微/奈米加工之化工技術應用於癌症治療與檢測
90
0
0
全文
(2) 行政院國家科學委員會補助專題研究計畫. ■成果報 告□期中. 進度 報告 (計畫名稱)微/奈米加工之化工技術應用於癌症治療與檢測 計畫類別:□ 個別型計畫 ■ 整合型計畫 計畫編號:NSC 94-2218-E-006-031- 執行期間: 94 年 1 月 1 日至 96 年 12 月 31 日 計畫主持人:周澤川 共同主持人:黃炳照、杜景順、許梅娟、楊明長、謝淑珠、林錫 璋 計畫參與人員:王盧正、遊子弘、張人文、曾莞容、陳建中、王 佩珊、蘇嘉弘、王思惠、黃佳琪、毛賢為、葉書瑄、蕭高軒、王 威智、邱秀珍、賴銘賢、李貞宜 成果報告類型(依經費核定清單規定繳交):■精簡報告 □完整 報告 本成果報告包括以下應繳交之附件: ■赴國外出差或研習心得報告一份 □赴大陸地區出差或研習心得報告一份 □出席國際學術會議心得報告及發表之論文各一份 □國際合作研究計畫國外研究報告書一份 處理方式:除產學合作研究計畫、提升產業技術及人才培育研究 計畫、列管計畫及下列情形者外,得立即公開查詢 □涉及專利或其他智慧財產權,□一年 □二年後可公開查詢 執行單位:國立成功大學 化學工程學系(所) 中 華 民 國 97 年 2 月 28. 0.
(3) 療劑磁性奈米載體之合成-子計畫一 黃炳照教授-國立台灣科技大學/化工系 1.. Introduction. Magnetic nanoparticles are attract many interest for a wide variety of applications such as: magnetic seals, printing, recording1-3 (technology aspect) and MRI agents4,5, cell tagging and sorting (biology aspect). Different areas of using requires different particle size, shape and surface properties. In this decade the special interest for core/shell structures arises because of the ability to enhance nanoparticles properties and functionality. This structure mainly used for stabilization as the shell will able to protect the core and furthermore the shell might use for surface modification and functionalization. There are a lot of work on magnetic core/shell nanoparticles such as core/shell Fe/Au6,7, Fe/Fe-oxide8, and Fe-oxide/Au9. The following research will utilize the reverse micelles synthetic as the main methods. The microemulsion was prepared using a quaternary surfactant system of cetyltrimethylammonium bromide (CTAB), n-butanol, n-octane, and water10. The aqueous reactants were introduced into the micelle solution and allow react11 within the confines of the micelles. The micelle synthesis method leads to a very uniform spherical particles of sizes determined by the ω, the molar ratio of surfactant to water12. This method allows the synthesis of iron particles that can be used as a nucleation source for the epitaxial growth of a gold shell13. 2.. Experimental. Fe/Au nanoparticles was synthesized by using reverse micelle reaction under the flow of N214. CTAB is use as surfactant, octane as oil phase, and 1-butanol as cosurfactant. The preparation of micelle solution is dissolve CTAB first in 1-butanol and then add n-octane slowly while keep stirring the solution, this will create micelle solution. After that prepare 4 aqueous solution consist of FeSO4 (Fe source, ω=8), HAuCl4 (Au source ω=12), and two NaBH4 solution (reducing agent ω=8,12). This aqueous solution then added to the micelle solution slowly by dropping it using pipette, and the white sludge-milky like micelle solution of surfactant mixed in the oil will slowly change become clear and homogeneous solution because the addition of water. This is what we called a reverse micelle solution. The water droplet in size of several 1.
(4) nanoparticles will disperse homogeneously throughout the oil-stabilize phase because the presence of surfactant. Then both four of solution is put in the water bath of maintained temperature 30° C and purged by N2 gas for two hours using a sparger to make the bubble smaller to achieve an environment O2-free. After 2 hours purging the solution with N2, one of NaBH4 reverse micelle solution was added to FeSO4 (both solution ω=8), this will react and form Fe-core nanoparticles for 5 minutes reaction time. After that the solution of HAuCl4 added and soon the other NaBH4 solution too (both solution ω=12). The larger ωis needed to expand the water droplet size in order to reduce HAuCl4 to the shell of Fe-nanoparticles. This solution is allowed to react overnight. The next day, the solution was taken and washed three times with mixture of methanol:chloroform in 1:1 ratio to remove the surfactant. The nanoparticles were precipitated by using centrifuge with 15000 rpm for 30 minutes. Then they were dried under vacuum which resulting a black powder. Figure 1 will tell the details of the experiment.. Figure 1. Detail of experiment Characterization of nanoparticles were carried by XRD, SEM equipped with EDX, and TEM. X-ray diffraction measurements were made on a XRD-Rigaku Dmax-B, the scanning carried out 2θrange from 20◦up to 90◦and using Cu K radiation Ni as filter, voltage 40 kV and current 100mA) with a scan rate of 10◦ /min and 0.05 as interval. The lattice parameter (a) was calculated and refined using a least-square method. The average particle size was estimated using the overlap (200) peak of the Au. 2.
(5) k and (110) peak of Fe diffraction pattern by using the Scherrer equation: D . B cos . where D is the average particle size in Å, k is a coefficient taken here as 0.9, λthe wavelength of the X-rays used (1.5418 Å), B the width of the diffraction peak at half height in radians, and θthe angle at the position of the peak maximum. The samples for xrd measurement were treated on stainless steel mold to form a pellet and the pellet was sticked on holder glass. The samples were later investigated using electron microprobe energy dispersive X-ray (EDX) emission analysis with a JSM 6500 energy dispersive X-ray analyzer. The Fe/Au nanoparticles were characterized by a JEOL JEM-1010 transmission electron microscope (TEM) that operated at an accelerating voltage of 200 kV. The TEM sample was prepared by placing a small droplet of as synthesized Fe/Au nanoparticles suspension onto a carbon coated copper TEM grid and leaving this to dry in an oven. 3. Result and Discussion As we can see from XRD result in Figure 2, the α-iron bcc peaks overlap/overshadow by gold fcc peaks resulting that as if only gold peaks appear in the XRD spectra. This result is consistent with previous works of other groups research6,14,15. The overlapped peaks indicates that from XRD is impossible to differentiate between iron and gold inside the system.. 3000. Intensity (A.U). Fe/Au nanoparticles Fe peaks Au peaks 2000. 1000. 0. 40. 60. 2 theta. 3. 80.
(6) Figure 2. XRD pattern of Fe/Au nanoparticles. Figure 3. EDX spectra of Fe/Au nanoparticles. Table 1. EDX analysis of Fe/Au nanoparticles Element. Weight (%). Atomic (%). OK. 24.22. 0.00. 71.78. 0.00. Fe K. 16.40. 20.96. 13.93. 48.33. Au M. 59.38. 79.04. 14.29. 51.67. Figure 4. TEM image From Scherrer equation by using the overlapped peak at 44 degree, the estimation particle size for Fe/Au nanoparticles are around 8 nm. This number indicating that it is slightly smaller than the experimental condition. The number of ωis indicating the size of the water droplet in nm (although this apply for ωaround 8 to 16). For the iron core nanoparticles formation, the water droplet size are 8 nm, and after addition of gold nanoparticles the water droplet were increase to the number around 9.5 nm. This step-reaction was conducted in order to obtain an 8 nm Fe-core with 2 nm Au-shell nanoparticles. TEM image from the prepared Fe/Au nanoparticles give result of uniform particle size around 9 nm, which is slightly larger than the XRD analysis. But the 9 nm is nearly 4.
(7) the same with what we expected from the experiment. The core/shell structure is not clearly seen in the TEM image. HRTEM is needed to see the differentiate the lattice fringes between iron and gold From EDX spectra, the oxygen element were recorded and presence in a high atomic ratio towards either iron or gold. This indicating even though from XRD there is no records of oxide formation of iron, but the oxidation of iron happened inside the nanoparticles. The explanation for this phenomena is might be because the oxidation of iron in the nanoparticles is creating a non-crystalline structure of iron-oxide resulting that the iron-oxide peaks will no appear in the XRD pattern. Why did the core/shell of the iron/gold nanoparticles cannot attain the pure iron phase inside its structure? Cho, et al.7 was succeded proven that the pure-iron phase did formed in the beginning of experiment, but after the time of storage it slowly oxidized and Carpenter, et al16. discuss about the probability of what happened inside the nanoparticles structure when it slowly oxidized. They suggest that it might happened for several reason such as: imperfect gold shell protection for iron resulting that some iron is able to expose to the outer environment and another probable explanation is grain boundaries of gold coating maybe larger enough and allow diffusion of oxygen through the gold shell to the iron core. We have suggestion that there is a chance that the some iron nanoparticles were protected well by gold shell, and some were not. So we try to separate them using the acid treatment (dissolve the particles back to acid solution 0.5% HCl). So the unprotected one might be dissolved into the acid solution creating iron salt, and the well-pretected nanoparticles will keep their structure. The result is that we can only obtain a pure gold nanoparticles, without any iron inside it. 4. Conclusion The reverse micelle methods successfully creating a core/shell iron/gold nanoparticles by subsequently reduction of iron in the core and follow by reduction of gold in the shell. The nanoparticles produce have the uniformly particle size around 9 nm and its value is close with what we expected from the experiment procedure. From EDX composition analysis the nanoparticles were oxidixed and cannot retain its pure iron form. This can be explain by the imperfect of gold shell protection which allow oxygen pass through the shell and oxidizing the iron core.. 5.
(8) References (1) (2) (3). Ross, C. Annu. ReV. Mater. Res. 2001, 31, 203-235. Murray, C. B.; Kagan, C. R.; Bawendi, M. G. Annu. ReV. Mater. Res. 2000, 30, 545-610. Martin, J. I.; Nogues, J.; Liu, K.; Vicent, J. L.; Schuller, I. K. J. Magn. Magn.. (4). Mater. 2003, 256, 449-501. Oswald, P.; Clement, O.; Chambon, C.; Schouman-Claeys, E.; Frija, G. Magn.. (5). Reson. Imaging 1997, 15, 1025-31. Kim, D. K.; Zhang, Y.; Kehr, J.; Klason, T.; Bjelke, B.; Muhammed, M. J.. (6). Magn. Magn. Mater. 2001, 225, 256-261. Lin, J.; Zhou, W.; Kumbhar, A.; Wiemann, J.; Fang, J.; Carpenter, E. E.;. (7). O’ Connor ,C.J .J. Solid State Chem. 2001, 159, 26-31. Cho, S.-J.; Kauzlarich, S. M.; Olamit, J.; Liu, K.; Grandjean, F.; Rebbouh, L.;. (8) (9). Long, G. J. J. Appl. Phys. 2004, 95, 6804-6806. Carpenter, E. E.; Calvin, S.; Stroud, R. M.; Harris, V. G. Chem. Mater. 2003, 15, 3245-3246. Lyon, J. L.; Fleming, D. A.; Stone, M. B.; Schiffer, P.; Williams, M. E. Nano. Lett. 2004, 4, 719-723. (10) Attwood, V; Mosquera, J; Rodriguez, M; Garcia, M.J; Suarez, Coll. Polym. Sci. 1994, 272, 584–591. (11) G.N. Glavee, C.R. Kernizan, K.J. Kalbunde, C.M. Sorensen, G.C. Hadjapanayis, Chem. Mater. 1991, 3, 967–976. (12) M. Boutonnet, J. Kizling, P. Stenius, Coll. Surf. A: Physicochem. Eng. Asp. 1982, 5, 209–225. (13) T. Prozorov, R. Prozorov, A. Gedanken, Adv. Mater. 1998, 10, 1529–1532. (14) Carpenter, E.E, et al. Mat. Sci and Eng. 2000, A286, 81-86 (15) Carpenter, E.E, et al. IEEE Trans on Magnetic. 1999, 35, 3496-3498 (16) Ravel, B; Carpenter, E.E; Harris, V.G. J. Applied Physics. 2002, 91, 8195-8197. 6.
(9) 療劑磁性奈米載體之合成-子計畫一 杜景順教授-東海大學/化工所. 摘要 本計畫在水相溶液系統中,利用共沈澱法在不同 Fe2+與 Fe3+濃度比與添加不 同界面活性劑濃度下,製備鐵氧化物,將鐵氧化物分散於溶液中,再利用還原法 將 Au 製備至鐵氧化物上。在不同莫耳濃度比(Fe2+/Fe3+)下所製備之鐵氧化物,其 粒徑大小隨著比值下降而減小,所獲得顆粒的型態與大小皆不均勻。當 Fe2+ / Fe3+ 濃度比為 2 M / 1 M、1 M / 2 M 與 1 M / 2.5M 時,其平均粒徑分別為 26、17 與 15 nm 。 而 當 添 加 固 定 濃 度 之 界 面 活 性 劑 SB12 (N-dodecyl-N,N-dimethyl-3ammonio-1-propane-sulfonate)時,在反應物 Fe2+與 Fe3+總濃度較低時,製備所得 之顆粒大小及其型態受界面活性劑的影響較大。製備所得之鐵氧化物的飽和磁化 量隨粒徑減小而下降。在水相中,當鐵氧化物與黃金的莫耳比為 1/1、1/2 與 1/3 時,製備所得之鐵氧化物@金顆粒大小分佈為 11-30, 12-30 與 15-40 nm。鐵氧 化物@金奈米粒子的飽和磁化量較鐵氧化物低。利用 Fe2+與 Fe3+之濃度比為 1 M: 2 M 製備所得之鐵氧化物進行磁熱效應分析,由結果發現磁熱效應之溫度上升量 與磁場強度、頻率以及磁性物質之含量皆成正比關係。改變反應前驅物的濃度比 例(Fe2+/Fe3+)1 M / 2 M、1 M / 2.5 M 與 1M / 3 M 製備所得之鐵氧化物,其飽和磁 化量分別為 68.5、49.68 與 16.11 emu g-1,磁熱效應中施加 65 Hz 及電壓 150 V 所獲得之溫度上升量為 1.1、0.85 與 0.6 ℃,其溫度上升量隨所獲得之飽和磁化 量上升而增加。核殼式鐵氧化物@金奈米粒子,在磁熱效應中溫度上升量隨著 Au 前驅物濃度增加而遞減,其原因為其飽和磁化量與磁滯區面積減小。. 關鍵字:鐵氧化物、磁性奈米粒子、鐵氧化物@金、磁性質、磁熱效應. 7.
(10) 第一章. 緒論. 1-1. 磁性奈米材料 磁性奈米材料不僅具有一般奈米材料所擁有的特性,還具備了其他物質所 沒有的磁性質,這個獨一無二的特性,使得其在許多應用領域有更進一步的突破 與發展。在週期表上 106 個元素中僅有 Fe、Co、Ni,3 個元素及稀土族的 Gd 在 室溫時具有鐵磁性之表現。可藉由此類磁性元素與不具磁性的物質結合或是相互 結合,形成不同特性的磁性材料,最常見也最常使用的磁性物質為 Fe、Co、Ni, 及其氧化物,如 Fe3O4、γ -Fe2O3、CoO 等。不過一般的磁性奈米材料若要擴大其 應用領域,則大多需要經過表面修飾後,才能使其具有選擇性的與其他分子、抗 體或 DNA 等物質結合,而達到不同的應用的目的。 在鐵系的氧化物系列中,大約可分為 16 種鐵氧化物。這些鐵氧化物通常以 2+ Fe 、Fe3+與其他元素結合所形成,一般較為常見的有 FeO、Fe2O3、γ -Fe2O3 與 Fe3O4,其中 γ -Fe2O3 與 Fe3O4 具有強磁性,為鐵系中最常使用的物質。而 Fe2O3 亦即 α- Fe2O3,由於其晶格的對稱性高,因此物質的磁矩大多互相抵銷而磁性很 小,但 α- Fe2O3 是鐵系氧化物中最穩定的。表 1-1 為鐵氧化物的分類與特性[1]。 Fe3O4(magnetite)的結構最早建立在 1915 年,其結構屬於反尖晶石(inverse spinel)型態,而其原子排列結構為面心立方結構 ( face- centered cubic structure, FCC)。Fe3O4 與一般鐵氧化物不同的地方在於同時包含了二價與三價鐵離子,一 般表示式可寫成 Fe2+Fe3+2O4。在其晶格結構中含有四面體與八面體的晶格,其中 四面體的位置分佈著 Fe2+與 Fe3+,而八面體的位置僅有 Fe3+的存在,主要結晶型 態有八面體、菱形、球狀等。 γ -Fe2O3(maghemite)的結構與 Fe3O4(magnetite)是很相似的,不同的地方只是 所組成的 Fe 元素皆為 3 價,且填滿四面體的位置後,剩餘的 3 價離子則隨意的 分佈於八面體的位置上,但未能填滿八面體的位置,因此可以 Fe8[Fe13.3□2.67]O32 表 示 , 其 中 □為 八 面 體 空 缺 的 位 置 。 一 般 認 為 γ -Fe2O3(maghemite) 是 Fe3O4(magnetite)轉變為 α- Fe2O3 的中間體。主要型態有立方狀、平板狀與細長狀 [1-2]。centered cubic structure, FCC)。Fe3O4 與一般鐵氧化物不同的地方在於同時 包含了二價與三價鐵離子,一般表示式可寫成 Fe2+Fe3+2O4。在其晶格結構中含有 四面體與八面體的晶格,其中四面體的位置分佈著 Fe2+與 Fe3+,而八面體的位置 僅有 Fe3+的存在。主要結晶型態有八面體、菱形、球狀等。. 8.
(11) 表 1-1 鐵氧化物的分類與特性[1] Mineral name. 化學式. 晶格結構. Goethite. α-FeOOH. orthorhombic. Lepidocro cite. γ -FeOOH. orthorhombic. Hematite. α-Fe2O3. Magnetite Maghemit e. Fe3O4. Wustite. γ -Fe2O3 FeO. rhombohedral hexagonal. cubic cubic or tetragonal cubic. 晶格常數 / nm a = 0.9956 b= 0.30215 c = 0.4608 a = 0.307 b = 1.253 c = 0.388 a= 0.50356(1) b= 1.37489(7) a = 0.8396 a= 0.83474 a=0.4302-0 .4275. 磁性種類. 物質顏 色. antiferrom ag.. yellow-b rown. 4.26. antiferrom ag.. orange. 4.09. red. 5.26. black reddishbrown. 5.18.. weakly-fer romang. or antiferrom ag. ferrimag. ferrimang antiferrom ag. black. 密度 / g cm-3. 4.87 5.9-5.9 9. 1-2. 磁性奈米材料鐵氧化物之製備與性質 合成鐵氧化物的方法很多,雖然到目前為止,其詳細的反應機構仍然不是 很清楚,但本質上形成氧化物最基本的兩個反應機制如下[1]; I. 含有 Fe2+或 Fe3+的溶液直接反應沈澱。 II. 以鐵氧化物作為前驅物,藉由溶解/共沈澱程序或是經由固體物質本 身內部的結構變化而獲得氧化物。 在反應機制 I.中,主要鐵氧化物生成及轉變途徑[1]: (1) 在不同的溫度及 pH 值下,進行 Fe3+的溶液水解(hydrolysis) FeCl3 + 6 H2O→ Fe ( H2O)63+ + 3Cl-. (1-1). 當 Fe3+與水分子形成錯合物,在鹼性環境下反應可形成 FeOOH。 Fe(H2O)63+ → Fe OOH+3H+ + 4 H2O 9. (1-2).
(12) (2). (3). (4). (5). Fe2+溶液進行氧化獲得鐵氧化物 當 pH ≧ 7 時,Fe2+會產生自催化反應而加速 Fe3+的生成,因 此溶液中可同時存在有 Fe2+與 Fe3+,在水溶液中可形成 Fe(OH)3 與 Fe(OH)2 形式之化合物,在不同條件下製備不同的鐵氧化物。 金屬化合物的熱裂解 鐵金屬化合物如五羰鐵(Iron pentacarbonyl, Fe(CO)5),其中結 構中心為 0 價 Fe 鍵結 5 個 CO 分子。利用高溫熱裂解的方式於 120 ℃裂解出 Fe 金屬,再利用氧化劑將其氧化形成鐵氧化物。[3] 在高溫下的相轉變[1] 由於鐵氧化物於 α-Fe2O3 之型態最為穩定,因此利用高溫熱處理, 可使其產生相轉變,如 γ -Fe2O3 在 150-180 ℃之水溶液中,其晶相 會由立方體(cubic)轉變為菱形六面體(rhombohedra) 溶解/再沈澱(reprecipitation)反應. 反應機制 II.中,以鐵氧化物作為前驅物,在適當的條件下,大部分的鐵氧 化物皆可轉變成至少兩種以上的其他鐵氧化物。. 1-3. 磁性奈米材料應用 表面經適當修飾,使其具有特定良好的化學性質的磁性奈米粒子可應用於 許多領域,如磁共振造影(Magnetic resonance imaging, MRI)的對比試劑、組織修 補、免疫分析、過高熱治療、藥物傳遞系統及細胞分離生物醫療上[4]。一般而 言,磁性奈米粒子應用於生物醫療上需考慮水溶性, 穩定性與生物相容性[5]。由 於磁性奈米粒子容易聚集,若表層沒有經過修飾,在水溶液或是組織液中聚集的 問題就無法解決,這種現象會使得在應用上受到限制,因此有些時候會藉由表面 修飾以減少聚集的現象,同時也可以在應用上增加其與其他物質的相容性。 奈米粒子表面的修飾可利用非聚合物的有機物、高分子聚合物與無機物進 行表面修飾。如 1999 年 Yee[6]利用 alkanesulfonic 與 octadecanephosphonic acids 非聚合物的有機物修飾非晶相的 Fe2O3,探討不同的修飾對鐵氧化物之影響及其 自組裝的特性。2001 年 Sahoo[7]等人以油酸、月桂酸等物質吸附在 Fe3O4 粒子上, 形成保護層避免進一步的氧化,同時也使得 Fe3O4 磁性粒子可以穩定的存在有機 溶劑中,此法在有機相中合成粒子時常被使用。1996 年 Lee [8]在 PVA 水溶液中 合成鐵氧化粒子,利用 PVA 修飾粒子表面等。這些都是為改善磁性奈米粒子聚 集,增加其應用價值的方法。 2005 年 Cheng [9]將 Fe3O4 粒子注入動物的腎臟細胞後,利用細胞毒素測試 與 NMR 來探討 Fe3O4 在生醫應用的可行性。研究結果發現細胞毒性檢驗結果顯. 10.
(13) 示,在不同 Fe3O4 粒子劑量下,沒有細胞毒性效應產生,且細胞生存力及增長能 力顯示,Fe3O4 有極佳的生物相容性。 到目前為止,磁性奈米粒子在生醫上的應用層面上相當廣泛[5], 包括作為 MRI 的對比試劑[5, 10];藥物傳遞系統(drug deliver system) [5, 10, 11]; 細胞的純 化分離[5, 10]; 過高熱治療(hyperthermia)[5], 西方醫學之父尊稱的 Hippocrates, 過去曾經提出利用熱鐵去灼燒癌細胞表面,藉由高溫消滅不正常細胞。磁性誘發 熱治療是利用磁性物質藉由交流電場治療癌症的方法之一,此治療方式不僅可以 避免副作用的產生,而且可以輕易的處理在較深層的癌細胞。將磁性奈米粒子埋 至癌細胞上,並且在變動的磁場作用下使溫度上升,而溫度上升的程度與材料的 磁性質、磁場強度、震動頻率以及在癌細胞上血液的冷卻能力有關。一般來說, 癌細胞所能承受的最高溫度約在 43 ℃ [5],而正常細胞則可承受更高的溫度。 1-4. 研究動機與目的 由衛生署公佈的國人十大死因結果,癌症已是第 23 年蟬連寶座了。除了改 善現代人的生活作息以及飲食習慣來降低癌症的發生機率外,在醫療上的診斷與 治療也需要不斷的改進與突破。在過去癌症治療皆以手術切除腫瘤細胞,之後再 進行化療,這樣的治療方式不但破壞了人體的免疫系統,且復發的機率高。而在 診斷上也不容易發現腫瘤細胞,通常檢驗報告發現的時候,大多接近末期了。因 此為了降低死亡以及提升診斷的準確度,近幾年,各領域的專家學者對於癌症治 療改良與診斷技術上突破不遺餘力。 磁性奈米粒子與一般材料不同,其具有特殊的磁性質,藉由表面的修飾, 如結合高分子、生物分子或者是金屬等形成多功能的複合材料。由於其具有磁性, 因此可利用外加磁場輕易的控制粒子的動向,而表面修飾使得粒子可以攜帶藥 物、細胞分離等,在生物醫療領域上的突破與發展更具有前瞻性。一般環境中由 於僅有 Fe、Co、Ni 具有磁性,但 Co 與 Ni 有致癌的可能,因此在生物醫療上的 應用受到相當多的限制。Fe 雖然其磁性相當強,但由於容易氧化,因此多數的 研究都在有機相中合成奈米粒子,藉由界面活性劑保護粒子避免氧化,但附著於 粒子表面的界面活性劑及殘留物,是否對人體造成危害尚未確定。 因此在本研究中吾人嘗試以 FeCl3 與 FeCl2 作為前驅物,利用共沈澱法在水 相系統中合成鐵氧化物,再利用還原劑將 HAuCl4 還原至鐵氧化物上,製備出核 殼式奈米粒子。目前此類材料多應用於生醫領域上,如細胞分離、藥物傳遞、癌 症熱治療等。由於核殼式奈米粒子可以少量的貴重金屬製作於磁性材料上,再藉 由表面修飾或分子結合等方式,利用磁場而達分離或藥物傳遞之目的,但其在癌 症熱治療上,利用高頻磁場的作用使磁性粒子產生足夠殺死癌細胞之應用則較 少。所以在本研究中,吾人利用 XRD 分析其晶相、TEM 與 SEM 分析其顆粒分 佈及表面型態、原子吸收光譜(AAS)與氧化還原滴定法分析確認其組成比例以及 使用超導量子干涉儀(SQUID)量測其磁性質,最後藉由變動磁場測量其磁熱效 應。. 11.
(14) 第二章. 實驗設備與程序. 2-1. 鐵氧化物之奈米顆粒製備 在鐵氧化物奈米顆粒的製備上分為添加與未添加界面活性劑兩種狀況,其製備 是利用 Fe2+與 Fe3+共沈澱進行製備。 鐵氧化物@Au 奈米複合材料之製備 首先如 2-1. 中所述先製備鐵氧化物奈米顆粒,再將 HAuCl4 水溶液中的金 離子利用 NH2OH 為還原劑將其還原製備於鐵氧化物奈米顆粒上,再利用離心方 法將製備所得之鐵氧化物@Au 奈米顆粒分離得之。 2-2.. 奈米顆粒及複合材料之鑑定與分析 製備所得之鐵氧化物與鐵氧化物@Au 奈米顆粒之晶相利用 XRD 分析;奈米 粒子之粒徑大小及分佈情形以穿遂式電子顯微鏡(analytical electron microscope, AEM)與場發式電子顯微鏡(field emission scanning electron microscope, FE-SEM) 觀察。在組成分析方面則利用氧化還原滴定加以分析,所利用之氧化劑為 KMnO4 水溶液;亦利用原子吸收光譜儀(AAS)分析製備所得奈米粒子之組成。製備所得 之奈米粒子的磁性利用超導量子干涉儀(superconducting quantum interference device, SQUID)加以分析。磁熱效應之量測乃利用 AC 電源供應器、高斯計、溫 度計、冷卻循環水系統,取樣品 7 mg 置入樣品瓶中,將其以保麗龍作為隔熱材 料,隔絕外界水對樣品瓶內部的溫度影響。樣品瓶外部之冷卻水以 30-35 ml/min 之速度通入氮氣以達攪拌作用,使電磁鐵中空管內的水溫降低且均勻分佈。利用 交流電所產生之交流電場的變化,研究自製之鐵氧化物之磁熱特性。下量測裝置 如圖 2-1 中所示。 2-3.. 圖 2-1 磁熱效應之儀器裝置圖. 12.
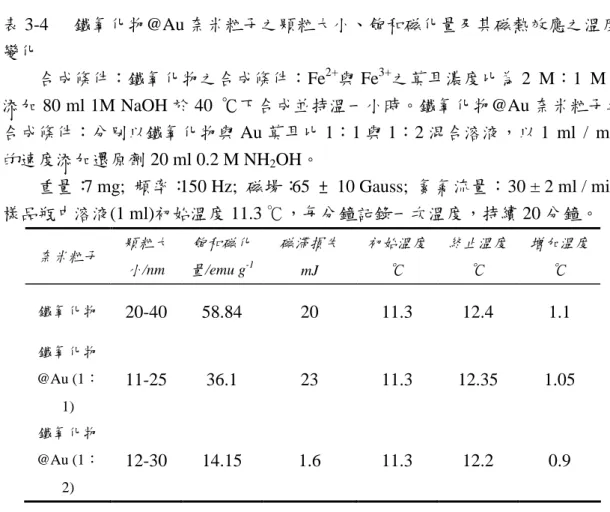
(15) 第三章. 結果與討論. 3-1 鐵氧化物奈米顆粒之性質分析 3-1-1 粒徑分析 未添加界面活性劑之鐵氧化物奈米粒子,在 Fe2+與 Fe3+之莫耳濃度比為 2 M: 1 M,溫度為 40 ℃下,持溫一個小時所合成之奈米顆粒,分散於無水乙醇中, 由 AEM 觀測所得粒子之粒徑介於在 15~40 nm 之間, 如圖 3-1 所示。. 30. Percentage / %. 25. 20. 15. 10. 5. 0 8. 12. 16. 20. 24. 28. 32. 36. 40. 44. Particle size / nm. 2+. 3+. 圖 3-1 Fe 與 Fe 濃度比為 2M:1M 時合成所得之鐵氧化物奈米粒子之 AEM 影 像與粒徑分佈圖。 製備條件:pH:13-14,反應溫度:40 ℃下,反應時間:1 小時,轉速:6000 rpm。 當 Fe2+與 Fe3+之莫耳濃度比為 1.5 M:1 M 時, 所合成之鐵氧化物奈米顆粒 粒徑範圍為 15~30 nm; 當改變 Fe2+與 Fe3+之莫耳濃度比為 1 M:2 M 時,其顆 粒明顯縮小,分佈範圍約 12-20 nm。進一步改變前驅物濃度為 1 M:2.5 M 與 1 M: 3 M 時,其顆粒分佈的 AEM 分析結果示其顆粒更小。當 Fe2+與 Fe3+莫耳濃度比 愈大時,其顆粒成長愈大,反之則愈小。這可能是由於 Fe3O4 奈米粒子形成之後, Fe2+會吸附於 Fe3O4 奈米粒子表面,且 Fe2+於鹼性環境中其氧化還原能力增加, 因此 Fe2+吸附於 Fe3O4 奈米粒子表面進行氧化反應,而使 Fe3O4 奈米粒子繼續成 長產生較大的顆粒。 由上述結果發現所合成之粒子在不添加界面活性劑下大小不均勻,因此嘗 試以添加 SB12 與 CTAB 兩種不同屬性界面活性劑的方式,試圖利用微胞的方式 限制顆粒的成長,因此選擇 Fe2+與 Fe3+之濃度比為 2 M:1 M 之條件作為反應液, 分別添加界面活性劑,使其濃度為 1.65、3.31 與 6.62 mM,在合成溫度為 40 ℃ 13.
(16) 的環境下,持溫 1 小時,反應完成之後,以轉速 6000 rpm 離心獲得之鐵氧化物 奈米顆粒。. 3-1-2 製備所得鐵氧化物的晶相分析 在不添加界面活性劑之條件下,改變反應物 Fe2+與 Fe3+之濃度比製備之鐵氧 化物,其繞射波峰與 JCPDS(19-0629)中的資料進行比對,結果如圖 3-2 所示,圖 中所顯示的鐵氧化物之主要晶面分別為(220)、(311)、(400)、(422)、(511)與(440) 此為典型的 Fe3O4 中的 fcc 結構。然而與 JCPDS(39-1346)中的資料進行比對後發 現,γ -Fe2O3 與 Fe3O4 具有共同的繞射晶面,且繞射角相當接近,因此推測所製 備的鐵氧化物可能同時含有 Fe3O4 與 γ -Fe2O3。. Fe 3 O 4 -Fe 2 O 3 -Fe 2 O 3. (311) (220). (511) (440) (422). Intensity (a.u). (400). (1.5:1) (1:2). (2:1). 20. 25. 30. 35. 40. 45. 50. 55. 60. 65. 70. 75. 80. 2. 圖 3-2. 在不添加界面活性劑之條件下,不同 Fe2+與 Fe3+濃度比製備所得之 鐵氧化物之 XRD 圖譜。製備條件:pH:13-14,反應溫度:40 ℃下,反應 時間:1 小時,轉速:6000 rpm. 圖 3-2 中顯示合成鐵氧化粒子時,其反應液中的 Fe2+與 Fe3+之濃度比愈低時,(311) 晶面之繞射波峰愈小,這可能是由於鐵氧化物之結晶性不佳,而圖 3-2 中,分別 可在繞射角為 44.74°、65.13°與 78.23°等三處,發現相當尖銳的繞射波峰,這是 由於吾人再分析 XRD 時所使用之基材(Al)的繞射波峰。吾人將由 SEM 分析中所 得之顆粒分佈,以及 XRD 所得之晶格常數及晶粒大小,整理於表 3-1 中。. 14.
(17) 表 3-1. Fe2+與 Fe3+濃度比對製備所得鐵氧化物特性之影響. pH13-14,反應溫度: 40 ℃,反應時間: 1 小時,轉速:6000 rpm [Fe2+] :[Fe3+] M:M. 晶格常數 / nm. 晶粒大小/ nm. 平均粒徑 / nm. 2:1. 0.8395. 17.0. 26. 1.5:1 1:2 1:2.5 1:3. 0.8394 0.8391 0.8371 0.8330. 13.6 9.9 6.4 5.5. 20 17 15 5*. 綜合 AEM 與 SEM 顆粒大小與顆粒表面型態分析及 XRD 晶相分析之結果, 可以發現在反應液中,水與界面活性劑之比例會影響微胞大小,而進一步影響顆 粒大小及型態。另外,除了水與界面活性劑之比例對顆粒大小與型態上會有影響 外,當前驅物之初始反應濃度與界面活性劑的濃度比例愈高,其對顆粒大小、型 態與晶粒大小的影響不顯著。而當前驅物之初始總反應濃度與界面活性劑的濃度 比例較低時,界面活性劑對製備所得鐵氧化物結晶特性的影響較大。因此在製備 鐵氧化物奈米粒子時,顆粒大小及其表面型態可藉由改變界面活性劑與水之比例 及前驅物初始莫耳濃度與界面活性劑之間的比例來控制所製備之奈米粒子大小 及其結晶表面型態。. 3-1-3 製備所得鐵氧化物之組成分析 吾人採用一般化學分析方法中,常使用的氧化還原滴定法分析鐵氧化物中之 組成。所使用的氧化劑為 KMnO4,利用 Mn7+還原成 Mn2+的顏色變化(滴定前溶 液中呈現無色,當達到滴定終點時溶液顏色會由透明轉變為粉紅色)作為指示劑, 滴定鐵氧化物中的 Fe2+含量,以確定鐵氧化物中之 Fe2+與 Fe3+之比例,而進一步 推算 Fe3O4 與 γ -Fe2O3 的莫耳百分比。 由實驗結果發現莫耳濃度比相同為 2:1,但其總濃度分別為 0.75 與 3 M 時, 其所製備之鐵氧化物中所含的 Fe2+重量百分比分別為 7.92 與 7.87 wt %,利用此 實驗數據可以推算所製備之鐵氧化物中 Fe3O4 與 Fe2O3 之莫耳百分比,分別為 25.18 % 與 74.82 % 以及 25.03 % 與 74.97 %。兩者所製備之鐵氧化物奈米粒子 中,其 Fe3O4 與 Fe2O3 之含量是相當接近。由顆粒大小、表面型態與晶相分析之 結果與組成分析之 Fe3O4 與 Fe2O3 之含量結果顯示,在相同的濃度比下,改變總 濃度製備鐵氧化物其顆粒大小、型態以及晶粒大小上有明顯差異外,在組成上的 差異不大(如表 3-2 中所示)。 製備鐵氧化物奈米粒子時,當 Fe3+濃度固定的情形下,改變 Fe2+與 Fe3+之 莫耳濃度分別為 2 M:1 M 與 1.5 M:1 M,製備所得之奈米粒子中,所含有的 Fe2+重量百分比分別為 7.87 與 9.26 wt %,據此計算粒子中 Fe3O4 與 Fe2O3 之莫耳 15.
(18) 百分比分別為 25.03 % 與 74.97 %,以及 30.03 % 與 69.97 %。由實驗結果顯示, 在製備鐵氧化物時,當反應中 Fe2+之莫耳濃度增加,不僅未對製備所得之奈米粒 子中,Fe2+之含量有提升作用,反而降低了奈米粒子中之 Fe2+之含量,這顯示降 低反應物中 Fe2+之初始莫耳濃度對於 Fe3O4 的形成是比較有利。在製備鐵氧化物 奈米粒子時,反應物中 Fe2+濃度固定的情形下 (1.0 M),改變 Fe2+與 Fe3+之莫耳 濃度分別為 1 M:2 M、1 M:2.5 M 與 1 M:3 M 製備所得之奈米粒子中,所含 有的 Fe2+重量百分比分別為 12.64、5.01 與 3.52 wt %,其相對之 Fe3O4 與 Fe2O3 之莫耳百分比分別為 43.14:56.86、15.30:84.70 與 10.55:89.45。當 Fe3+之莫 耳濃度增加時,相對的 Fe2+所佔的比例就會比較少,因此鐵氧化物奈米粒子中之 Fe3O4 莫耳比例也就大幅度的衰減。. 表 3-2 添加界面活性劑條件下反應物初始濃度比對製備所得之鐵氧化物奈米 粒子組成之影響 鐵氧化物之製備條件:依據不同初始濃度比配製 10 ml 反應液並於氮氣下曝氣除 氧 1 小時後,添加 80 ml 已除氧之 1M NaOH,並加熱至 40 ℃持溫 1 小時。 滴定條件:以 20 ml 的 6M 硫酸與數滴磷酸混合,待金屬氧化物完全溶解後稀釋 成 1M,以標定過濃度約 0.02 M 的 KMnO4 進行氧化還原滴定分析。 2+. [Fe ] : [Fe3+] M:M. 鐵氧化 物樣品 重 / mg. KMnO4 滴 定莫耳數 /μmol. Fe 莫耳 數 / mmol. Fe2+重 量百分 比 / wt %. 0.5:0.25. 14.5. 4.1. 0.0205. 7.92. 20.12: 79.88. 25.18: 74.82. 2:1. 22.9. 6.56. 0.0322. 7.87. 20.02: 79.98. 25.03: 74.97. 1.5:1. 17.6. 5.82. 0.0291. 9.26. 23.09: 76.91. 30.03: 69.97. 1:2. 22.7. 25.65. 0.0513. 12.64. 30.14: 69.86. 43.14: 56.86. 1:2.5. 16.2. 2.898. 0.01449. 5.01. 13.27: 86.73. 15.30: 84.70. 1:3. 18.1. 2.277. 0.01136. 3.52. 9.54: 90.46. 10.55: 89.45. 2+. 鐵氧化物中之成分莫 耳比 FeO: Fe3O4: Fe2O3 Fe2O3. 反應物 Fe2+與 Fe3+初始濃度比為 2 M:1 M 的系統中,分別添加濃度為 1.656、 3.31 與 6.62 mM 的 SB12 界面活性劑所製備之鐵氧化物奈米粒子,利用氧化還原 滴定法分析其組成之結果,如表 3-1.6 所示。由分析結果顯示,界面活性劑對於 16.
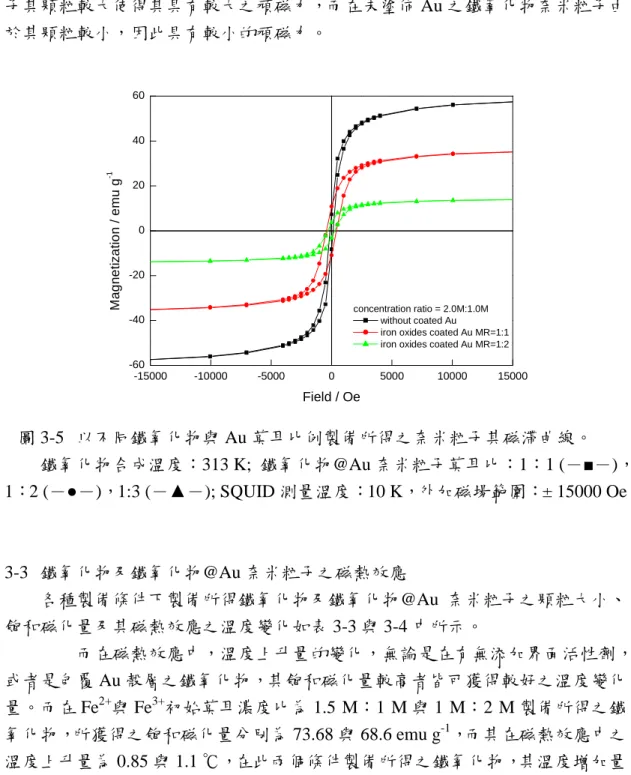
(19) 組成的影響並不明顯,其所獲得之 Fe3O4 與 Fe2O3 之莫耳百分比分別為 26.86: 73.14、28.78:71.22 與 29.70:70.30,雖然 Fe3O4 所佔的比例隨界面活性劑濃度 的增加而增加,但其值相較於與未添加界面活性劑之 25.03:74.97 來說,差距並 不大。接著吾人降低總濃度使得反應物 Fe2+與 Fe3+初始濃度比改變為 0.5 M:0.25 M 時。由實驗結果發現 Fe3O4 與 Fe2O3 之莫耳比例其分別為 27.87:72.13、28.26: 71.74 與 30.35:69.65,其成分與界面活性劑添加量間的關係與初始濃度比為 2 M: 1 M 條件下所製備之鐵氧化物之分析結果相同;即 Fe3O4 之組成百分比隨界面活 性劑添加量的增加而增加,但當反應物的初始濃度較低時其組成的變化受界面活 性劑的影響較為顯著。. 3-1-4 製備所得鐵氧化物之磁性質量測 未添加界面活性劑與反應溫度 40 ℃條件下,在不同反應物初始莫耳濃度中 製備所得之鐵氧化物奈米粒子,置放於 10 K 的環境中,並施加一外加磁場 ± 15000 Oe,利用超導量子干涉儀測量其磁滯曲線,結果如圖 3-3 所示。圖中發現 所製備之鐵氧化物在 10 K 之低溫環境下,當反應物 Fe2+與 Fe3+初始莫耳濃度比 為 2 M:1 M、1.5 M:1 M、1 M:2 M、1 M:2.5 M 與 1 M:3 M,其飽和磁化 量分別為 58.84、73.68、68.50、49.68、與 16.11 emu g-1。一般 Fe3O4 塊材的飽和 磁化量為 92 emu g-1,與吾人在此所製備之鐵氧化物其值皆低於塊材的飽和磁化 量,這可能與材料大小、結構及型態有關之外,在本論文中製備所得之鐵氧化物 中不完全為 Fe3O4,大部分為 γ -Fe2O3,故其飽和磁化量較低。. 80 60. Magnetizaion / emu g. -1. 40 20 0 -2 0 2+. 3+. [F e ]:[F e ]/ 2+ 3+ [F e ]:[F e ]/ 2+ 3+ [F e ]:[F e ]/ 2+ 3+ [F e ]:[F e ]/ 2+ 3+ [F e ]:[F e ]/. -4 0 -6 0 -8 0 -1 5 0 0 0. -1 0 0 0 0. -5 0 0 0. 0. 5000. M :M M :M M :M M :M M :M. 10000. 1 .0 :2 .0 1 .0 :2 .5 1 .0 :3 .0 2 .0 :1 .0 1 .5 :1 .0. 15000. M a g n e tic F ie ld / O e. 圖 3-3. 不添加界面活性劑條件下所製備之鐵氧化物奈米粒子其磁滯曲線。. 製備條件: pH13-14,40 ℃, 反應 1 小時。SQUID 測量條件:298 K,外加磁場: ±15000 Oe. 17.
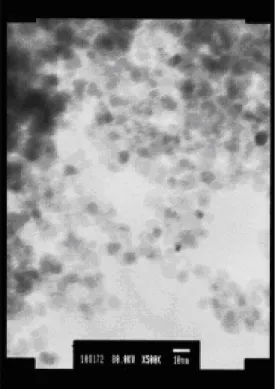
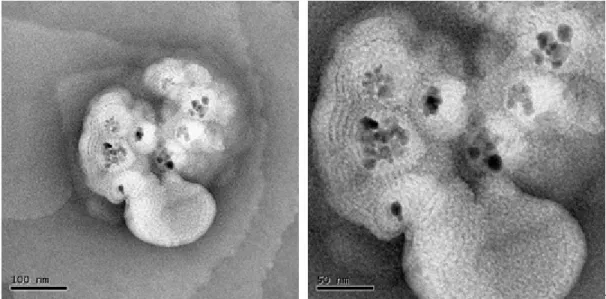
(20) 由實驗結果發現製備過程中,添加有界面活性劑 SB12 之鐵氧化物奈米粒子, 其飽和磁化量皆比沒有添加界面活性劑時所獲得的飽和磁化量大。由 AEM 粒徑 分析之結果顯示,在 Fe2+與 Fe3+初始莫耳濃度比為 2 M:1 M 之情況下,添加不 同濃度之界面活性劑 SB12 製備所得之鐵氧化物奈米粒子其顆粒大小相當接近, 而飽和磁化量似乎也有隨著顆粒大小變化而變化的趨勢。. 3-2 鐵氧化物@Au 奈米粒子之性質分析 不添加界面活性劑條件下,固定反應物 Fe2+與 Fe3+初始濃度比為 2 M:1 M 時,製備所得之鐵氧化物奈米粒子作為核種,先以四甲基氫氧氨與檸檬酸鈉溶液 進行粒子表面的前處理,再將其分散於含有界面活性劑 SB12 之水溶液中。添加 HAuCl4 使鐵氧化物與 HAuCl4 之莫耳比分別為 1:1 與 1:2,添加還原劑 NH2OH, 使 Au 還原至鐵氧化物表面,即可獲得鐵氧化物@Au 奈米粒子。 3-2-1 粒徑分析 當 Fe2+與 Fe3+莫耳濃度比為 2 M:1 M 時,製備所得之鐵氧化物奈米粒 子之平均粒徑為 20-40 nm。利用此條件所製備之奈米粒子作為核種,製備鐵氧 化物@Au 奈米粒子。調配溶液組成使鐵氧化物與 Au 之莫耳比為 1:1,製備所 得之顆粒大小影像分析如圖 3-4 中所示。在圖 3-4(a)中,吾人發現其顆粒尚稱均 勻,且與無 Au 之鐵氧化物粒子大小比較,發現在此之粒子大小沒有很大的改變, 推測可能是當鐵氧化物與 Au 之莫耳比 1:1 時,附著於鐵氧化物表面的黃金粒 子數量不大,另外,亦有可能部分黃金自我成核形成黃金顆粒,並未附著於鐵氧 化物表面,因此發現原有鐵氧化物之粒徑顆粒大小沒有明顯的改變。將鐵氧化物 與 Au 之莫耳比提高為 1:2 時,其顆粒大小分析如圖 3-4(b)所示,由圖中結果可 發現當鐵氧化物與 Au 之莫耳比提高時,所獲得之鐵氧化物@Au 奈米粒子顆粒 粒徑相較於莫耳比 1:1 時來得大,但仍有聚集的現象,其平均粒徑分佈介於 12-30 nm 之間;但莫耳數比為 1:1 時,其粒徑大小為 11-25 nm 之間,因此進一步的 將莫耳比例增加至 1:3 顆粒大小分佈如圖 3-4(c)所示,由圖中影像可知粒子的 型態較為規則,而粒徑分佈範圍亦擴大,其值介於 15-40 nm。 由於大多數製備核殼式奈米粒子大多是在有機相中合成,因此嘗試利用有機 金屬乙醯丙酮鐵(Fe(acac)3)作為前驅物,與還原劑 1,2-十六二醇及界面活性劑油 胺和油酸於高溫下製備鐵氧化物,再與有機金屬醋酸金(Au(OOCH3)3)混合製備鐵 氧化物@Au 奈米粒子,其粒徑分析結果如圖 3-4(d)中所示。分析結果顯示,與 在水溶液系統中製備所得之奈米粒子相比較,可以明顯的發現在有機相中,當鐵 氧化物與 Au 之莫耳比為 1:3 時,製備所獲得之奈米顆粒大小約 7 nm 且顆粒大 小分佈均勻而其分散性也較佳。. 18.
(21) (a). (b). (c). (d). 圖 3-4 製備所得之鐵氧化物@Au 奈米粒子 AEM 分析影像. 水溶液中鐵氧化物與 Au 莫耳比: (a)1:1 (b)1:2 (c)1:3; (d) 有機相中鐵氧化物 與 Au 莫耳比為 1:3 時製備所得.. 3-2-2 磁性質之量測 由於鐵氧化物@Au 奈米粒子其核心為具磁性之鐵氧化物,因此為了了解覆 蓋黃金殼層後,黃金殼層是否對其磁性特性造成影響,所以利用 QUANTUM DESIGN 所設計之型號為 MPMSXL7 之超導量子干涉儀測量其磁性質。測量條 件為 10 K 下,施加一外加磁場± 15000 Oe,量測其在不同外加磁場作用下,粒 子所產生的磁化量變化,利用外加磁場對磁化量做圖則可獲得鐵氧化物之磁滯曲 線,並藉由磁滯曲線了解殼層對於核心粒子之磁性影響。鐵氧化物@Au 奈米粒 子之初始莫耳比為 1:1 與 1:2 時,利用超導量子干涉儀分析其磁性,結果如圖 3-5 所示。由圖中之結果可以發現沒有覆蓋 Au 殼層之鐵氧化物奈米顆粒其具有 19.
(22) 最佳之飽和磁化量,其值為 58.84 emu g-1,且其磁滯現象也是最不明顯的。當覆 蓋上 Au 殼層時,其與鐵氧化物之莫耳比為 1:1 時,由圖中之結果可以清楚的 發現其飽和磁化量明顯變小,其值為 36.1 emu g-1,且有明顯的磁滯現象。當莫 耳比改變為 1:2 時,其飽和磁化量已經低於 20 emu g-1,同樣也有磁滯的現象發 生。這可能是由於所製備之鐵氧化物@Au 奈米粒子中,其鐵氧化物之含量較少(由 原子吸收光譜之元素分析可知),使得其飽和磁化量減弱。另一方面磁性材料鐵 氧化物覆蓋 Au 時,Au 具有磁遮蔽效應,也是造成其飽和磁化量較低的原因。 另外,在鐵氧化物與 Au 之初始莫耳比為 1:1 與 1:2 時,兩者所顯示之頑磁力 皆大於未覆蓋 Au 之鐵氧化物,這可能是由於塗佈有 Au 之鐵氧化物@Au 奈米粒 子其顆粒較大使得其具有較大之頑磁力,而在未塗佈 Au 之鐵氧化物奈米粒子由 於其顆粒較小,因此具有較小的頑磁力。. 60. Magnetization / emu g. -1. 40. 20. 0. -20 concentration ratio = 2.0M:1.0M without coated Au iron oxides coated Au MR=1:1 iron oxides coated Au MR=1:2. -40. -60 -15000. -10000. -5000. 0. 5000. 10000. 15000. Field / Oe. 圖 3-5 以不同鐵氧化物與 Au 莫耳比例製備所得之奈米粒子其磁滯曲線。 鐵氧化物合成溫度:313 K; 鐵氧化物@Au 奈米粒子莫耳比:1:1 (-■-), 1:2 (-●-),1:3 (-▲-); SQUID 測量溫度:10 K,外加磁場範圍:± 15000 Oe. 3-3 鐵氧化物及鐵氧化物@Au 奈米粒子之磁熱效應 各種製備條件下製備所得鐵氧化物及鐵氧化物@Au 奈米粒子之顆粒大小、 飽和磁化量及其磁熱效應之溫度變化如表 3-3 與 3-4 中所示。 而在磁熱效應中,溫度上升量的變化,無論是在有無添加界面活性劑, 或者是包覆 Au 殼層之鐵氧化物,其飽和磁化量較高者皆可獲得較好之溫度變化 量。而在 Fe2+與 Fe3+初始莫耳濃度比為 1.5 M:1 M 與 1 M:2 M 製備所得之鐵 氧化物,所獲得之飽和磁化量分別為 73.68 與 68.6 emu g-1,而其在磁熱效應中之 溫度上升量為 0.85 與 1.1 ℃,在此兩個條件製備所得之鐵氧化物,其溫度增加量 20.
(23) 與飽和磁化量間的趨勢不同。綜合粒徑、組成與飽和磁化量之結果,並與磁熱效 應中溫度上升量交互比對下,推測可能在飽和磁化測量上有些許的實驗誤差,而 產生 Fe2+與 Fe3+初始莫耳濃度比為 1.5 M:1 M 下製備所得之鐵氧化物,其飽和 磁化量與溫度上升量沒有呈現相同的趨勢。 製備過程中添加不同濃度之 SB12 界面活性劑之鐵氧化物奈米粒子,其所 測得之飽和磁化量皆比沒有添加 SB12 界面活性劑時所獲得的飽和磁化量大,由 於所獲得之顆粒大小差異並不大,因此推測可能與奈米粒子的形態有關,且飽和 磁化量隨顆粒改變而有些許的改變。在磁熱效應上,亦可發現在溫度上升量隨飽 和磁化量上升而上升,此與前述之趨勢相同。 在鐵氧化物@Au 奈米粒子系統中,當鐵氧化物奈米粒子覆蓋 Au 殼層時,鐵氧 化物奈米粒子與 Au 之前驅物莫耳比例為 1:1 與 1:2,所測得之飽和磁化量明 顯的小於未覆蓋 Au 殼層所獲得之飽和磁化量,這是由於鐵氧化物在整體磁熱效 應實驗中,總添加量(7 mg)所佔之比例,隨著 Au 所佔比例的增加而減少,使得 其飽和磁化量下降,另外亦可能是 Au 的存在對鐵氧化物有遮蔽效應。 表 3-3 各種製備條件下製備所得鐵氧化物之顆粒大小、飽和磁化量及其磁熱效 應之溫度變化 合成條件:依照不同的 Fe2+與 Fe3+之莫耳濃度比為反應液,添加 80 ml 1M NaOH 於 40 ℃下合成並持溫一小時。重量:7 mg; 頻率:150 Hz; 磁場:65 ± 10 Gauss; 氮氣流量:30 ± 2 ml min-1; 樣品瓶中溶液(1 ml)初始溫度 11.3 ℃,每分鐘記錄 一次溫度,持續 20 分鐘。 [Fe2+] :[Fe3+]. [SB12] /. 顆粒大. 飽和磁化量 -1. M:M. mM. 小 / nm. / emu g. 2:1 1.5:1 1:2 1:2.5 1:3 2:1 2:1 2:1. 0 0 0 0 0 1.656 3.31 6.62. 26 20 17 15 5 23 25 24. 58.84 73.68 68.50 49.68 16.11 66.35 60.54 63.86. 21. 初始溫度. 終止溫. 增加溫度. /℃. 度 /℃. /℃. 11.3 11.3 11.3 11.3 11.3 11.3 11.3 11.3. 12.4 12.15 12.4 12.15 11.9 12.45 12.35 12.4. 1.1 0.85 1.1 0.85 0.6 1.15 1.05 1.1.
(24) 表 3-4 鐵氧化物@Au 奈米粒子之顆粒大小、飽和磁化量及其磁熱效應之溫度 變化 合成條件:鐵氧化物之合成條件:Fe2+與 Fe3+之莫耳濃度比為 2 M:1 M, 添加 80 ml 1M NaOH 於 40 ℃下合成並持溫一小時。鐵氧化物@Au 奈米粒子之 合成條件:分別以鐵氧化物與 Au 莫耳比 1:1 與 1:2 混合溶液,以 1 ml / min 的速度添加還原劑 20 ml 0.2 M NH2OH。 重量:7 mg; 頻率:150 Hz; 磁場:65 ± 10 Gauss; 氮氣流量:30 ± 2 ml / min; 樣品瓶中溶液(1 ml)初始溫度 11.3 ℃,每分鐘記錄一次溫度,持續 20 分鐘。 奈米粒子. 鐵氧化物. 顆粒大. 飽和磁化 -1. 磁滯損失. 初始溫度. 終止溫度. 增加溫度. mJ. ℃. ℃. ℃. 小/nm. 量/emu g. 20-40. 58.84. 20. 11.3. 12.4. 1.1. 11-25. 36.1. 23. 11.3. 12.35. 1.05. 12-30. 14.15. 1.6. 11.3. 12.2. 0.9. 鐵氧化物 @Au (1: 1) 鐵氧化物 @Au (1: 2). 22.
(25) 第四章 1.. 2.. 3.. 4.. 5. 6.. 7.. 8.. 結論. 本研究之結論如下: 利用不同的 Fe2+與 Fe3+初始莫耳濃度比製備所得之鐵氧化物奈米顆粒其顆粒 大小隨 Fe2+ / Fe3+初始莫耳濃度比增加而變大,當 Fe2+ / Fe3+初始莫耳比濃度 為 2 M / 1M、1M / 2 M 與 1 M / 2.5M,其粒徑分別為 26、17 與 15 nm。 鐵氧化物顆粒大小與型態受製備過程中界面活性劑之添加量,與反應物初始 濃度等之影響,在界面活性劑添加濃度為 1.656、3.31 與 6.62 mM 下,Fe2+ 與 Fe3+莫耳濃度比為 2 M:1 M 所製備之顆粒大小相當接近,分別為 23、25 與 24 nm 左右,在顆粒型態上仍有不規則的現象。當將反應物總濃度下降, 使 Fe2+與 Fe3+莫耳濃度比為 0.5 M:0.25 M 時,其顆粒成長受到界面活性劑 之影響較大,當界面活性劑濃度分別為 1.656、3.31 與 6.62 mM 時,其顆粒 大小分別為 17、15 與 13 nm,在型態上亦較均勻。 在 Fe2+與 Fe3+反應物初始莫耳濃度比為 2 M:1 M,界面活性劑添加濃度為 1.656、3.31 與 6.62 mM 時,添加兩性離子型界面活性劑 SB12 時,所獲得之 奈米粒子粒徑分別為 23、25 與 24 nm;而在添加陽離子型界面活性劑 CATB 之奈米粒子粒徑分大小為 20、18 與 18 nm,故陽離子型的界面活性劑可更有 效的降低鐵氧化物奈米粒子的粒徑。 在組成分析上,Fe2+與 Fe3+初始莫耳濃度比為 1 M:2 M 時,製備所得之鐵以 化物奈米粒子其含有之 Fe3O4 最高,其值為 43.14 %,而當 Fe3+莫耳濃度愈高, 製備所得之 Fe3O4 含量愈少,添加不同濃度之界面活性劑對鐵氧化物之組成 比例影響不大。 在黃金殼層的包覆方面,在有機系統中製備所得之鐵氧化物@Au 奈米粒子之 分散性,相較水溶液系統者佳,且粒徑分佈均勻且較小,約 7 nm。 在磁性質分析中,於室溫下測量鐵氧化物之磁滯曲線可以發現不具有磁滯現 象,而當溫度下降至 10 K 時,有些微的磁滯現象。而飽和磁化量隨著顆粒大 小的減小隨之上升,其中以 Fe2+與 Fe3+莫耳濃度比為 1.5 M:1 M 製備所得之 鐵氧化物奈米粒子其具有最佳之飽和磁化量,其值為 73.68 emu g-1。而在添 加濃度為 1.656、3.31 與 6.62 mM 之 SB12 界面活性劑時,所獲得之鐵氧化物 飽和磁化量,分別為 66.35、60.54 與 63.86 emu g-1。 在鐵氧化物@Au 奈米粒子方面,所製備奈米粒子其飽和磁化量隨著 Au 所佔 比例的增加而降低,其當鐵氧化物與 Au 之莫耳比為由 1:1.07 提高至 1:2.59 時,所獲得之飽和磁化量由 36.1 降低至 14.15 emu g-1。 以 Fe2+與 Fe3+莫耳濃度比為 1 M:2 M 之鐵氧化物奈米粒子進行磁熱效應探 討中,在固定頻率與磁場強度下,發現奈米粒子的添加量愈高則所獲得磁熱 效應愈好,以 7 mg 為最佳。而在固定頻率的情況下,磁熱效應會隨供給之電 壓增加而增加。另外在固定磁場強度下,改變不同的頻率所獲得之磁熱效應 在 175 Hz 時,具有較佳的溫度上升量。 23.
(26) 9. 當 Fe2+與 Fe3+莫耳濃度比為 1 M:2 M、1 M:2.5 M 與 1M:3 M 製備所得之 鐵氧化物,其飽和磁化量分別為 68.5、49.68 與 16.11 emu g-1,磁熱效應中所 獲得之溫度上升量為 1.1、0.85 與 0.6 ℃,溫度上升量隨飽和磁化量上升而增 加,在其他製備條件下製備所得之鐵氧化物與鐵氧化物@Au 奈米粒子大致上 皆有相同的趨勢(參考表 3-3.5 與 3-3.6)。另外,由磁滯曲線上,所包圍的磁滯 區域面積愈大,其所損失的能量愈多,因此磁熱效應中溫度上升量也較多。. 參考文獻 1.. 2. 3.. 4.. 5. 6.. 7.. 8.. 9.. Cor ne l l ,R. M. ,Sc hwe r t ma nn,U. ,“ The I r on Oxi ds ---Structure, Properties, Re a c t i on,Oc c ur r e nc e sa ndUs e s ” ,s e c ond,Completely Revised and Extended Edition, WILEY-VCH. 周永洽,“ 現在磁性材料原理與應用” ,化學工業出版社, 1st, 北京, 2002, 14-22, 120-127, 633-674 Lingyan Wang, Jin Luo, Mathew M. Maye, Quan Fan, Qiang Rendeng, Mark H. Engelhard, Chongmin Wang, Yuehe Lin and Chuan-Jian Zhong, “ Iron oxide-gold core-s he l lna nopa r t i c l e sa ndt hi nf i l ma s s e mbl y ” ,J. Mater. Chem., 2005, 15, 1821-1832. Hy e on,T. ,Le e ,S. S. ,Pa r k,J . ,Chung ,Y.a ndNa ,H. B. ,“ Sy nt he s i sofHi g hl y Crystalline and Monodisperse Maghemite Nanocrystallites without a Size-Se l e c t i onPr oc e s s ” ,J. Am. Chem. Soc. 2001, 123, 12798-12801. Gupt a ,A. K. ,Gupt a ,M. ,“ Sy nt he s i sa ndSur f a c eEng i ne e r i ngofI r onOxi de Na nopa r t i c l e sf orbi ome di c a la ppl i c a t i on” ,Biomaterials, 2005, 26, 3995-4021. Yee, C., Kataby, G., Ulman, A., Prozorov, T., White, H., King, A., Rafailovich, M. , Sokol ov, J . ,a nd Ge da nke n, A. ,“ Self-Assembled Monolayers of Alkanesulfonic and-phosphonic Acids on Amorphous Iron OxideNanoparticles” , Langmuir, 1999, 15, 7111-7115. Sahoo, T., Pizerm, H., Fried, T., Golodnitsky, D., Burstein, L., Sukenik, CN., a nd Ma r kovi c h,G. ,“ Al ky lPhos phona t e / Phos pha t e Coa t i ng on Ma g ne t i t e Na nopa r t i c l e s : A Cmopa r i s on wi t h Fa t t y Ac i ds ” , Langmuir, 2001, 17, 7907-7911. Le e ,J . ,I s obe ,T. ,Se nna ,M. ,“ Pr e pa r a t i on ofUl t r a f i neFe 3O4 Particles by Pr e c i pi t a t i oni nt hePr e s e nc eofPVAa tHi g hpH” ,J. Coll. Interf. Sci, 1996, 177, 490-494. Cheng, F.Y., Su, C.H., Yang, Y.S., Yeh, C.S., Tsai, C.Y., Wu, C.L., Wu, M.T., Sh i e h,D. B. ,“ Cha r a c t e r i a z a i o nofAque ousdi s pe l r s i onsofFe 3O4 Nanoparticles a ndt he i rBi ome di c a lAppl i c a t i ons ” ,Biomaterials, 2005, 26, 729-738. 24.
(27) 10.. 劉吉平 郝向陽,“ 奈米科學與技術” ,世貿出版社, 1st, 台北, 2003, 18-25,. 11. 12.. 28-50 尹邦躍, “ 奈米時代” ,五南圖書出版公司, 1st, 台北, 2002, 1-20, 63-70 劉浩澧,“ 相位陣列式超音波熱治療系統之加熱策略設計與熱劑量控制” ,國 立台灣大學電機工程研究所博士論文, 2003.. 利用負載ICG 之磁性奈米粒子檢測癌細胞-子計畫二 何國川教授-台大化工所. 前言 在二十一世紀的初期,高科技發展一日千里,而科技發展的方向,將是以能 帶給人類更便捷迅速的資訊傳遞與更舒適安全的生活品質為首要目標。為達成這 些目的,新材料的開發與應用研究扮演著極為重要的角色。奈米材料科學領域在 近幾年來發展的非常迅速,部份關於奈米粒子特性的研究,已經到達成熟階段, 並且應用到相關領域。例如:金奈米粒子的性質與應用,是目前最成熟亦是最廣 泛的,不論在哪個領域都有相當的發展;硒化鎘(CdSe)之類的奈米粒子,在半導 體領域的應用近年來亦備受矚目,扮演著一個重要的角色;其他如碳奈米管 (Carbon Nanotube)、二氧化鈦奈米粒子(TiO2)、合金奈米粒子(PdRu)等,分別在 材料、光觸媒殺菌、燃料電池方面都是當前熱門的研究課題,其中氧化鐵粒子 (Fe3O4)應用於癌症治療與檢測的技術,更是目前極具發展潛力的新技術之一。因 此利用奈米粒子的特有性質,或是製備新型的奈米粒子來發展嶄新技術,將是未 來的趨勢。 磁性粒子可應用於記錄媒質及鐵流體,其最新的應用包括了高密度的資訊儲 存,以及許多生物醫學方面之應用,這些最新領用應用,需要極佳的特性控制, 如粒子粒徑大小及影響磁性特性之變數,皆須做深入的探討與研究。磁性粒子一 25.
(28) 般包含有多磁性領域,其旋轉群皆指向同一方向,且在作用上相互合作,這些磁 性領域被領域界璧分開,其長度規模約 100nm,奈米級的微粒不夠大,因此不足 以支撐多領域之形成,所以他們具有獨特的特性。 單一領域之磁性微粒於特定溫度顯示超順磁性行為,單一粒子由於結晶結構 內旋轉之排列具有磁動量,然而組合體個別粒子之磁動量並無調整,因此組合體 並不一定顯示出淨磁動量,外部磁場可施加使得粒子組合體磁化(或使所有磁動 量調整)。磁動量方向改變會形成能量障礙,使得粒子組合體動量即使在磁場消 除後仍成調整狀態,這是剩磁感應,是鐵磁性材料之特性之一。降低磁性到 0 之磁場強度被稱之為矯頑磁性(Coercivity),於高溫下、磁性奈米材料具有足夠熱 能克服磁場動態方向改變之能量障礙,因此當外部磁場消除後,於組合體個別微 粒動量可回歸到未調整之方向,這種行為被稱為超順磁性。 本研究選用 ICG 作為追蹤因子,藉由表面修飾生物相容性良好的磷脂質之 氧化鐵奈米粒子,藉由外加磁場進行標定與熱治療以及藥物釋放之研究。. 研究目的 利用改變電磁場使磁性奈米粒子產生熱,並可控制在 42~43℃,以此殺死癌 細胞[1,2]。此種癌症熱治療方式的表現及效率,與癌細胞的種類及磁性奈米粒子 的特性有關。因此,如何掌控上述磁性奈米粒子的結構、大小、表面官能基物質、 粒徑分佈等是很重要的因素。除了直接注射磁性奈米粒子於腫瘤上之細胞給於治 療外,此磁性奈米粒子也可以利用人體循環系統使此粒子到達並累積於癌細胞或 腫瘤上,治療後,亦可經由肝臟的代謝而排出人體,因此有益於應用在生物體內 具相容性的癌症治療。 將含有 Fe2+與 Fe3+的鐵鹽離子溶液混合後,以加入鹼的共沉澱法形成粒徑約 20nm 的四氧化三鐵粒子[3,4],再將甲基丙烯酸甲酯的單體,利用種子乳化聚合的 方式將其聚合於四氧化三鐵粒子外層[5],由於磷脂質成膜後外型無法硬化,容易 破裂變形,故特別選用聚甲基丙烯酸甲酯做為第二層磷脂質修飾時的支架[6,7], 然而,循血綠分子則可藉由磷脂質成膜於粒子上時,以鑲嵌的形式進入磷脂質膜, 使粒子成為可具有螢光追蹤性質。另一研究方向則針對氧化鐵以聚丙烯酸修飾, 以供進一步接上藥物或螢光劑進行實驗探討[8]。 若在人體內循環,藉由外加磁場累積於癌細胞組織中,達到追蹤檢測的功能, 之後,若可將藥物以包埋或接枝的形式加入粒子,更可進一步達到藥物釋放治療. 26.
(29) 之功能,而本身內部的氧化鐵核心,也可藉由外加磁場達到磁阻生熱的熱治療功 能[9-16],期望能達到一藥多用的功能。. 文獻探討 四氧化三鐵的結構最早建立在 1915 年,其結構屬於反尖晶石,其原子排列 則為面心立方結構。四氧化三鐵與一般鐵氧化物的不同在於其同時具有二價與三 價鐵離子,故其晶格結構中含有四面體及八面體的晶格,四面體結構的位置分布 著二價及三價鐵離子,而八面體中僅有三價鐵離子的存在,其主要結晶型態就有 八面體、菱形、球形等。 合成氧化鐵的方法很多,到目前為止,其反應機構尚未完全明朗,但基本上 都不外乎是以下兩種方法: (一) 含有二價及三價鐵離子的溶液直接反應沉澱 (二) 以鐵氧化物做為前驅物,藉由溶解及共沉澱程序或是經由固體物質本身 內部的結構變化而得 本實驗基本上都是採用共沉澱法的方式進行合成,因此,文獻回顧將針對 此法進行探討。首先是在 1956 由 David 使用 FeSO4 溶液為前驅物,在曝氮之下 加熱至 90℃,加入 KOH 與 KNO3 數滴,持溫攪拌ㄧ個小時候冷卻可得黑色之四 氧化三鐵。利用所合成之四氧化三鐵粒子氧化成γ-三氧化二鐵型態,發現結構 中之水無法去除,因此提出須以 OH-取代 O 原子使結構穩定之理論。 1929 年 Schikorr 提出在鹼性環境下將 FeSO4 溶液水解反應生成 Fe(OH)2 後 加熱至 100℃可得四氧化三鐵,此反應稱為 Schikorr reaction。1981 年 Regazzoni 等人提出以 α-三氧化二鐵作為前驅物,在 400℃、5%H2 與 95%Ar 環境下通入 無氧飽和水蒸汽可獲得四氧化三鐵。另外,利用二價鐵與三價鐵離子比例 2:1 的 混合溶液,在曝氮與鹼性環境下 80℃下亦可得四氧化三鐵。 1996 年 Kang 利用共沉澱法合成約 10nm 的四氧化三鐵,其中 pH 值須控制 在 11~12 之間,並以 NaOH 為沉澱劑,使四氧化三鐵奈米粒子沉澱,在離子收 機沉澱物。而 2001 年 Pardoe 將氯化鐵與氯化亞鐵之結晶水鹽類以莫爾數比 1:1 的關係混合,並加入氨水使 pH 值維持在 11.5 並加熱至 60℃持溫 15min,以去離 子水洗至 pH 值 6.5,離心即可得到四氧化三鐵,並透過合成過程中加入高分子 使其降低聚集狀況。 2005 年 Fong-Yu Cheng 等人同樣透過氯化鐵與氯化亞鐵之結晶水鹽類共沉 澱法方式,唯一改變的是將氨水置換為四甲基氫氧化銨,而將 pH 值控制在 13, 27.
(30) 此一合成方式,不再需要進一步修飾及可使四氧化三鐵粒子穩定懸浮於溶液中而 不產生沉澱,其原理乃是利用四甲基氫氧化銨可在四氧化三鐵表面形成立體排斥 障礙,使得粒子聚集狀況降低,因而得到性質良好的磁性流體,以進一步應用於 顯影劑以及熱治療等醫療用途。 本實驗在利用聚甲基丙烯酸甲酯來修飾氧化鐵,主要目的則在於使得磷脂質 可進一步修飾於上而不產生塌陷。合成之方式則參考 2004 年由 PING-CHIEH WANG 等人所提出之聚甲基丙烯酸甲酯與氧化鐵複合粒子之合成。將共沉澱法 合成之氧化鐵粒子穩定懸浮後,加入甲基丙烯酸甲酯單體後,曝氮並同時升溫至 80℃後加入起始劑進行種子乳化聚合,使聚甲基丙烯酸甲酯包覆於氧化鐵粒子表 面形成 Core-shell 型態,以進行下一步修飾。 磷脂質的修飾則參考了 2005 年由 Jyotsnendu Giri 等人提出的方法,將磷脂 質以及膽固醇加入氯仿中,待溶解後,再以旋轉濃縮機進行濃縮使其在瓶璧上成 膜後,再將已經修飾過聚甲基丙烯酸甲酯後的磁性流體加入濃縮瓶中,以超音波 震盪使磷脂質膜由瓶璧震落,再以自組裝聚合的方式在粒子表面重新成膜,其中 ICG 與膽固醇皆會在成膜時,藉由鑲嵌或包覆的形式進入膜層或粒子中,即可進 一步應用於追蹤治療。. 研究方法 四氧化三鐵奈米粒子的合成 方法一 部份還原共沉澱法 將氯化鐵與亞硫酸納以去離子水各自配製成水溶液後,以不同莫爾數比的方 式進行混合以探討其四氧化三鐵粒子純度,在 700rpm 之速率下攪拌,同時加入 10N 之氫氧化鈉水溶液 100ml,並以加熱包控制升溫至不同溫度,以探討不同溫 度反應下四氧化三鐵之性質。一小時後反應結束可得四氧化三鐵的磁性粒子。 方法二 鐵鹽溶液的配置與前處理 將氯化鐵與氯化亞鐵之結晶水鹽類以 2M 的鹽酸溶液各自配製成溶液後,以 莫爾數比 2:1 的方式進行混合,之後曝氮並以 400rpm 之速率進行預攪拌,同時 以加熱包加熱至 70℃。 四氧化三鐵的粒子合成. 28.
(31) (1)當溫度達設定之反應溫度後,開始逐滴緩慢加入不同的鹼液來進行共沉澱法, 同時,氮氣與機械攪拌仍然持續,一小時後結束反應即可得到四氧化三鐵的磁 性粒子,並探討不同鹼液所形成之四氧化三鐵其性質差異。 (2)將粒子以磁性分離的方式將粒子與溶液分離,並透過多次加入去離子水離心 的方式將粒子表面殘餘的其他物質洗淨純化後,加入分散媒使得磁性粒子能 穩定懸浮於水中而不產生嚴重的聚集沉澱現象,並以磁性流體之狀態保存, 以待接下來步驟的合成與修飾。 聚丙烯酸殼層的修飾 將方法一所合成之四氧化三鐵以分子量 2000 的聚丙烯酸寡聚物進行修飾。 其修飾條件:將聚丙烯酸與四氧化三鐵粒子以重量比 3:2.6 的比例,在反應溫度 90℃,700rpm 下反應二十分鐘 聚甲基丙烯酸甲酯殼層的修飾 甲基丙烯酸甲酯單體的前處理 由於購買的 MMA 單體內,均含有 50ppm 的抑制劑,因此在用來聚合之前,需 利用減壓蒸餾在 40℃下純化,把抑制劑與單體溶液分離,收集後保存在 4℃冰箱 中冷藏以待聚合使用。 界面活性劑及起始劑之配置 (1) 秤取 NaHCO3 0.02g 及 PEG(M.W=2000) 0.48g 以去離子水配置於 100ml 容量 瓶中,超音波震盪使其完全溶解,以作為聚合反應時之界面活性劑及 pH 值 緩衝液。 (2) 秤取 K2S2O8 0.06g 以去離子水配置於 10ml 容量瓶中,超音波震盪直到完全 溶解,以作為聚合反應之起始劑。 甲基丙烯酸甲酯的種子乳化聚合 將上述(1)之溶液與方法二之磁性流體 70ml 混合後,配合曝氮與機械攪拌及 加熱包加熱直到 80℃後,加入甲基丙烯酸甲酯單體 20ml,直到溫度再度平衡達 80℃時,將起始劑加入以起始聚合反應,此時可以停止曝氮,但需將反應器密封 以防止氧氣再度進入,一小時後反應結束,即可得到聚甲基丙烯酸甲酯-四氧化 三鐵的 core-shell 結構。 磷脂質膜之製備與修飾並同時鑲嵌 ICG 磷脂質成膜 秤取磷脂質 0.06g 溶解於 5ml chloroform 溶液中,以超音波震盪完全溶解後, 再以旋轉減壓濃縮機於 60℃下於濃縮瓶璧進行成膜。 磷脂質修飾至粒子並同時鑲嵌 ICG 將已用聚甲基丙烯酸甲酯修飾之磁性流體 50ml 與含有 ICG 之溶液同時加入 至減壓旋轉濃縮機之濃縮瓶中,以超音波振盪方式將濃縮瓶璧上磷脂質膜震落, 29.
(32) 此時,由於磷脂質具有自組裝聚合的性質,所以將重新於粒子表面成膜,並同時 將 ICG 鑲嵌至膜層中,即可得修飾完成之粒子。 X 光晶體繞射分析 X 光晶體繞射分析法的光源為 X 光射線,X 光是一種短波長電磁輻射, 其波長約為 1A°左右。由於如此波長之電磁波是無法以透鏡聚焦,使得僅能用來 分析一較大區域,而獲得一整體平均的結果,然而也因如此,其為提供一非破壞 性的分析方式,甚至可在不同的分析條件,如高溫、低溫、真空、高壓……等特 殊環境下進行分析工作。如此不但試樣製作容易,也比較能獲得接近其原製造環 境或使用狀況下的結果。 X 光晶體繞射可提供材料晶體結構的重要資訊。由其繞射峰的位置,可判斷晶 格面間距大小及晶格面排列情形,據此鑑定該晶體結構及晶相純度。檢視其繞射 強度的高低,則可判斷各原子於晶格中的排列位置;透過其繞射峰之寬度則可分 析奈米粒子的大小及應變程度。X 光繞射技術為人類提供了窺測奈米尺寸原子 排列的最有力工具。 首先將針對四氧化三鐵粒子在不同操縱便因下所得之粒子進行 X 光繞射之 分析,藉由比較一般商用的四氧化三鐵之標準特徵峰,以尋求最佳之合成條件。 超導量子干涉磁量儀 Superconducting Quantum Interference Device (SQUID)分 析 超導量子干涉元件(Superconducting Quantum Interference Device,簡稱 SQUID)是一種能夠偵測極低磁場的感測器,由具有兩個約瑟芬接點超導電流環 組成。上述被二個奈米厚的絕緣層所隔開的超導環,當外界磁場變化時,磁力線 會穿過絕緣層進入超導環之中,並會對超導環感應產生整數倍的超導電子對,因 為電子對的電量與磁通量均極小,所以超導量子干涉磁量儀是能夠偵測極低磁場 的感測器。超導量子干涉磁量儀目前已經廣泛用在研究室中,以量測材料的磁性, 也用來探測地下礦物資源。而其獨特的磁通與電壓的週期特性,使得 SQUID 也 被使用在精密測量,成為微弱物理量如磁場、磁場梯度、電流、電壓、電阻、電 感及磁化率等測量上最領敏的感測元件。 我們將不同溫度下所得之磁性奈米粒子進行 SQUID 之分析,同樣藉由與商用 的粒子比較其飽磁值與磁滯現象以獲得最佳之加熱性質。 動態光散射粒徑分析 由物質與光的交互作用可以得到物質結構和力學的重要資訊。此交互作用可 以從光散射實驗研究。根據半古典的光散射理論與光照射在物質上時,光的電場 會引起分子中電子偏振振盪。此分子就像是第二個光源而散射出光。頻率的改變、 角度的分佈、偏振、散射光強度都由散射物質的大小、形狀、和分子交互作用來 決定,所以從一個系統的光散射特性,並加上電動力學和時間有關的統計力學理 論,可以得到有關散射介質的結構和分子力學資訊。 30.
(33) 將所合成的磁性奈米粒子以動態光散射粒徑分析儀進行粒徑分析,確認其修 飾狀況是否成功,並找尋最適合之合成條件以達到最佳粒徑分布。 TEM 及 SEM 攝影 將所合成之磁性奈米粒子拍攝 TEM 及 SEM 以確認修飾狀況是否成功,並 了解其聚集狀況以待實驗條件進一步修正。 傅立葉轉換紅外線光譜儀 Fourier Transform Infrared Spectroscopy (FTIR)分析 F.T.I.R.光譜儀是利用化合物分子中的官能基吸收特定波長的紅外光原理而 用來觀察分子的基本結構,其功用是以紅外光入射樣品,樣品受照射所產生之光 電流經過傅立葉轉換之運算而形成樣品之紅外光吸收頻譜,藉以得知樣品對紅外 線之反應及其光電與生物特性。 將經過聚丙烯酸修飾與未經修飾的氧化鐵粒子以及聚丙烯酸三種試樣進行 光譜分析,以確認修飾是否成功。 熱重量分析儀 Thermo-gravimetric Analysis (TGA)分析 熱重量分析儀可同時定量測定樣品的重量和熱量隨溫度的變化,並在完全相 同的測試條件下研究某一樣品的化學變化和物裡相變所引起的質變與熱變。 將經過不同比例聚丙烯酸修飾的氧化鐵粒子進行熱重量分析,藉此得知聚丙 烯酸是否修飾成功,並了解其修飾上去粒子的含量比例以調控最佳修飾厚度。 磁性流體加熱性質測試 將所合成之磁性奈米粒子進行加熱性質測試,在不同的 AC 磁場頻率下,記 錄其升溫曲線,以了解是否適合進行熱治療之應用。 螢光光譜檢量線分析 特別針對 ICG 染料進行螢光光譜分析,以了解其激發與放射波長性質以及 其濃度範圍內檢量線之分析。. 結果與討論 X 光晶體繞射分析. 31.
(34) 實驗結果如圖一所示。圖一為不同莫爾數比下所合成之磁性奈米粒子其繞射 峰之比較。由 X 光晶體繞射分析可知,在氯化鐵與亞硫酸納比例為 2:1 時,其五 個繞射峰均明顯較其他比例明顯,即粒子之晶體結構較為完整。. 6:1 4:1 2.5:1 2:1. (311) (200). Intensity. (400). 20. 30. 40. (440). (511). 50. 60. 70. 2 Theta (degree) 圖一、不同氧化鐵與亞硫酸鈉莫爾數比下之 X 光晶體繞射分析圖。當氯化鐵與 亞硫酸納比例為 2:1 時,有較明顯之繞射峰。. (311). 300. Intensity. 250. 40℃ 50℃ 60℃ 70℃ 80℃ Standard. (220). (400). (511) (422). 200. (440). 150 100 50 0 20. 30. 40. 50. 60. 70. 2 Theta 圖二、不同反應溫度下合成的磁性奈米粒子與商用(Aldrich)粒子其繞射峰之比 較。反應溫度 70℃時,其主要繞射峰與商用粒子較為相近。 圖二則改變不同操縱變因。將不同反應溫度下所合成之磁性奈米粒子,與商 用(Aldrich)之四氧化三鐵奈米粒子其繞射峰進行比較。由 X 光晶體繞射分析可得 知,在反應溫度 70℃時,有最明顯的繞射峰,且與商用粒子較為相近,但有些. 32.
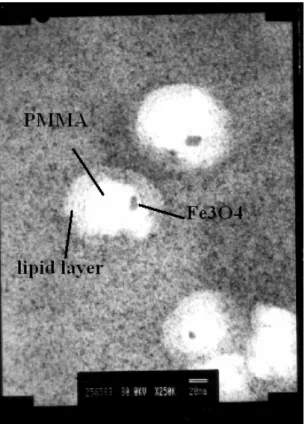
(35) 繞射峰並不是非常明顯,討論其原因,可能還有部分雜質披覆於晶體結構上導致 其 X 光繞射之繞射峰強度受到影響。 超導量子干涉磁量儀 Superconducting Quantum Interference Device (SQUID)分 析. Magnetization (emu/g). 圖三為不同反應溫度下所合成之粒子與商用之氧化鐵粒子其 SQUID 分析, 由圖可知,商用粒子之飽磁值約可達到 60emu/g,而自行合成的粒子,則在 70℃ 下可得較佳的飽磁值,約 40emu/g。其他溫度下所合成的粒子,大約都僅能達到 20emu/g,因此,合成之最佳溫度,應選在 70℃。. 40 ℃ 50 ℃ 60 ℃ 70 ℃ 80 ℃ standard. 60 40 20 0 -20 -40 -60 -30. -20. -10. 0. 10. 20. 30. Field (kO e) 圖三、不同反應溫度下所合成之粒子與商用之氧化鐵粒子其 SQUID 分析圖 動態光散射粒徑分析 圖四為氧化鐵粒子未經修飾及經過聚丙烯酸修飾後之粒徑大小分佈,由圖可 知,在未經修飾前,粒子大小的大約在 100μm,這是因為未經修飾的粒子,其傾 向聚集以降低表面自由能,而在聚丙烯酸修飾過後,粒子大小降低至約 82nm 左 右,然而,若經由聚丙烯酸修飾之粒子,由於其會使粒子表面帶有 COO-因而產 生電荷排斥作用,使粒子降低聚集狀況而穩定懸浮,並有助於進一步接上藥物或 螢光劑等相關應用的開發。. 33.
(36) Modified with PAA without modification. Volume (%). 20. 15. 10. 5. 0 100. 1000. 10000. 100000 1000000. Particle diameter (nm) 圖四、氧化鐵粒子未經修飾及經過聚丙烯酸修飾後之粒徑大小分佈圖 圖五則針對以另一方式合成氧化鐵之粒子,其經過聚甲基丙烯酸甲酯修飾後 以及再進一步以磷脂質修飾後之粒徑分布圖,由圖上可發現,在未經修飾前,粒 子粒徑大約是 80nm 左右,與圖四有很大的差異,其原因在於本實驗中所合成氧 化鐵的鹼液為四甲基氫氧化銨,其可於氧化鐵粒子表面形成立體排斥效應,可助 於氧化鐵穩定懸浮於溶液中,因此聚集狀況教不嚴中,而經過修飾後,粒徑均有 顯著增加,表示修飾的步驟是順利成功的。. 35 Fe3O4 Fe3O4 +PMMA Fe3O4 +PMMA+Lipid. 30 25 % in class. 20 15 10 5 0 -5 0. 200. 400. 600. diameter(nm). 34. 800. 1000.
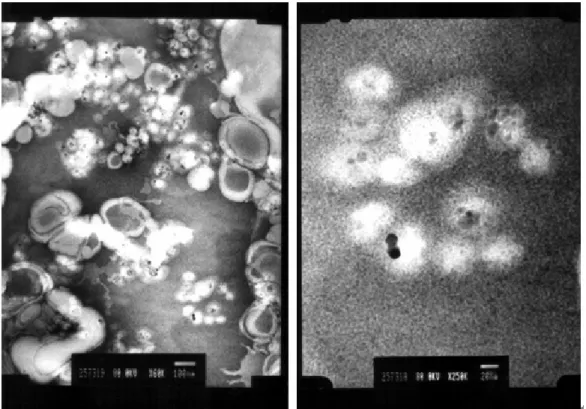
(37) 圖五、未經修飾之氧化鐵粒子與經過聚甲基丙烯酸甲酯修飾後以及再進一步以 磷脂質修飾後之粒徑分布圖。 TEM 及 SEM 攝影圖 圖六為 TEM 及 SEM 攝影圖,(a)為氧化鐵未經修飾的照片,由於表面自由 能關係,使其傾向聚集,故圖中可見其聚集狀況。(b)為經過 PMMA 修飾過後的 氧化鐵粒子,由於對比色不明顯,故無法明確看出氧化鐵之包覆,但經由修飾後, 卻可減少其聚集狀況,只是粒徑大小增加。(c)再經過 lipid 的修飾,由圖可見, 外層有明顯的包覆狀況,但由於 lipid 自組裝聚合時可能聚合不均,導致區域聚 集狀況反而嚴重,但基本上能可達到懸浮良好的效果。(d)為經過聚丙烯酸修飾 過後之氧化鐵粒子 SEM 攝影圖,外層包覆狀況良好,但高分子鏈互相糾纏的狀 況嚴重也會導致粒子聚集。. (a). (b). (c). (d). 圖六、合成及經過修飾過後之粒子之 TEM 及 SEM 攝影圖。(a)圖之平均粒徑約 20nm。(b)圖之平均粒徑約 260nm。(c)圖之平均粒徑約 380nm。(d)圖之平均粒徑 約 82nm。 35.
(38) 傅立葉轉換紅外線光譜儀 Fourier Transform Infrared Spectroscopy (FTIR)分析. PAA oligomer Fe3O4/PAA=1:1 Fe3O4 only 580. 1405 1450. 1710. 580. 1405 1450. 1710. Transmittance (a.u.). 圖七為傅立葉轉換紅外線光譜圖。由圖可發現,聚丙烯酸之特徵峰主要有四 個:Aliphatic acid : 1150-1280 cm-1。Stretch of C-O and deformation vibration of OH : 1400-1450 cm-1。Carbonyl group stretch 1710 cm-1。而氧化鐵粒子則有 Fe-O bond : 580 cm-1。而圖中紅色曲線所顯示的為氧化鐵與聚丙烯酸 1:1 比例修飾下 的光譜圖。我們可以發現其特徵峰皆符合聚丙烯酸以及氧化鐵個別之特徵峰,因 而可確定修飾狀況是成功的。. 1150-1280. 4000 3500 3000 2500 2000 1500 1000 500 -1. Wavenumber (cm ) 圖七、聚丙烯酸與氧化鐵以及經由聚丙烯酸修飾的氧化鐵粒子其紅外線光譜分 析圖。 熱重量分析儀 Thermo-gravimetric Analysis (TGA)分析 圖八為針對不同比例之聚丙烯酸修飾的氧化鐵粒子其熱重量分析,由圖可 知,當只有氧化鐵而沒有修飾聚丙烯酸時,粒子的重量經過高溫後,並不會有質 量上的變化,而只有聚丙烯酸時,由於為高分子聚合物,經高溫後會裂解為逐漸 裂解為單體並燒毀,故質量會有明顯變化。而圖上不同比例修飾之粒子均有質量 上的損耗,因此可確認,聚丙烯酸有成功修飾於粒子表面,且我們可發現一個現 象,不管是任何比例下,其質量損耗均為定值,因此可推估聚丙烯酸修飾於氧化 鐵表面上可能有最大厚度之限制。. 36.
數據


![表 3-1 Fe 2+ 與 Fe 3+ 濃度比對製備所得鐵氧化物特性之影響 pH13-14,反應溫度: 40 ℃,反應時間: 1 小時,轉速:6000 rpm [Fe 2+ ]:[Fe 3+ ] M:M 晶格常數 / nm 晶粒大小/ nm 平均粒徑 / nm 2:1 0.8395 17.0 26 1.5:1 0.8394 13.6 20 1:2 0.8391 9.9 17 1:2.5 0.8371 6.4 15 1:3 0.8330 5.5 5 * 綜合 AEM 與 SEM 顆粒大小與顆粒表面型態分析及](https://thumb-ap.123doks.com/thumbv2/9libinfo/9324602.536243/17.892.137.761.110.667/Fe++濃度比對製備所得鐵氧化物特性之影應溫℃反應時間.webp)
+7

相關文件
Proprietor can discover the problems either do not have efficiency value at input internal resources or the output operation revenue does not reach operation effectiveness for
The major qualitative benefits identified include: (1) increase of the firms intellectual assets—during the process of organizational knowledge creation, all participants
This research is to integrate PID type fuzzy controller with the Dynamic Sliding Mode Control (DSMC) to make the system more robust to the dead-band as well as the hysteresis
This paper integrates the mechatronics such as: a balance with stylus probe, force actuator, LVT, LVDT, load cell, personal computer, as well as XYZ-stages into a contact-
This project integrates class storage, order batching and routing to do the best planning, and try to compare the performance of routing policy of the Particle Swarm
由於本計畫之主要目的在於依據 ITeS 傳遞模式建構 IPTV 之服務品質評估量表,並藉由決
As for current situation and characteristics of coastal area in Hisn-Chu City, the coefficients of every objective function are derived, and the objective functions of
Subsequently, the relationship study about quality management culture, quality consciousness, service behavior and two type performances (subjective performance and relative
