., 1990). It is a protein with 620 amino acid residues with molecular weight of 68 kDa and appears to function as a tetramer. It is very stable, and will tolerate many detergents, widely varying ionic conditions, and general abuse. It is most active in the presence of reducing agents such as β-mercaptoethanol or DTT. It may be assayed at any physiological pH, with an optimum between 5.2 and 8.0 (Jefferson et al., 1987).
1.4 Soybean
The Fabaceae (or Leguminosae) commonly known as the bean family or pea family, is the third largest family of flowering plants with over 19,400 species, and
11
second only to Poaceae (grasses) in terms of agricultural and economic importance (Wojciechowski et al., 2006). They are important components of natural ecosystems and play an essential role in the global nitrogen cycle by establishing symbiotic interactions with soil bacteria of the Rhizobiaceae family to convert atmospheric nitrogen into ammonia and amino acids, which later can be used by other organisms. Legumes include a large number of domesticated species harvested as crops providing the world’s largest source of vegetable protein as well as for oils, fiber, fuel, fertilizers, timber, medicinals, chemicals, and horticultural varieties. In addition, the family includes several species studied as genetic and genomic model systems (e.g., pea, Pisum sativum, barrel medic, Medicago truncatula, and trefoil, Lotus corniculatus).
Soybean (Glycine max) is one of the most important crop plants of the world market in oil crops and serving as an important source of protein for both human consumption and as fodder. The seed of soybean contains 35-40% protein, serving as a potential source of recombinant protein production.
Soybean contains the highest concentration of isoflavones among the foods consumed by human. These soy isoflavones (e.g., daidzein and genistein) are implicated in some health-enhancing properties such as the prevention of certain cancers (Cappelletti et al., 2000; Miura et al., 2002; Ravindranath et al., 2004), lowering the risk of cardiovascular diseases (Anthony et al., 1996; Goodman-Gruen and Kritz-Silverstein,
12
2001), and an improvement of bone health (Cotter and Cashman, 2003; Weaver and Cheong, 2005).
1.5 Research aim
With considerably high protein content, soybean is considered a suitable plant host for expressing recombinant proteins. Combining the capability of hairy roots to grow rapidly in a hormone free liquid culture, soybean hairy roots is thought to be an ideal system for metabolic engineering and a valuable material for specialty chemical production. Previous studies on soybean hairy roots focused on high efficiency hairy roots induction and using soybean hairy roots as a model for soybean root defense system (Cho et al., 2000; Xiang et al., 2005). A more recent study attempted to modify phenolic metabolic pathways in soybean hairy roots by transforming two key enzymes into soybean separately, it resulted in a down regulation due to homology-dependent gene silencing (Lozovaya et al., 2007).
In this study, we attempt to construct an A. rhizogenes-mediated soybean hairy roots induction system. Further, using soybean hairy roots as an expression system for studying the efficiency of multiple genes co-expression. This co-expression system is thought to be a useful tool for soybean isoflavone synthetic pathway research.
13
Chapter 2
Materials and Methods
14
2.1 Plasmid constructions
2.1.1 Binary vectors
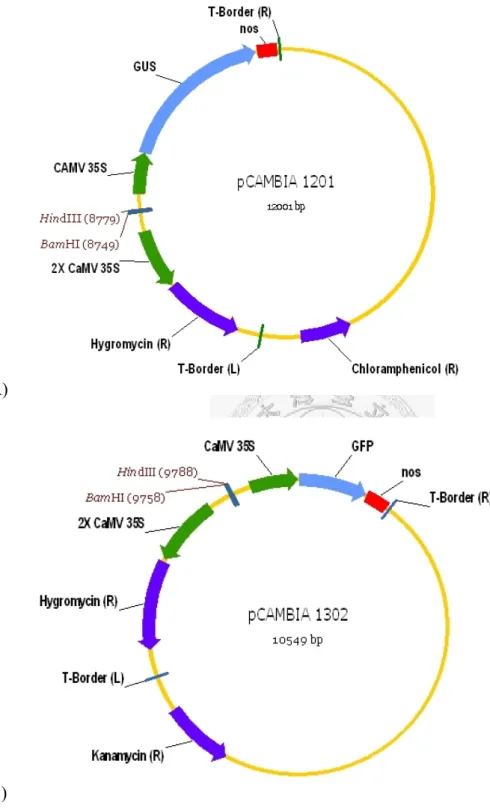
Binary vectors pCAMBIA 1201, pCAMBIA 1301, and pCAMBIA 1302 were purchased from CAMBIA (Canberra, Australia). Vector pCAMBIA 1201 and pCAMBIA 1301 both have gusA in the T-DNA region, with chloramphenicol and kanamycin resistance genes on the backbone, respectively. Vector pCAMBIA 1302 contains mgfp5 and kanamycin resistance gene (Figure 2).
For vector conservation and high-copy production of the vector, all vectors were transformed by heat shock into E. coli JM-109 competent cells (Yeastern Biotech, Taipei, Taiwan). Binary vectors were then purified from an over-night incubated E. coli cultures. Vectors were then digested with respective restriction enzyme, the digestion products were resolved on 1.2% agarose gel, and the digested fragments were confirmed after ethidium bromide (EtBr) staining.
Vector pCAMBIA 1201 and pCAMBIA 1302 were used directly without modification in A. rhizogenes transformation and then A. rhizogenes-mediated soybean co-transformation study. The two vectors were transformed into A. rhizogenes individually or simultaneously (the A. rhizogenes transformation method will be described later), resulting in A. rhizogenes containing each one, respectively, or both of the vectors. A. rhizogenes 1201 and A. rhizogenes 1302 were used together for
15
co-transformation of soybean (the soybean transformation method will be described later), this experiment construction was named 2AR for two strains of transformed A.
rhizogenes inducing hairy roots simultaneously (Figure 3). The A. rhizogenes
containing both pCAMBIA 1201 and pCAMBIA 1302 was also used in soybean co-transformation study and named as 2BV for one A. rhizogenes containing two binary vectors (Figure 4).
2.1.2 Two reporter genes in one T-DNA region
The gfp gene containing the upstream CaMV35S promoter and downstream nos terminator was cloned from binary vector pCAMBIA 1302. The forward primer contained a restriction site BamHI, and the reverse primer contained a HindIII site (This primer-pair was designed according to NCBI nucleotide database AF234298). The 1679 base pairs target sequence was amplified by PCR reaction.
F-35SGFPNOS:5’-TTTGGATCCTTTCCGCCTTCAGTTTAGCTTCATGGAGTCAAAGATTCA-3’
R-35SGFPNOS: 5’-TTTAAGCTTTTTCTTTTCTCTTAGGTTTACCCGCCAATATATCCTGTC-3’
The composition of PCR reaction solution:
16
Volume (μl) Final concentration
Template: pCAMBIA 1302 1.0 < 0.1 μg
4 mM dNTP 2.5 400 μM each
10X Reaction buffer 2.5 1X
10 μM primers 2.5 1 μM
Super-Therm Polymerase 0.5 2.5 U
Double distilled H2O 13.5 -
Total volume 25.0 -
PCR amplification was performed for 30 cycles in a thermal cycler (PCR Px2 Thermal Cycler Personal, Thermo Fisher Scientific Inc., Waltham, MA, USA). The PCR program for gfp was: 95°C, 5 min for denaturation; 94°C, 1 min, 55°C, 1 min, 72°C, 2 min for 30 cycles; 72°C 15 min and paused at 4°C.
The PCR product and vector pCAMBIA 1301 were both digested by restriction enzyme BamHI and HindIII, the two digested fragments were then ligated by T4 DNA ligase according to the user manual. The ligation product was resolved on 1.2% agarose gel, followed by EtBr staining to check the ligated vector. The ligated vector was transformed into E. coli JM-109 by heat shock method, then purified from an over-night incubated culture and sent for DNA sequencing (Tri-I Biotech Co., Taipei, Taiwan). This newly constructed binary vector was named plasmid 2RG for containing two reporter genes, gfp and gus, in a single T-DNA region (Figure 5).
17
2.1.3 Two T-DNA regions in one binary vector
The whole T-DNA region between the left-border and right-border of binary vector pCAMBIA 1302 was cloned by PCR amplification with primers containing restriction site SacII (This primer pair was designed according to NCBI nucleotide database AF234298). The 4336 base pairs target sequence was amplified by ExSel High Fidelity DNA Polymerase (JRM Holdings, Kent, UK).
F-TDNAGFP: 5’-TTTTCCGCGGTGGCAGGATATATTGTGGTGTAAACAAATTGACGCTTAG-3’
R-TDNAGFP: 5’-TTTTCCGCGGTAAACGCTCTTTTCTCTTAGGTTTACCCGCCAATATATC-3’
The composition of PCR reaction solution:
Volume (μl) Final concentration Template: pCAMBIA 1302 1.0 < 0.1 μg
4 mM dNTP 2.5 400 μM each
10X Reaction buffer 2.5 1X
10 μM primers 1.0 0.4 μM
ExSel Polymerase 1.0 5 U
Double distilled H2O 16.0 -
Total volume 25.0 -
18
PCR amplification was performed for 30 cycles in a thermal cycler. The PCR program for the whole T-DNA region containing gfp was: 95°C, 5 min for denaturation;
94°C, 1 min, 65°C, 30 sec, 72°C, 5 min for 30 cycles; 72°C 15 min and paused at 4°C.
The PCR product and the vector pCAMBIA 1301 were digested by restriction enzyme SacII, and then both dephosphorylated with alkaline phosphatase according to the user manual. The two fragments were then ligated with T4 DNA ligase, and the ligated vector was resolved on 1.2% agarose gel followed by EtBr staining to check the ligated vector. The ligated vector was transformed into E. coli JM-109 by heat shock method, then purified from an over-night incubated culture and sent for DNA sequencing. This newly constructed binary vector was named plasmid 2TD for containing two T-DNA regions in a single binary vector (Figure 6).
2.2 Bacteria transformation methods
2.2.1 Heat shock method for E. coli transformation
E. coli heat shock transformation method was carried out according to the user
manual, describe as follows. Prepare Luria-Bertani (LB) plate containing antibiotics 50 μg ml-1 kanamycin, and 50 μg ml-1 chloramphenicol. Competent cells were thawed from -70°C storage by holding under running tap water for about 20 seconds until half thawed. Two μl of pre-chilled vector or ligation product was added to 100 μl competent
19
cells, mixed by vortex then placed the tube on ice for 5 minutes. Cells were heat shocked for 45 to 90 seconds in a water bath at exactly 42°C, then immediately placed back on ice for 10 minutes. Cell recovery was carried out by incubating the competent cells in 2 ml LB medium without antibiotics in an orbital shaker at 150 rpm, 37°C for 1 hour. Cells were plated on LB plate containing respective antibiotics and incubate at 37°C overnight. Finally, single colony was selected and conserved.
2.2.2 Electroporation for A. rhizogenes transformation
2.2.2.1 Preparation of A. rhizogenes cells
A. rhizogenes 1610 wild-type was cultured in a Hinton flask with 100 ml LB
medium in an orbital shaker at 125 rpm, 30°C. A. rhizogenes was cultured until the cells grown to the middle-log phase with OD600 at 0.6-0.8, the cells were collected by centrifugation at 1000x g, 4°C. The pellet was re-suspended by 100 ml pre-chilled sterile double distilled water, and then collected by centrifugation again. This step was repeated four times and decreasing the re-suspension volume by 50 ml, 25 ml, 10 ml, then 5 ml. The pellet was finally re-suspended in 1 ml pre-chilled sterile double distilled water, placed on ice, and the A. rhizogenes cells were prepared.
20 2.2.2.2 Electroporation of A. rhizogenes
Two micro liter purified binary vector (~200 ng) was mixed with 48 μl A.
rhizogenes cells, the mixture was then placed in a pre-chilled sterile Pulser Cuvette
(Bridge Tech., Palo Alto, CA, USA) with a 1 mm electrode gap. The electroporation was performed at 25 μF, 200 Ω, 2.5 kV with a Gene Pulser (Bio-Rad Laboratories Inc., Hercules, CA, USA). The transformed A. rhizogenes was then recovered by cultivating 1 hour in an orbital shaker at 100 rpm, 30°C. After cultivation, the cells were plated on LB plate containing corresponding antibiotics and incubated at 30°C for 48 hours.
Single colony was selected and transformation was confirmed by PCR described later.
2.2.3 Transformation efficiency of A. rhizogenes
Transformation efficiency is the number of transformed cells (transformants) generated by 1 µg of plasmid DNA in a transformation reaction. For wild-type A.
rhizogenes single binary vector transformation, binary vector pCAMBIA 1201 was first
transformed into wild-type A. rhizogenes. The transformed cells were series diluted and plated on LBC plate. After 48 hours of cultivation, colonies on each plate were counted and the transformation efficiency was calculated by dividing the number of colonies on plate by micro grams of plasmid DNA plated. For pCAMBIA 1302, the same process was carried out by using LBK plate instead of LBC plate.
21
Successfully transformed A. rhizogenes 1201 and A. rhizogenes 1302 were then used as recipient strains for the transformation efficiency test of the second binary vector. The transformed cells were planted on LBCK plate, and the transformation efficiency was calculated as mentioned above.
Finally, both pCAMBIA 1201 and pCAMBIA 1302 were transformed simultaneously into wild-type A. rhizogenes for the calculation of transformation efficiency of two plasmids. The transformed cells were planted on LBCK plate, and the transformation efficiency was calculated as mentioned above.
2.2.4 PCR confirmation
Transformants of A. rhizogenes were confirmed by colony PCR with respective primer pairs specific for gfp and gus (Primer pairs were designed according to NCBI nucleotide database AF234293 and AF234298 ).
F-gfp: 5’-GAGAGAACACGGGGGACTCTTGAC-3’
R-gfp: 5’-ACTTTATTGCCAAATGTTTGAACG-3’
F-gus: 5’- TGATGATGATAGTTACAGAACCGACGA-3’
R-gus: 5’- CAGTCAACAGACGCGTGGTTACAGTC-3’
22 The composition of PCR reaction solution:
Volume (μl) Final concentration
A. rhizogenes - -
4 mM dNTP 2.5 400 μM each
10X Reaction buffer 2.5 1X
10 μM primers 2.5 1 μM
Super-Therm Polymerase 0.5 2.5 U
Double distilled H2O 14.5 -
Total volume 25 -
PCR amplification was performed for 30 cycles in PCR Px2 Thermal Cycler Personal (Thermo Fisher Scientific Inc., Waltham, MA, USA). The PCR program for both gfp and gus was: 95°C, 5 min for denaturation; 94°C, 1 min, 60°C, 30 sec, 72°C, 1 min for 30 cycles; 72°C 15 min and paused at 4°C.
PCR products were then resolved on a 1.2% agarose gel, followed by EtBr staining to check the 638 base pairs gus fragment and 833base pairs gfp fragment.
2.3 Establishment of hairy root clones
2.3.1 Soybean sterile plants germination
Six genotypes of soybean were used in this study to select the soybean as the suitable material for A. rhizogenes-mediated hairy roots induction. Soybean Tainan #2,
23
Tainan #5, Tainan #7, and Kaohsiung #5 were kindly provided by Tainan District Agricultural Research & Extension Station (Tainan, Taiwan). Soybean BS90T60 Minnesota was kindly provided by American Soybean Association IM (Taipei, Taiwan).
In addition, one commercial non-transgenic soybean was also used (Figure 8 (A)).
The surface-sterilization solution was prepared by adding 3 ml commercial hydrogen peroxide (~35%) into 21 ml commercial ethanol solution (~97%) in a sterile 50 ml tube. Soybean seeds were dipped in the solution and surface-sterilized by inverting the tube for 2 minutes. The seeds were washed with sterilized distilled water for 10 times, and then dipped in sterilized distilled water for 1 hour to remove residue detergents. Sterilized seeds were placed on sterilized vermiculite, and sterilized distilled water was added to moisturize the vermiculite. Seeds were germinated in the culture box with a cycle of illumination at 28°C for 12 hours, and at 25°C for another 12 hours in the dark.
2.3.2 Preparation of A. rhizogenes for induction of soybean hairy roots
A. rhizogenes harboring the binary vector was cultured overnight in LB medium
containing corresponding antibiotics in an orbital shaker at 125 rpm, 30°C.
Subsequently, 20 μl of the A. rhizogenes culture was inoculated to 2 ml fresh LB medium containing antibiotics, A. rhizogenes was cultured for 12 hours. Acetosyringon
24
was added to a final concentration of 200 μM to enhance the virulence of A. rhizogenes.
This culture was then cultured for another 12 hours, and then A. rhizogenes was ready for soybean infection.
Before using A. rhizogenes to induce soybean hairy roots, cells of A. rhizogenes strains were measured. Both strains 1201 and 1302 of A. rhizogenes underwent series dilution and their OD600 was measured by spectrophotometer (Hitachi, Pleasanton, CA, USA). The same diluted series were also plated on LB plates, colonies were counted after overnight cultivation. Calibration curve of A. rhizogenes OD600 versus colony number was used to estimate the colonies for A. rhizogenes-mediated soybean hairy root induction (Figure 7).
2.3.3 Induction of soybean hairy roots
One-week old soybean plants were about 8-10 cm in height and the cotyledons were green and expanded, soybean explants at this time are most suitable for A.
rhizogenes infection (Kereszt et al., 2007). The soybean shoots were cut into ~2 cm
segments, and cotyledons were cut at both ends to make wounded exposure.
Subsequently, A. rhizogenes was applied to one end of the shoot or cotyledon, then the segments were placed on fresh 1/2 MS (Murashige and Skoog, 1962) plate with the
25
“clean” end. A. rhizogenes and soybean tissue were co-cultivated at dark, 22°C for hairy root induction.
2.3.4 Sterilization of hairy roots
After co-cultivation for 14 to 21 days, the induced roots were picked up individually and placed on fresh 1/2 MS plates containing 300 μg ml-1 cefotaxime for sterilization of A. rhizogenes. After 7 days, roots were transferred to a fresh plate also containing cefotaxime and incubated for another 7 days. After two rounds of sterilization, roots were transferred to liquid cultures.
2.3.5 Liquid cultures of hairy roots
The roots were transferred individually to 50 ml 1/2 MS medium containing cefotaxime in 250 ml flasks and cultivated in an orbital shaker at 70 rpm, 25°C in the dark. Root clones were weighted at day 0, and weighted again after 21 days of cultivation. The root clones were then transferred to fresh 1/2 MS medium without cefotaxime and cultivated for another 21 days.
26
2.4 Confirmation of foreign DNA in root clones
2.4.1 Extraction of genomic DNA
Fresh root tissues were grounded into powder with liquid nitrogen, and genomic DNA was extracted using Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the user manual with a few modifications: (1) Place 50 mg of root tissue powder in a 1.5 ml microcentrifuge tube, add 600 μl of Nuclei Lysis Solution and mix by vortexing 3 seconds. (2) Incubate the tube at 65°C for 15 min and invert the tube every 3 to 5 minutes. (3) Add 3 μl RNase Solution to cell lysate and mix by inverting the tube, incubate the tube at 37°C for 15 minutes. (4) After cooling the sample to room temperature, add 200 μl Protein Precipitation Solution and vortex vigorously for 20 seconds. (5) Centrifuge for 10 minutes at 12,000 rpm and then carefully transfer the supernatant to a clean tube containing 600 μl isopropanol. (6) Mix the solution by inversion until DNA form thread-like visible mass, centrifuge at 12,000 rpm for 3 minutes. (7) Carefully remove the supernatant then add 600 μl 70% ethanol and wash the DNA by inverting the tube, centrifuge at 12,000 rpm for 3 minutes. (8) Carefully remove the supernatant, invert the tube on clean absorbent paper and air-dry the pellet for 15 minutes. (9) Add 100 ml Rehydration Solution and incubate the tube at 65°C for 1 hour. (10) Store Genomic DNA at -30°C.
27
2.4.2 Confirmation of foreign genes with PCR
Genomic DNA extracted from transgenic root clones were used for PCR analysis.
For foreign genes confirmation, conditions of PCR reactions were the same as A.
rhizogenes. To detect the hairy roots specific gene, rolC specific primers were used for
PCR reaction. (Primers were designed according to the NCBI nucleotide database E03275.)
F-rolC: 5’- ATGGCGGAATTTGACCTATGTGCTCTCTTTTCC – 3’
B-rolC: 5’- CCTCACTCCATTCCAAATTTGCATTCGCCATGCC – 3’
The composition of PCR reaction solution:
Volume (μl) Final concentration
Genomic DNAextracts 2.5 < 1 μg
4 mM dNTP 2.5 400 μM each
10X Reaction buffer 2.5 1X
10 μM primers 2.5 1 μM
Super-Therm Polymerase 0.5 2.5 U
DMSO 1.0 5.0%
Double distilled H2O 12 -
Total volume 25 -
28
PCR amplification was performed for 30 cycles in a thermal cycler. The PCR program for rolC was: 95°C, 5 min for denaturation; 94°C, 1 min, 56°C, 30 sec, 72°C, 40 sec for 30 cycles; 72°C, 10 min and paused at 4°C. The amplified fragment was resolved on a 1.2% agarose gel, followed by EtBr staining to check the PCR products.
2.5 Functional GFP and GUS confirmation
2.5.1 Direct observation of GFP
Green Fluorescence Protein (GFP) has two excitation peaks, a major one at 395 nm and a minor one at 475 nm. Its emission peak is at 509 nm in the lower green portion of the visible spectrum. Fluorescence of roots was observed directly on 1/2 MS plate by fluorescence microscopy (Nikon E600,Tokyo, JP) combined with a digital camera. The fluorescence microscope has a fluorescence filter combination of excitation wavelength:
450-490 nm; dichroic mirror wavelength: 495 nm; barrier wavelength: 500-550 nm. The digital camera was Olympus C7070 wide-zoom (Olympus, Tokyo, Japan) set to full manual mode with aperture at 4.8, shutter at 30 sec, and ISO at 80. The image was taken with 1600 x 1200 pixels, and then resized without further modification of the image.
29 2.5.2 GUS activity assay
β-Glucuronidase (GUS) was detected by a histochemical assay using 5-bromo-4-chloro-3-indolyl glucuronide (X-Gluc) for qualitative assay (Jefferson et al., 1987). Fresh roots were washed several times with 50 mM NaH2PO4 (pH 7.0) to clean up culture medium, then transferred to 24 well plates. The 1 mM indigogenic substrate X-Gluc in 50 mM NaH2PO4 (pH 7.0) was added and incubate at 37°C overnight. After staining, roots were rinsed in 70% ethanol for 5 min, then roots were examined both bare eyes and under microscopy.
2.6 Protein quantification
2.6.1 Extraction of cytosol protein
Fresh roots were grounded into powder in liquid nitrogen, and 50 mg was taken for extraction of total intracellular protein by a homogenizer with 500 μL protein extraction buffer. After centrifugation at 12000x g for 10 min at 4°C, supernatant was collected, and then total intracellular protein extract was stored at -30°C.
30 2.6.2 Quantification of total protein
The total protein content was measured with Bradford’s reagent (Bio-Rad Laboratories Inc., Hercules, CA, USA) with bovine serum albumin (BSA) as the standard.
2.6.3 Western blotting for detection of GFP
2.6.3.1 Gel electrophoresis
Protein extract was mixed with 2X loading buffer for SDS-PAGE analysis. After the sample was boiled at 95°C for 10 min, it was loaded into the well of SDS-PAGE (resolving gel: 15.0%, stacking gel: 5.0%). The electrophoresis was performed in 150 V for 1 hour. For western blot analysis, the prestained molecular weight marker was utilized in SDS-PAGE in order to evaluate the efficiency of protein transfer.
2.6.3.2 Protein transfer
After electrophoresis, the gel was taken for protein transfer. It was transferred to Amersham Hybond polyvinylidene difluoride (PVDF) membrane (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) membrane in 80 V for 1 h by PowerPac HC Power Supply (Bio-Rad Laboratories Inc., Hercules, CA, USA). The presence of prestained molecular marker ensured that the protein transfer was successful.
31 2.6.3.3 Immunoblotting
The membrane was blocked at room temperature for 1 h by blocking reagent buffer.
Anti-penta His HRP conjugate was added on the membrane, incubated at room temperature for 1 h. The membrane was then washed with TBS-T buffer twice and TBS buffer once. Chemifluorescence reagent was used at a ratio of 1:1 for the chemilumogenic substrate and oxidizing reagent, and then analyzed by AutoChemi BioImaging System (UVP LLC, Upland, CA, USA).
32
33
Chapter 3
Results
34
3.1 A. rhizogenes transformation
The transformation efficiency for transforming pCAMBIA 1201 into wild type A.
rhizogenes (harboring only Ri plasmid) was 5.5 x 105, and 8.25 x 105 for transforming pCAMBIA 1302 (Table 3). The transformation efficiency was 8.5 x 102 for transforming pCAMBIA 1201 into A. rhizogenes 1302 (harboring Ri plasmid and pCAMBIA 1302), and 1.05 x 103 for pCAMBIA 1302 into A. rhizogenes 1201 (harboring Ri plasmid and pCAMBIA 1201). The efficiency for transforming two binary vectors, pCAMBIA 1201 and pCAMBIA 1302, simultaneously into wild-type A.
rhizogenes was 2.75 x 102.
3.2 Selection of soybean materials
Among the six soybean genotypes tested, after 7 days of germination, soybean Tainan
#2 and Tainan #7 showed the highest germination rate of 96.5% and 90.2%, average plant height of 3.05 cm and 4.35 cm, respectively (Figure 8
(Utilization factor = total usable tissue fragments per seed planted)
Figure 8 (B)). Because of the need of cutting soybean plant into segments for Agrobacterium infection, the higher the plant is, the more usable materials are. We
created a “utilization factor” here combining soybean germination rate and plant height,
35
which was calculated by dividing total usable fragments by total planted soybeans, resulting 2.62 and 2.64 for soybean Tainan #2 and Tainan #7.
Although soybean Tainan #7 showed slightly better results on calculation, but its plant shoot was much thinner than that of Tainan #2, considering this fact soybean Tainan #2 was the most suitable soybean material for fast and efficient obtaining materials for further studies.
3.3 Establishment and selection of transgenic root clones
After A. rhizogenes were applied to the wounded soybean tissues, roots were induced from tissue wound sites. The wild-type A. rhizogenes-mediated soybean Tainan
#2 root induction rate was 48.0% on cotyledons and 15.4% on the lower-shoots (Table
4). Root induction rate of A. rhizogenes 2BV (harboring pCAMBIA 1201 and 1302) was 39.6% and 14.8% on cotyledons and lower-shoots, respectively. Root induction rate raised to 68.2% and 32.6% on cotyledons and lower-shoots respectively when infecting simultaneously with A. rhizogenes 1201 and A. rhizogenes 1302. The results above were calculated each with over 50 cotyledons and shoot segments in each construction. Most
A. rhizogenes infected tissues induced more than one root. The fast elongating, branched
roots were considered candidates for transgenic hairy roots (Table 4), these roots were removed from the soybean tissue and placed on plates with cefotaxime.
36
After detached from the original soybean tissues, several root clones didn’t’
propagate anymore after two rounds of A. rhizogenes sterilization. Thirty candidate root clones were picked from each group according to the growth on solid 1/2 MS plate, transferred to liquid 1/2 MS medium containing antibiotic for mass cultivation.
3.4 Liquid cultures of transgenic root clones
Root clones induce by wild-type A. rhizogenes showed very poor growth after 21 days liquid cultivation in medium containing antibiotic, averaged in a 1.22 fold growth in fresh weight (Figure 9). The 2BV group root clones showed similar growth as WT root clones, with an average growth of 1.20 fold in fresh weight (Figure 10). The only exception after the first 21 days of cultivation was root line 2AR-C31-1, this line grew 8.09 fold and gained 790 mg in fresh weight, but the other lines in 2AR group showed poor growth with average growth at 1.32 fold (Figure 11). Our first assumption was that the antibiotic added for sterilization of A. rhizogenes suppressed root growth, so we transferred all 90 root clones into fresh medium containing no antibiotic.
Wild-type roots grew better after 21 days of cultivation in antibiotic-free medium, averaged at 1.97 fold growth (Figure 9). On the other hand, most of the 2BV and 2AR root clones didn’t show improvement after transferring to antibiotic free medium. One root line 2AR-C16-2 did result in rapid growth of 3.57 fold and gained 540 mg in fresh
37
weight. The previously rapid growing line 2AR-C31-1 grew 1.98 fold and gained 390 mg in fresh weight. Excluding these two lines, the average growth of 2BV and 2AR root were 1.15 fold and 1.16 fold in fresh weight, respectively (Figure 10 and Figure 11).
We also calculated carbon source consumption by analyzing residue sugar content in the culture medium after the second cultivation using HPLC. The system was Shimadzu LC-3A (Shimadzu Science Co., Tokyo, Japan) combined with Bio-Rad Model 1770 Differential Refractometer (Bio-Rad Laboratories Inc., Hercules, CA, USA), using Thermo Hypersil® APS2 column (Thermo Scientific Inc., Waltham, MA, USA), and a mobile phase at 0.8 ml/min consist of 85% acetonitrile and 15% H2O. The average carbon source consumption of WT root clones was 20.01% (Figure 9), which was quite low comparing to the two rapid growing 2AR root clones, C16-2 and C31-1, each with carbon source consumption at 61.11% and 58.77% (data not shown).
The morphology of root clones was quite variant between poor growing lines and rapid growing lines. All root clones showed fast extension on the taproot and low branching number, but the rootlets showed no further branching. The overall growth was considered fast while still attached to the original soybean tissue (14 to 21 days post induction, see Figure 12 (A) and (B)). After detaching the original tissue and transferred to 1/2 MS plate containing antibiotic, growth of all root clones were dramatically lowered. For some root clones, after two rounds on plate for over 14 days,
38
the growth was not even visually identifiable. The chosen 30 root clones from each group were comparably faster growing clones on plates. However, after transferring to liquid medium and cultivate for two rounds of a total 42 days, most of the root clones remained the morphology with only little growth on the taproot and no visually identifiable growth on rootlets (Figure 12 (D), (E), and (F)). On the other hand, the two fast growing root clones, 2AR-C16-2 and 2AR-C31-1, showed plagiotropic growth and were highly branched after culturing in liquid medium (Figure 12 (G) and (H)).
Plagiotropic growth is the common morphology for hairy roots, which means the longer axes of rootlets or branches from rootles slanting away from the vertical line (Cho et al., 2000).
3.5 Transgenic root clones gene confirmation
The 90 root clones underwent 42 days of liquid cultivation were ground in liquid nitrogen and genomic DNA were extracted, DNA confirmation was then carried out by PCR amplification. For all candidate root clones, the first target in DNA confirmation is the 543 base pairs rolC sequence. In WT construction, rolC was not observed on gel for all thirty root clones (data not shown). For 2BV and 2AR construction, rolC was not observed either. Even the two root clones with the hairy roots like morphology did not
39
show rolC band as well (Figure 13). Because of the inexistence of rolC in all candidate root clones, we could not verify hairy roots and adventitious roots at this point.
The other two targets for 2BV and 2AR construction in DNA confirmation was gfp and gus, each sized 833 and 638 base pairs. For gfp confirmation, two of the root clones, 2BV-C30-2, and 2AR-C31-1 showed gfp band on gel (Figure 13). All root clones from these two constructions were then examined under fluorescent microscopy for the confirmation of functional GFP. For the two root clones that gfp was observed, GFP was functionally expressed and observed. In addition, root clone 2AR-C16-2 also showed GFP expressing (Figure 14).
For gus confirmation, all sixty candidate root clones from 2BV and 2AR construction did not show gus band on gel. With the experience of false estimating gene transcription of gfp, all root clones underwent examination of GUS activity. We found no root clones functionally expressing GUS.
The two fast growing root clones were the only ones that had sufficient material for protein extraction, and the 2BV-C16-2 clone expressing GFP was also extracted. We extracted total protein from the three root clones as mentioned above. After quantification, the protein extracts were further concentrated 5 fold because of the low protein content. SDS-PAGE was first carried out (Figure 15). According to the result on gel, both roots 2AR-C16-2 and 2AR-C31-1 had a 27 kD GFP band as expected. Root
40
2BV-C16-2 had protein content too low for observing GFP band on gel. For GUS (68 kD), a light band of around 70 kD which might be GUS was observed for root 2AR-C31-1 and 2BV-C16-2. We then further examined the three protein extracts with Western blotting, using the 6x His tag on both GFP and GUS as target. We did not observe any band on the membrane, which might be due to the low protein content of root protein extracts (data not shown).
All of the gene confirmation results are summarized in Table 5.
41
Chapter 4
Conclusions and Discussions
42
4.1 A. rhizogenes transformation
In this study, we attempted to construct an A. rhizogenes-mediated multiple gene transformation system in soybeans. The strategies could be divided into two parts concerning the binary vector number being used. The first strategy is transforming two genes in separate binary vectors simultaneously, either harbored by one A. rhizogenes transformant or by two separate transformants. The second strategy is transforming two genes in a single binary vector, either in a single T-DNA region or in separate T-DNA regions. We have only managed to test the first strategy so far in this study.
Using the strategy of transforming genes in two separate binary vectors, we first have to understand the efficiency of constructing A. rhizogenes transformants.
Transforming a single binary vector into wild-type A. rhizogenes had a transformation efficiency averaged at 6.88 x 105, which had little difference depending on the vector transformed. This protocol is considered quite efficient of gaining A. rhizogenes transformants. Further transforming the second binary vector into transformed A.
rhizogenes had a efficiency averaged at 9.5 x 102. The protocol was still capable of gaining sufficient transformants but the transformation efficiency was a thousandth of transforming the first vector into A. rhizogenes. The transformation efficiency transforming two vectors simultaneously into wild-type A. rhizogenes was even lower at 2.75 x 102. This decrease in the acceptability of A. rhizogenes might be related to
43
plasmid incompatibility due to the considerable similarity between the two binary vectors being transformed.
Plasmid incompatibility is due to the sharing of one or more element of the plasmid replication or partitioning systems (Novick, 1987). In this case, pCAMBIA 1201 and pCAMBIA 1302 share the backbone sequence which contains the replication origin and a hygromycin resistance gene, a total of 8737 base pairs (pCAMBIA 1201 and pCAMBIA 1302 are sized 12,001 and 10,549 base pairs, respectively). Vectorial plasmid incompatibility, in which case one plasmid is lost exclusively or with higher probability, might happened in this study. When transforming the second binary vector into a pre-transformed A. rhizogenes, most cells retained only the original vector harbored. Only 1 x 10-9 of the transformed cells lost the original vector and gained the newly transformed vector. Although plasmid incompatibility seems to played an important role affecting the transformation process, it is considered not to have its affect after the cells are cultured under selection stress, which is antibiotics in this case (Novick et al., 1976).
4.2 Soybean and transgenic soybean roots
We examined six genotypes of soybean for selecting the most suitable material for further soybean researches, and we found soybean Tainan #2 showed the highest
44
germination rate and a high utilization factor compared to the other five genotypes of soybean tested. The selection strategy was based on the germination rate and also considering the growth rate, plant height and shoot thickness at an early period of germination as the index of selecting the suitable soybean material. With this strategy, it seemed we have overlooked the difference of soybean genotype characteristics between the six soybean genotypes tested.
Using soybean Tainan #2 as the material for A. rhizogenes mediated gene transformation, we gained plenty candidate root clones which showed fast growth while still attached to the A. rhizogenes infected soybean tissue. However, from these candidate root clones we only gained two root clones which maintained fast growth on solid plate and in liquid medium after detaching the original tissue. Based on the morphology and the growth, these two clones had a high possibility being transgenic hairy roots, and the rest of the candidate root clones might be adventitious roots.
The PCR results for gene confirmation were not quite as we expected. For the poor growing root clones which were not expected to be hairy roots, rolC was not detected as expected. But for the fast growing clones which were expected to be hairy roots based on the morphology and growth, rolC was not found, either. Secondly, there was a root clone which had GFP functionally expressed but gfp not detected in the PCR confirmation. The two confusing results gave us a conclusion that the problem might
45
lies in PCR. We then tried many modifications with the PCR reaction mixture and the PCR sequence, but still gained the same results. As the PCR problem still not solved, for now we will make our judgments based mainly on the morphology and on the protein level. As a conclusion, we had gained two transgenic hairy root clones, 2AR-C16-2 and 2AR-C31-1, both expressing GFP but not GUS. We also gained one adventitious root 2BV-C30-2 expressing GFP, and we gained no wild-type hairy roots.
As with the PCR problem, because of the massive numbers of gene within soybean genomic DNA pool and the unknown site of T-DNA insertion (Zambryski, 1988), it is somehow difficult to amplify some certain genes. To avoid this problem, reverse-transcription PCR can be carried out. Because not all genes in soybean genomic DNA are being transcript in hairy roots, RT-PCR can enhance the chance of finding the desired genes.
The two hairy root clones we gained is considered that both gained two of the three T-DNA we transformed, which in this case was a T-DNA region containing rolC and a T-DNA region containing gfp. Furthermore, the adventitious root expressing GFP can be considered which gained only one of the three T-DNA being transformed. From the above results, we suspect that A. rhizogenes might have a transformation limit for T-DNA numbers being transformed simultaneously into soybean. On the other hand as we mentioned above, plasmid incompatibility has a high probability of happening when
46
A. rhizogenes is cultured under no selection stress. After A. rhizogenes was applied to
the wounded soybean tissue, A. rhizogenes and soybean were co-cultivated for a 14 to 21 days period on antibiotic free 1/2 MS plates. In this period, there was no selection stress for A. rhizogenes to retain the two nonessential extra-chromosomal binary vectors.
So, plasmid incompatibility might be another reason causing some of the genes not being transformed into soybean.
This low efficiency of gaining soybean transgenic hairy roots, compared to previous studies in our lab of inducing tobacco hairy roots, might be related to the soybean characteristics. Soybean Tainan #2 is a crossbred genotype bred by the Tainan District Agricultural Research and Extension Station (Tainan, Taiwan) in 1993. This soybean genotype has a strong disease-resistant ability to soybean downy mildew caused by Peronospora manshurica and soybean rust caused by Phakopsora pachyrhizi, it also has a medium disease-resistant to bacterial leaf spots disease caused by
Xanthomonas axonopodis and bacterial pustules caused by Xanthomonas campestris (連
大進 等,1993). These disease-resistant abilities of soybean Tainan #2 might have an effect on the A. rhizogenes-mediated gene transformation. Agrobacterium, as mentioned above, is a soil born plant pathogen, so Tainan #2 might also have the ability to resist or partially-resist the infection of A. rhizogenes, which might be the reason that we were not able to gain sufficient transgenic hairy roots as expected.
47
Also, we suspected that the low efficiency of inducing soybean hairy roots might also have relation with the tissues used for induction. In this study, we used cotyledon and the lower shoot for induction of hairy roots. From our results, we suspected that many of the root clones inducted from lower shoot and cotyledon might just be soybean root which will grow even without A. rhizogenes induction. One member of our lab is currently studying A. rhizogenes-mediated soybean Tainan #5 gene transformation, most of the root clones induced from the upper shoot of Tainan #5 showed hairy roots morphology on plates and in liquid cultures. We then tested the induction rate of soybean Tainan #2 upper shoot with wild-type A. rhizogenes. The induction rate was 51.4%, and 32.6% of the induced roots showed fast growth while still attached to the upper shoot tissues. Roots induced from upper shoot generally showed more branching than those inducted from lower shoot and cotyledon (Figure 12 (C)), more likely to be hairy roots. Although this assumption is not yet confirmed, it is a point that should be considered in the following studies.
4.3 Perspectives
Our current strategy of transforming multiple genes into soybean is using multiple binary vectors (Figure 3 and Figure 4), this strategy is thought to be easier to construct.
But since plasmid incompatibility might have occurred and had a negative effect on the
48
transformation efficiency, other transformation strategies should be considered. The other strategy we constructed but not yet utilized in this study, transforming two genes in a single binary vector (Figure 5 and Figure 6), is thought to be more suitable for multiple genes transformation. Construction of two genes in a single binary vector, either constructing the two genes in a single T-DNA region or in separate ones, can avoid plasmid incompatibility happening during the co-cultivation period of
Agrobacterium and plant tissues. This will ensure the two genes both have its chance to
be transformed into plant cells. So to transform soybean with the two constructions that use only one binary vector is the first step for the follow ups of this study. And the binary vector with the two genes in separate T-DNA regions can also be a model for the study to find out if there’s a transform limit of T-DNA number for A. rhizogenes.
We had an assumption that soybean genotype characteristic and the tissue being induced might affect soybean susceptibility to A. rhizogenes-mediated transformation.
This assumption needs a broad scanning of different tissues of plenty soybean genotypes, testing sterile plant germination rate, wild-type A. rhizogenes induction rate, and the percentage of hairy roots over induced roots. This test may take long to finally select the most suitable soybean tissue, but it should be an important step to construct a complete soybean gene transformation system.
49
The A. rhizogenes-mediated soybean multiple genes transformation is not yet fully constructed. Although this study leaves a lot of unsolved questions behind, it still give us some knowledge about A. rhizogenes-mediated soybean hairy roots induction system:
1. the A. rhizogenes our lab uses can successfully induce soybean hairy roots and the hairy roots can propagate under solid and liquid medium; 2. the foreign gene transformed is successfully inserted into soybean hairy roots genome; 3. the foreign gene transformed can be functionally expressed in soybean hairy roots. These knowledge assure us that A. rhizogenes-mediated soybean gene transformation system can be used to become a foreign protein expression system, and this system can also be used for Genomics like promoter trapping and gene tagging.
50
51