Predi
國立臺
Gr Coll
從一 icting ta
臺灣大學
raduate In lege of Ele
N
一級結構 arget seq
指導 Adv
學電機資 碩
nstitute of ectrical En
National T Ma
構預測 D quences
prim
Ch
導教授:
isor: Ye
Chien-Y
中華民 Ju
資訊學院 碩士論文
Computer ngineering
Taiwan U aster The
DNA 結合 of DNA mary stru
林志瑋 hih-Wei
歐陽彥 陳倩瑜 en-Jen O
Yu Chen
民國 100 uly, 201
院資訊工 文
r Science g and Com
Universit esis
合蛋白之 A-bindin ucture
瑋 Lin
彥正 博 瑜 博士 Oyang, P
n, Ph.D.
年 7 月 11
工程學研
Engineeri mputer Sci
ty
之標的序 ng prote
博士 士 Ph.D.
.
月
研究所
ing ience
序列
eins based on
兩年 決定 幫助 跨領 長、
還要 一起 小姐 面的
年前憑著喜歡 定加入了歐陽 助。最重要的 領域計畫的研 翊鍾學長在 要謝謝元鴻以 起打拼是很棒 姐,雖然在去 的確成長了許
歡生物的一 陽彥正老師 的就是歐陽 研究過程中 在我摸索生 以及又仁,
棒的事情,
去年的時候 許多,感謝
一股傻勁,就 師的實驗室 陽彥正老師 中,我學到了 生物資訊的過 雖然我們研 非常開心可 候離開了我 謝你。
誌謝
就決定進入
。在念碩士
、陳倩瑜老 了許多東西 過程中不時 研究的主題 可以認識兩
,讓我低迷
入生物資訊 士班的這兩 老師以及張 西。另外還 時的給我許 題不同,但 兩位。最後 迷了一陣子
這個領域的 年之間,得 天豪老師的 要感謝廷因 多指教,令 是有兩位夥 要感謝我的
。但是在這
的研究,因 得到了許多 的教誨,在 因學長、鈺 令我成長許 夥伴在這兩 的前女友湯 這之後我在
因此就 多人的 在參與 鈺峰學 許多。
兩年中 湯穎婷 在各方
結合 設計 何進 (posi 種而 樣的 準確 常關 物結 的序 結構
這篇 起來 樣以 所提 處,
DNA 究之
合特定 DNA 計生物實驗尋 進行,並解釋
ition freque 而言,截至目 的模型。由於 確預測位置頻 關心的研究議 結構與紀錄不 序列的新方法 構,接著利用
篇論文使用了 來,這篇論文 以序列資訊為 提之方法表現 本論文所提 A 複合物結 之進行。
A 序列的蛋白 尋找這些 D
釋基因組中 ncy matrix) 目前為止,
於生物實驗 頻率矩陣,
議題之一。
不同胺基酸 法。當我們 用該樣板與
了兩組資料 文提出的新 為基礎且利 現略差。由 提之方法,
結構之蛋白質
中文
白質在基因 NA 結合蛋 中序列的變
) 是最常被 只有一小部 驗往往需要高 加速這個研 這篇論文針 酸和核酸之間 們拿到一條蛋 與所得之知識
料去評估新方 新方法可以達 利用已知位置 由於現存這些 仍可幫助一 質序列預測
文摘要
因調控中扮演 蛋白質的標的
變異如何擾 被拿來描述這
部分的轉錄 高資金與人 研究領域的 針對這個問
間結合偏好 蛋白質序列 識庫進行位
方法的表現 達到和它們 置頻率矩陣 些以序列資 一些相關的 測其標的序列
演重要的角 的序列可以 擾亂正常的基
這些標的序 錄因子已經 人力成本,
的進展,一 問題,提出一
好的知識庫去 列,會先挑 位置頻率矩陣
現,和其他 們一樣的預 陣訓練所得 資訊為基礎 的研究,針 列,所得之
角色,利用計 以幫助我們了 基因表現。
序列的模型 從相關生物 因此,如何 直以來是生 一個利用蛋白
去預測 DNA 選一個適當 陣的預測。
利用三級結 測效果;但 之預測模型 的預測方法 對其同源序 之預測結果將
計算方法預 了解基因調
。位置頻率
,對大部分 物實驗中取 何利用計算 生物資訊學 蛋白質 DNA
A 結合蛋白 當的樣板複
結構的方法 但若與另一 型相比,本 法仍各有其 序列已有蛋 將有助於相
預測或 調控如 率矩陣
分的物 取得這 算方法 學家非 複合 白之標 複合物
法比較 一個同 本論文 其侷限 蛋白質 相關研
Prote expre how norm used only PFM desir progr the k nucle Whe prote incor The comp comp that i meth since summ predi facili
eins that bi ession. Iden genes are r mal gene exp
models to a small fra Ms. Since bi red to deve
ress. Here, knowledgeb eotides is pr n given a q eins is selec
rporated wit proposed putational m pared struct is trained by hod perform
e different mary, it is icted based itates relate
ind specific ntifying targ regulated in pression. Po
represent su action of the
ological ex elop compu
a new met base contai roposed to p query protein cted and th th the built method is methods. It
ture-based m y collected ms slightly w
predictors concluded d on its prim
ed studies i
AB
c DNA seq get sequenc n cells and e osition frequ
uch target s e transcripti periments u utational m thod based ning the pr predict quan n sequence, he PFM pre
knowledgeb evaluated turns out th methods. O experiment worse. Even
usually h that a DN mary structu n the future
BSTRA
quences pla ces of a DN explain how uency matri sequences. H
ion factors usually requ methods with on existing reference o ntitative spe , a protein-D ediction is
base.
by two d hat the prop On the other tally determ n though, th have their NA-binding
ure using th e because t
CT
ay importan NA-binding
w genetic va ices (PFMs) However, u (TFs) have uire much t h satisfied g protein-DN
of contacts ecificities o DNA compl made base
datasets and posed metho
hand, whe mined PFMs
he proposed own advan protein’s he complex target seque
nt roles in protein help ariations cau
) are one of up to now, f experimen time and co accuracies NA comple between a of protein-D lex structure
d on the se
d compare od can predi
n a sequenc s is compar
d method s ntages and binding pre xes of its ho ences of pr
regulating ps to under use disrupti f the most w
for most sp ntally determ
ost, it is stro to speedu ex structure amino acids DNA interac
re of homolo elected tem
ed with ex ict as well a ce-based m red, the prop still has its d limitation eference ca omologues.
roteins with gene rstand ion of widely ecies, mined ongly p the s and s and
tions.
ogues mplate
isting as the ethod posed value ns. In
an be This hout a
solveed structure could be prredicted noww.
口試 誌謝 中文 ABS CON Table Figur Chap Chap
Chap
試委員會審定 謝 ...
文摘要 ...
STRACT ....
NTENTS ....
es ...
res ...
pter 1 In
pter 2 L
2.1 Algo 2.1.1 2.1.2 2.1.3 2.2 Algo
2.2.1 2.3 Algo
2.3.1
pter 3 M
3.1 Mat 3.1.1 3.1.2 3.1.3
定書 ...
...
...
...
...
...
...
ntroduction Literature R orithms for
Predicting Predicting Predicting orithms of s
BLAST ..
orithms of s TM-align Methods ...
erials ...
Collection Collection Relating P
CO
...
...
...
...
...
...
...
n ...
Review ...
predicting p g the bindin g PFM by h g PFM base sequence ali ...
structure ali n ...
...
...
n of protein n of PFMs . PFMs to pro
ONTEN
...
...
...
...
...
...
...
...
...
protein-DN ng preferenc homologues
ed on structu ignment ...
...
ignment ...
...
...
...
n-DNA com ...
otein-DNA
NTS
...
...
...
...
...
...
...
...
...
NA binding s ce of DNA- s’ annotated
ural model ...
...
...
...
...
...
mplex structu ...
complex st
...
...
...
...
...
...
...
...
...
specificities binding pro PFMs ...
and potentia ...
...
...
...
...
...
ures ...
...
ructures ...
...
...
...
...
...
...
...
...
...
s ...
oteins ...
...
al functions ...
...
...
...
...
...
...
...
...
... #
...i
... ii
... iii
... v
.... vii
... viii
... 1
... 4
... 4
... 4
... 5
s ... 6
... 10
... 10
... 11
... 11
... 13
... 13
... 13
... 14
... 14
Chap
Chap REFE
3.2 Buil 3.3 Pred 3.3.1 3.3.2 3.3.3
pter 4 R
4.1 Mea 4.2 Vali
4.2.1 4.2.2 4.3 Perf
4.3.1 4.3.2 4.4 Eval 4.5 Disc 4.5.1
4.5.2 4.5.3 4.5.4 4.5.5 4.5.6
pter 5 C
ERENCE ..
lding the kn diction fram Template Building Refining Results ...
asuring perf dation sets . Training d Protein-D formance ....
Training d Protein-D luating SAB cussion ...
Differenc structures The effec How to se Similar pr Using the The frequ Conclusions ...
nowledgeba mework ...
selection an the predicte the PFM by ...
formance ....
...
data of SAB DNA comple ...
data of SAB DNA comple BINE ...
...
ces betwee s and their a t of differen elect a temp rotein seque e number of uency of am s ...
...
se ...
...
and contact r ed PFM by y knowledg ...
...
...
BINE ...
exes with an ...
BINE ...
exes with an ...
...
en DNA s annotated PF
nt contact d plate ...
ences bind s f contact ato mino acids an
...
...
...
...
residue subs DNA seque ebase ...
...
...
...
...
nnotated PF ...
...
nnotated PF ...
...
sequences FMs ...
distance cut- ...
similar DNA oms of conta
nd nucleotid ...
...
...
...
stitution ...
ence in the t ...
...
...
...
...
FMs ...
...
...
FMs ...
...
...
in protein ...
-off ...
...
A sequence act residues des ...
...
...
...
...
...
template ....
...
...
...
...
...
...
...
...
...
...
...
n-DNA com ...
...
...
es ...
s ...
...
...
...
... 14
... 16
... 18
... 18
... 18
... 21
... 21
... 21
... 21
... 22
... 22
... 22
... 25
... 30
... 30
mplex ... 30
... 32
... 33
... 33
... 34
... 35
... 38
... 40
Table Table Table Table Table
e 4-1 PFM b e 4-2 Differ e 4-3 Simila e 4-4 Simila e 4-5 Avera
become mo rence before arities of 24 arities of 12 age similarit
ore similar w e and after P 4 proteins of 2 proteins w ty under diff
Tables
with annotat PFM refinem
f different a with similari fferent distan
tion after re ment by the algorithms ..
ty > 0.7 ...
nce cut-off .
efinement. . e knowledge ...
...
...
...
ebase ...
...
...
...
... 23
... 26
... 29
... 32
... 33
Figur Figur Figur Figur Figur Figur Figur Figur Figur
re 2-1 Predi re 2-2 Work re 3-1 Prefe re 3-2 Predi re 4-1 Simi re 4-2 Distr re 4-4 Corre re 4-5 Cont re 4-6 Score
iction frame kflow of the erence betw iction frame larities und ribution of s
elation betw tact counts b es between
ework of Sc e study of A ween amino ework of th er training d similarities b ween protein
between am amino acid
Figure
chroder, A., Alamanova, acids and n his study. ...
data set of S between DN n sequence mino acids an
ds and nucle
es
et al’s work et al. ...
nucleotides ..
...
SABINE...
NA sequenc similarities nd nucleotid eotides unde
k ...
...
...
...
...
ces and anno and PFM s des ...
er the older
...
...
...
...
...
otated PFM similarities.
...
scheme ...
... 6
... 10
... 16
... 17
... 24
Ms .. 31
... 34
... 36
... 37
Ch
Prote regul binds cause assig comp DNA progr prote bindi
Posit conse of tra nucle
Ther Surfa affin intera on th mole
hapter 1
eins that bi lation of ge s can help t e disruption gned to a c putational m A-binding s
ress in the ein-DNA in ing specific
tion freque ensus of tar anscription eotides A, C
re are exper ace plasmo
ity of a pro actions, but he fact that ecular attach
Intr
nd to speci ne expressi to understan n of normal certain mol methods eff specificities developme nteraction p city informa
ency matrix rget DNA s factors (TF C, G and T o
rimental me on resonanc otein-DNA i t it can also t the angle hed to the o
oductio
ific DNA s on. Identify nd how gen gene expre ecular func ficiently. Ho
) remains ent of high-
arameters, t ation is still
x (PFM) is sequences th Fs) [1-3]. PF occur at eac
ethods for ce (SPR) is interaction d o be used to
of light re other side of
on
equences a ying the targ ne regulatio ession withi ction (e.g.,
owever, qu insufficient -throughput the determi laborious.
s a simple hat can be r FMs indica ch position w
determining s one of th
directly. SP o measure p eflection fro
f the surfac
are extremel get sequenc
n proceeds n cells. In r transcriptio antitative fu t for the re t technologi
ination of h
probabilist recognized ate for a cer
within the b
g the bindi e methods PR is often u protein-DNA om a surfac e. DNA can
ly importan es of DNA- and how g recent years on factor) b functional in
equirement.
ies for the highly resol
tic model t by a DNA- rtain TF how binding site.
ng specific for measur used to stud A interaction
ce depends n be attache
nt for the p -binding pro genetic varia s, proteins c
by biologis nformation . Despite r measureme lved quanti
to represen -binding do w frequentl .
city of a pr ring the bin dy protein-l
ns. SPR is b on the ma ed to the sur
proper oteins ations can be sts or
(e.g., recent ent of
tative
nt the omain
ly the
otein.
nding igand based ass of rface,
and t the f meas (PBM array prote amou antib prote synth deter
Usin than meth seque and subse are c some appro are s conse appli for th
then protein formation of sured, and t Ms) are ano ys of over 4 ein is added unt of prote body to the eins still ne hesized in rmine the bi
ng computat using expe hods for PF ences to wh determine equences. S currently on e structure- oaches are shown to p erved and d ied the met he PFM sim
ns are added f protein-D then the bin ther techno 44,000 spots d into the ar eins that bin e protein. D eed to be
vitro. So inding spec
tional meth erimental m FM inferenc hich the tran the frequen Such metho nly availabl -based app based on a perform we do not allow thod suppor milarity of b
d to change NA comple nding affinit logy for me s which con rray, and is nd to a spe Despite the prepared fo it still spen ificity of pr
hods to mod methods doe ce of a tra nscription f ncy of each ods require
le for a sma proaches w analyses of ell in tellin w degenerat rt vector reg between pro
e the reflect ex and the o
ties can be m easuring bin ntain all pos then washe ecific DNA e significan for these m nds some rotein-DNA
del binding es. One of anscription
factor can b h position sufficient s all amount were also p
protein-DN ng which p tion [4-7].
gression (S oteins based
tion angles off-rate for
measured. P nding specif
ssible comb ed to remov
spot is det nt progress methods, eit
time using A interaction
specificity f the most factor is to bind, and th
among the sequences fo
of DNA-bi presented t NA complex
positions in On the othe SVR) to pre d on their pr
of the light its dissocia Protein bind ficity of pro binations of ve nonspeci termined wi of experim ther purifie g experimen ns.
can spend widely use o collect a en to condu e detected o for pattern d
inding prote to predict x structures n a PFM s er hand, Sch edict a quan
rimary struc
t. The on-ra ation can th ding microa oteins. PBM f DNA 10-m ific binding with a fluore mental met ed form cel ntal metho
d much less ed computat
set of prom uct motif fin
over-repres discovery, w teins. Previo
PFMs. Se s These me
should be chroder, A., ntitative me cture, and fu
ate for hus be arrays M uses mer. A g. The escent thods, lls or
ds to
s time tional moter nding sented which ously, everal ethods more et al.
easure urther
predi
How nucle to ha at pr from prote prote struc as a proba migh were that d PFM
In Ch prote meth meth inclu
ict the PFM
wever, in [8 eotides was ave an annot roviding an m primary s ein-DNA co ein based o ctures is pro
PFM that e ability equa ht bind to s e used to inf describes th Ms built by th
hapter 2, se eins and pre hod is introd hod is show uded in Chap
M of a protei
8] , the pr not consid tated PFM,
alternative structure, p omplex stru on its prim oposed. The
every colum als to one.
similar DNA fer the PFM he contact fr he DNA seq
everal metho evious studi
duced in Ch wn and som
pter 5.
n [8].
reference b ered. In add which is no to predict provided th ucture. In th mary structu e DNA sequ mn contains Based on t A bases, th M of a given requency be quence in a
ods propose ies of protei
hapter 3, an me discussio
between am dition, it req ot usually a
target DNA hat any hom his thesis,
ture and a uence in a p
only one o the idea tha he sequence n protein seq
etween ami protein-DN
ed for predi in-DNA inte nd in Chapt on is made.
mino acids quires homo available. In A sequences
mologue o a novel app
collection protein-DNA of the nucle
at similar c es in protei quence. Fur no acids an NA complex
icting target eractions ar ter 4, the pe
Finally, co
in protein ologues of t this regard s of a DNA f the query proach to p
of protein A complex eotides (A, contact resid in-DNA com rthermore, a nd bases is b x structure.
t sequence o re introduce
erformance onclusions o
ns and bas the query pr d, this thesis A-binding pr y protein h predict PFM n-DNA com
can be reg T, C, or G) idues of pro
mplex struc a knowledg built to refin
of DNA-bin ed. The prop of the prop of this thes
es in rotein s aims rotein has a M of a
mplex arded ) with oteins ctures ebase ne the
nding posed posed is are
Ch
In se intro speci solve speci exam comp
The seque while PFM sectio
2.1
2.1.
In th 168 h obtai differ
hapter 2
ection 2.1, duced. Giv ificity can b ed structure
ificity can b mple, protei
plex structu
technique o ence alignm e comparin M prediction
on 2.3.
Algor speci
1 Predic
he study of homeodom ined using prent mach
2 Lite
different m ven a protein
be inferred e (i.e. prot be inferred in binding ures.
of sequence ment is intro ng the propo
n, so an alg
rithms ificities
cting the b
Berger, et a ains agains protein bin hine learninrature
methods for n with a so by structur tein with o
by protein- microarray
e alignmen oduced in se
osed metho gorithm of
for p
binding pre
al. [9] the Z t all the pro ding microa ng algorithReview
predicting olved protei ure-based PF
only primar -DNA bindi y (PBM) da
nt was used ection 2.2. I od in this th f structure a
predictin
eference o
Z-score tran obabilities o arrays (PBM hms to prw
protein-DN in-DNA com FM predict ry structure ing informa ata, annotat
d in this the In addition, hesis with t alignment,
ng prot
of DNA-bi
nsformed re of 32,896 8 Ms). After redict theNA binding mplex struc
ions. For a e informati ation of its ted PFMs,
esis, thus th structure a the existing TM-align
tein-DNA
inding pro
elative sign 8-mer DNAthat, Alleyn Z-scores b
specificitie cture, its bin a protein wi
ion), its bin homologue or protein-
he basic id alignment is g structure-b
is introduc
A bind
oteins
nal intensitie A sequences ne, et al. ap between mes are nding ithout nding es, for -DNA
dea of s used based ed in
ding
es for were pplied mouse
homo In th into conv binar 20 o binar vecto fores funct Z-sco algor revea bindi
2.1.2
Schro predi struc featu The p i)ii)
oedomains he study of feature set verted into n
ry vectors o rthogonal v ry vectors or of 20 zer st regression
tion kernel ore. It turn rithms. The als the fact ing preferen
2 Predic
oder, et al.ict PFMs of ctural and p ures to train
prediction f Feeding trained S (the SVR
and indivisu [10], the c s for the tr numerical e of length l vectors and
correspond ros. With de n, support l, and prin ns out that e root mea t that, the nce of prote
cting PFM
. applied th f proteins ba physicochem n the SVR mframe work the sequen SVR models R model) re
ual DNA 8- ontact regio raining of p encodings r
×20 digits, an amino ing to resid erived featur
vector regr ncipal comp t nearest n an square e
amino acid eins.
M by homo
he regressi ased on thei mical prope models in oris described nce, organis
s;
porting the
-mers [10].
on of these prediction m representing
i.e. the 20 acid seque dues at eac res, classifi ression with ponents reg neighbor p error (RMS ds within c
ologues’ an
ion methodir primary s erties, and rder to pred d as followe sm, and stru
PFMs with
proteins w models. Th g amino aci different am nce was re ch position ers includin h linear, po gression we erformance SE) of near ontact regio
nnotated P
d support v structure [8]phylogenet dict the PFM
ed (Figure 2 uctural supe
h similarity h
were aligned he sequence id sequence mino acids w
presented b . Gaps wer ng nearest n olynomial,
ere applied best com rest neighbo
ons do hav
PFMs
vector regre]. Pairwise a tic distance M similarity
2-1):
erclass of th
higher than
d and transf e alignment es of length were encod by concaten re encoded neighbor, ran
and radial d to predic mparing to or is 0.76.
ve influence
ession (SVR alignment s es, were us y of two pro
he protein t
n a threshold ferred
t was h l as ded as nating d as a
ndom basis ct the
other This es on
R) to scores, ed as oteins.
to the
d;
iii) iv)
The a scale rando signi
2.1.3
Moro PFMFiltering Merging
Fig
average abs e from 0 to omly sampl ificantly hig
3 Predic
ozov, et al.Ms [5, 6]. T
g outlier PFM g the remain
gure 2-1 Pre
solute error o 2. In com led is 0.64, gher than th
cting PFM
developed This modelMs;
ning PFMs t
ediction fram
(AAE) of t mparison, th indicating e performan
M based on
a simple n only caresto get the fin
amework of
this framew he average s
that the pr nce expecte
n structura
null model s about thenal predictio
Schroder, A
work with de similarity b redicted per ed by random
l model an
called ‘con e number oon;
A., et al’s wo
efault param between two rformance o
m guesses.
nd potenti
ntact’ mode of atomic cork
meter is 0.12 o PFMs tha of SVR mo
ial functio
el for predi contacts bet2 on a at are del is
ns
icting tweenprote colum
, wh prote
d param phys and h Diffe the a amin know Amo of ea funct To c dista wher of th dista prote comb
ein side cha mn is cal
here is th ein and DN denotes the meter. This ics-based p has the adva erent to the all-atom kn no acid typ wledge-base ong the serie asy implem
tions, cFIRE onstruct the ance falling
re = 3, 4, he values of ance falling ein-DNA co binations, th
ains and DN lculated as f
1 4 1 1
4 1 3
he observed NA respectiv
base type o model has potential fun antage of co contact mo nowledge-ba pes into con ed potential
es of all-ato mentation an E and vcFIR e knowledg g within a , 5, 6, 7, 8, f . In this
within a sp omplex stru
he potential
NA bases. Th follows:
⁄
⁄
d number o vely is defin observed in been shown nction when
onsuming m odel, which ased potent nsideration.
function th om scoring f nd is shown RE, in predi gebase, the
specified r 9, and 10 (Å
study, the pecified ran ctures colle between at
he probabil
of atomic c ned to be in n the positio
n to be gene n a native pr much less co h considers tial function . The FIRE hat consider functions pr n to be gen icting PFMs
number of range Å), and ∆
number of nge,
ected from P tom types i
ity of the ba
, 0 , 1
contacts (a n contact if on i in the c
erally as go rotein-DNA omputation
only the co n presented E potential
rs interactio resented in nerally as g
s.
f pairs of a
∆ , wer is set as 3 pairs of ato , , , are c PDB [11]. W
and j is repr
ase pair typ
heavy atom they are cl omplex, od as the st A complex i
cost.
oordinates o d by Zhou, function [ ons between
[7], FIRE h ood as two
atom types re denoted
for = 3 a om types calculated b
With resented as
pe in the
m pair from loser than 4
and is a tatic model
s considere
of residue a et al. take 7] is a suc n all atom t has the adva o of its exte
i and j wit
as ,
and 1 for th and wit based on the
, , of a follows:
PFM
m the 4.5Å),
a free using d [5],
atoms, es the
ccinct types.
antage ended
th the , , , he rest th the e 990 all the
wher 10Å dista given of th
Assu and t
wher comb PFM
wher Alam
wher atom
re , , . The va ance-depend n complex, e observed
ume that inf thus G can
re ∆ is t bining two Ms as follow
re is a fr manova, et a
re is the m types.
, alue of dent number
the binding atom pairs
fluences on n be represe
the binding equations a s:
ree paramet al. used ano
set of atom and
, ,
, ⁄∑ is set as r of ideal-g g free energ
[5]:
∆
binding fre ented as foll
∆
g free energ above, we
∑ ∈ ter.
ther potenti
ln |
mic distance are the nu
ln 0
, , , 1.61 becau gas points in gy, ∆ , is d
,
ee energy fro lows:
∆ ∆
gy of a bas can estimat
∆
∈ , , ,
ial function
s betw umber of a
, , , ,
use it best n finite pro defined as t
, ,
om differen
e (A, T, te the prob
∆
∆
proposed in
ln |
ween interfac atoms in th
∆ ∑⁄ fits of otein-size sp
he sum of a
nt positions
C, or G) a abilities in
n [12] to pre
, ,
ce atoms, he protein
∆ , to the a pheres [7].
all the pote
are indepen
at position each colum
edict PFM
and ar and DNA.
actual For a entials
ndent,
i. By mn of
[4].
re the . The
proba
wher proba expre
Whe separ For seque calcu estim
, wh matri DNA linea colum
The w
ability of at
re |
ability, esses the lik
re
rated by dis each DNA ences were ulated by th mated by sol
ere is a ix of dimen A sequence.
ar equation i mn of PFM
workflow o
tomic conta
, , is is the B kelihood of
, ,
, , is stance . A sequence e generated,
he formula lving the lin
vector of 4 nsions (4N,
. In additio is solved by s were estim
of this study
acts was mo
| , ,
the likeli Bayesian pr observation
s the numb
of length N , and their as above. T near equatio
4N dimensi 4N+X), wi on, is a y least squa mated as fol
∑ ∈ y is describe
deled as the
ihood func rior and wa n a native-li
, ,
mber of co
N in the p potentials w The weight on:
ions of the ith each row a vector con ares optimiz
llows:
∆
∈ , , ,
ed as Figure
e likelihood , , |
, , ction,
as set to o ike interatom
∑
ntacts obse
protein-DNA when boun s of each
estimated w of the ma nsists of 4N zation. Final
∆
∆ e 2-2
d:
, , is ne. The fo mic distance
, , , , erved betw
A complex, nd by the q position in
weights, an trix A corre N+X poten lly, the prob
s the mar ollowing for
e:
ween an
, 4N+X ran query protei n the PFM
nd A is a b esponding t ntial values babilities in
rginal rmula
nd
ndom in are
were
binary o one . The n each
2.2
Sequ BLA uses wher seque hand time
2.2.
BLA
Algor
uence alignm AST are the
dynamic p re n and m
ences. In th d, BLAST u than Smith
1 BLAS
AST (BasicFigure 2-2
rithms o
ment is a fu most famou programmin m are the l his regard, i uses a heuri h-Waterman
ST
Local Align
2 Workflow
of sequen
undamental us algorithm
g to solve t length of th
t is not use istic approa while align
nment Sear
of the study
nce align
problem o ms for local the problem he sequenc ful while se ach to searc ning long se
rch Tool) is
y of Alaman
nment
f bioinform l sequence a m, and its ti ces. So it t
earching a h ch the solut equences.
s a local seq
nova, et al.
matics. Smith alignment. S ime comple takes lots o huge databa tion, and it
quence align
th-Waterman Smith-Wate exity is O(n of time to ase. On the takes much
nment algo n and erman nm), align other h less
orithm
for b align and f BLA to its solut being The m
2.3
Prote rangi prote struc DAL
2.3.
TM-a
biological s nment [13].
further find AST is one o
s high spee tion as Smi g widely us major steps Making a sequences;
Constructin Matching q Scanning th Extending
Algor
ein structur ing from pr ein function cture alignm LI [16] and S
1 TM-al
align onlysequences, We can al its similar s of the most ed for searc
ith-Waterma ed.
s of BLAST list of nei
ng a diction query with s he database
the match in
rithms o
ral compari rotein fold n annotatio ment [14]. I SAL [17].
lign
employs tand we can ign a query sequences i
commonly ching a solu
an does, its
T are listed a ighborhood
nary for all w score higher e for words;
n both direc
of structu
sons are em classificatio on. TM-alig It is ~4 tim
the informa
n use it fo y sequence in the databa y used progr
ution. Altho s high spee
as followed:
d words, fo
words in the r than a thre
ctions when
ure align
mployed in on, protein gn is one mes faster th
ation of ba
r protein se with a sequ ase.
rams in the ough it doe ed still mak
:
or example,
e query;
eshold;
n a match is
nment
n many bra structure m of the mo han CE [15
ackbone
equence or uence datab
field of bio es not guara kes it very
, length 4
found;
nches of st modeling to ost famous 5] and 20 t
coordinate
r DNA sequ base by BL
oinformatic antee an op useful, and
for amino
tructure bio o structure-b s algorithm times faster
es of the uence LAST,
cs due ptimal d thus
acid
ology, based ms for
r than
given
prote 1.
2.
ein structure Initial struc There are alignment proteins us on the gap against the programmi initial align Heuristic it In this proc initial align
where residue of 1.24 alignment S , . The a new sco converges.
e. The algor cture alignm
three initi is obtained ing dynami pless matchi e larger p ing, but the nment.
teration cedure, stru nment [18].
is the dista the second
15 1.
can be obta en a new TM
re matrix i Finally, the
rithm execu ment
ial alignme d by assign ic programm
ing of two protein. The e scoring m
uctures are r The score s
S ,
ance betwee d structure .8, i ained by im M-score rota is obtained e alignment
utes the follo
ents being ning second ming. The se
proteins. T e third in matrix is a co
rotated by T similarity is
1 ⁄
en the th r under the s the leng mplementing
ation matrix . The proc
with the hi
owing steps
exploited.
dary structu econd type The smaller itial alignm ombination
TM-score r s defined as
1
⁄
esidue of th TM-score gth of the
g dynamic p x is obtained edure is re ghest TM-s :
The first ure to each
of initial ali protein is g ment is al
of the first
otation mat :
he first struc superpositi
smaller st programmin d by the new epeated unti score is retu
type of i h residue of ignment is b gapless thre lso by dyn t and the se
trix based o
cture and th ion.
tructure. A ng on the m w alignmen til the align urned.
initial f two based eaded namic econd
on the
he th
new matrix t, and nment
Ch
Altho espec allow struc struc In th these
As in speci PBM prote by us
3.1
3.1.
Prote colle given prote seque criter
hapter 3
ough the st cially in tel w degenera cture-based cture solved his regard, t e proteins.
ntroduced i ificities bas M or PFM.
ein-DNA bin sing protein
Mate
1 Collec
ein-DNA co ected [11]. P n query pro ein-DNA coences of th ria: a) it i
3 Met
tructure-bas lling which ation, it is PFM pred d. However, this thesis a
in Chapter sed on thei However, nding infor n-DNA com
erials
ction of pr
omplex stru Protein-DNA otein sequenomplex str he query. Th
s an X-ray
hods
sed PFM pr h positions s strongly diction can
there are s aims to dev
2, there are ir homolog
protein-D mation. Her mplex structu
rotein-DN
uctures fro A complexe nces. Since ructure of he template y structureredictors ha in a PFM s
desired to be applied still many p velop a me
e algorithm gues’ protei DNA compl re a novel m ures of its h
NA comple
om the 27 es were col similar pro the query’e structures with resol
ave been sh should be m o design a d only whe proteins do n
thod to pre
ms for predi in-DNA bin lex structur method for homologues
ex structur
February 2 llected for f oteins binds s homolog were requi lution betteown to per more conse a new alg en the quer not have a edict the tar
cting prote nding inform res provide
inferring PF is proposed
es
2009 releas finding a pr s similar DN
ue helps p red to satis er than 3.0Å
rform well erved and d gorithm be ry protein h
solved stru rget sequen
ein-DNA bin rmation, suc e some kin FMs of a pr d.
se of PDB roper templa NA sequenc predicting t sfy the follo Å, b) the
[5-7], do not
cause has a cture.
nce of
nding ch as nd of rotein
were ate of ces, a target owing DNA
mole chain prote abov
3.1.2
Anno anno of hu3.1.3
After struc comp prote supp After anno3.2
Befo hiera repor simil
ecule has n has 5 c ein chain h ve.
2 Collec
otated PFM otated PFMs uman, 802 o3 Relatin
r collecting ctures the c plex structu ein have theosed to bin r mapping P otated PFM
Build
ore building archical clus rted by BL larity to the
6 paired b ontact resid has 40 res
ction of PF
Ms of humas of yeast w of mouse, an
ng PFMs
g the data collected P ures by the e same entrynd to the D PFMs to the are remaine
ding the
g the know stering algo LAST [13].
e other prote
ases and ha dues (residu sidues. The
FMs
an, and mo were collectend 117 of ye
to protein
above, the PFMs are a eir entry nay name, they DNA sequen
e protein-DN ed, correspo
knowled
wledgebase, orithm Hom
In each cl ein chains is
as less than ues within 4 ere are 990
ouse were c ed from MY
east were co
n-DNA com
next step associated.ames in Un y are associ nces similar NA complex onding to 26
dgebase
we cluster moClust [22
luster, the p s assigned a
n 30% non- 4.5Å to the
protein ch
collected fr YBS [20]. T
ollected.
mplex stru
is to know PFMs wer niProt datab iated with er with thos x structures 6 different p
red the col ] based on protein chai as the repre
paired base DNA mole hains satisfy
om TRAN Totally, 592
uctures
w with whicre related base [21]. I ach other, i se of the sa s, 119 protei proteins.
lected prot pair-wise s in with the sentative of
es, c) the pr ecule) and d fying the cr
SFAC [19]
annotated P
ch protein- to protein- If a PFM a i.e. this prot ame entry n
in chains w
tein chains sequence ide e largest av
f the cluster rotein d) the riteria
, and PFMs
-DNA -DNA and a tein is name.
ith an
by a entity verage r. The
seque In th The 260 t types for a discr are c nucle
,whe obser betw
wher and 0.25 and n The that Lysin On th
ences of co e end, 260 r contact resi templates w s of amino a amino acid riminate dif considered.
eotides are c
ere mean rved contac ween and
re f is t f is the in this stud nucleotides.
scores betw nucleotides ne) and am he other han
ollected 990 representati idues of pro were defined
acids and fo ds, only the fferent amin After cou calculated b
ns a type of ct counts o d . The exp
the frequenc e frequency
dy and i .
ween amino s prefer am ino acids w nd, it is inte
protein-DN ives were us otein chains
d first. With our types of
e atoms of no acids. Si unting the by the follow
S ,
f amino acid of and
pected conta , cy that amin y of the app is the obser
o acids and mino acids with polar (A
eresting that
NA complex sed to build s (residues w
h this inform f nucleotide f the side imilarly, for contacts th wing equati
log
d , mean , and , act number
f ∙ f no acid a pearance of rved total c
d nucleotide with positi Asparagine t Serine pref
xes were us d the knowle with 4.5Å to mation, con es were repo
chains are r nucleotide he scores ion.
, ,
ns a type o is the e is calculate
∙ appears in t f the nucleo
ontact coun
es are show ive charges and Glutam fers Thymin
sed as input edgebase.
o any nucle ntact counts
orted respec considered es, only the
between am
f nucleotide expected co ed by
he Swiss-Pr otide , w nts between
wn as Figure s (Arginine mine) to oth ne to other n
t for HomoC
eotide prese s between tw
ctively. Not d, because e atoms of
mino acids
e. , ontact frequ
rot database which was s n all amino
e 3-1. It ap e, Histidine her amino a nucleotides
Clust.
ent) in wenty e that they bases s and
is the uency
e [21], set to acids
ppears , and acids.
.
3.3
Whe follo i) ii) iii) . The p
‐3
‐2.5
‐2
‐1.5
‐1
‐0.5 0 0.5 1 1.5 2 2.5
F
Predi
n a query p wing three
Template Building Refining
prediction f
igure 3-1 Pr
iction fr
protein sequ steps.
e selection a g a predicted g the PFM b
framework o
reference be
amewor
uence is giv
and contact d PFM by D by the know
of this study
etween ami
rk
ven, the PFM
t residue sub DNA sequen wledgebase
y is shown a
ino acids an
M predictio
bstitution nce in the te
as Figure 3-
nd nucleotid
on will be p
emplate
-2
des
performed bby the
A T C G
Figur datab Gene know PFM
re 3-2 Pred base by B erating a pr wledge base M refinement
diction fram LAST and redicted PFM
e and the d t by DNA s
mework of th d find out M from the distance bet
equence in
his study. (A the contac e DNA sequ tween nucl the templat
A) Selectin ct residues uence. (C) C
leotides and te structure.
ng a templat that are Calculating d the conta
te from tem substituted
the PFM b act residues
mplate d. (B)
by the s. (D)
3.3.
Give chain selec BLA nucle repla will perfo
3.3.2
Since temp selec be ob other3.3.3
Befo PFM struc struc matri betw1 Templ
en a query pns in temp cted as tem AST, there w
eotide) of th aced by. No
be perform ormed.
2 Buildi
e similar pr plate structu cted, a PFM btained. If rwise,3 Refini
ore refining M is built bcture, we ca cture is , th
ix M is ca ween amino
late select
protein sequlate databa mplate of th will be mism
he template ote that not f med. If there
ing the pre
rotein seque ure gives us M with probathe position
, 0
ing the PF
the PFM b by the knowan calculate hen a PFM alculated by acids and n
ion and co
uence, a pr ase using Be query. A matches on
protein. Th for all the g e’s no temp
edicted PF
ences bind s s importantability of ei n of the 0
FM by kno
built by the wledgebase a column o of length y the contaucleotides.
ontact resi
roper homol BLAST. Th According tothe contact he contact re given query
plate with e
FM by DN
similar DNAinformation ither one or
DNA sequ
owledgeba
DNA sequfirst. For of the PFM is constru act residues
idue subst
logue is sel he protein wo the seque t residues ( esidues of th
protein seq e-value < 0
NA sequen
A sequence n to infer P r zero based ence is basase
ence in the each DNA M. If the DN ucted. To co s of each b
titution
lected from with the lo ence alignm residues wi he template quences, the .001, no PFnce in the t
s, the DNA PFMs. After d on the DN se , thentemplate st A base pair NA sequence onstruct the base pair an
m the 990 pr owest e-val ment reporte
ithin 4.5Å t e protein are e PFM predi FM predicti
template
A sequence ir a template NA sequenc
,
tructure, an in the tem e in the tem e PFM, a sc
nd the dist rotein lue is ed by to the e then iction ion is
in the e was ce can 1,
nother mplate mplate coring ances
M , the s base, conta an in defin temp resid After
wher know
For know toget by th done
, i conta
M
is the sc score betwe
, i.e. , act residues nverse ratio ned as the sh plate. Here dues closer t r calculating
re is a fr wledgebase.
refining th wledgebase, ther. If the c he DNA seq e by the follo
, is the ratio act residues
M , ∑
core of nuc een contact
. For each s Γ of this o of the dis hortest atom
is a fre to the nucle g M, we can
,
ree paramet .
he PFM bu , the two P contact resi quence in th
owing equa
, of the num s of the p
∈ ,
∑ ∈ leotide a
residue h position
position. T tance t m pair distan ee paramete
otide are fa n obtain a P
∙
∑ ∈ , , , er. Now we
uilt by the PFMs built dues of a ba he template ation.
∙ 1 mber of subs
osition a
∙
at position and the nu
in the te The weight to the DNA nce between er that can avored as PFM by
,
∙ , e have a sec
e DNA se t ( a ase pair are structure sh
stituted con
and 0
1~ , ∈
and it equ ucleotide emplate stru
of each co A molecule n amino aci be adjuste gets large
1~ ,
cond PFM,
quence by and ) e conserved, hould be kep
, ∙ ntact residu
if there is
∈ , , ,
uals to the w defined by ucture, we ntact residu in the temp id and DNA d by the u r.
∈ , ,
which is co
the PFM ) are going , then the in pt, so the re
1~ , es over the
no contac
weighted su y the know
have the s ue is giv plate, and A molecule i user. The co
,
onstructed b
built with g to be m nformation efinement c
∈ , , , total numb ct residue a
um of wledge set of ven in
is in the ontact
by the
h the merged
given can be
ber of at the
posit
For t resid these 0.25.
provi tion.
the position dues, they ar e positions, . These pos ide any use
ns that are c re regarded
four types sitions are t ful informa
close to the d as unimpo of nucleotid trimmed bef ation.
e starting or ortant positio
des would b fore reporti
r ending po ons in this p be assigned ing the pred
ositions and protein-DN with the sa diction beca
d without co NA interactio ame probab ause they d
ontact on. In bility : do not
Ch
In th Intro In se other
4.1
To c was e calcu
wher are th
4.2
4.2.
In [8 differ SAB
hapter 4
his chapter oduction of ection 4.3 a r methods is
Meas
ompare the employed, r ulate the sim
re is the he probabili
Valid
1 Traini
8], 1239 pr rent databa BINE. These4 Resu
r, a measu validation s and 4.4, the
s introduced
suring pe
e predicted referring to milarity betw
1
number of ities of nucl
dation se
ng data of
rotein seque ases. 453 o e protein seqults
ure to com sets that us e performan d. Finally, d
erforma
PFM and t the similar ween two PF
1 1
√2
f positions th leotides
ts
f SABINE
ences with of them we quences wermpare PFMs ed to test p nce of the p discussions a
ance
the annotate rity function
FMs, say
∈ , , ,
hat two PFM at position
E
their annot ere public re used to e
s is first i performance proposed m
are made in
ed PFM, a n used in [2 and , th
, ,
Ms aligned in PFM
tated PFMs and attach evaluate the
introduced e is followe ethod and c n section 4.5
measure ca 23] for comp
ey are align
together, an and
s with were hed in the proposed m
in section ed in section
comparison 5.
alled ‘Simil paring PFM ned to maxim
nd , and , respective
e collected source cod method.
n 4.1.
n 4.2.
n with
larity’
Ms. To mize
d , ely.
from de of
4.2.2
As m with prote for p BLA prote evalu4.3
Two 4.3.1 in th requi propo comp as we
4.3.
453 p the p with PFM simil
2 Protein
mentioned iknown PF ein sequenc prediction. W AST with oth
eins cannot uation.
Perfo
validation 1, we can se he template
ire a prote osed meth plexes with ell as struct
1 Traini
protein sequ proposed me-value <
Ms built by e larity of 0.6
n-DNA co
n section 3 FMs, and th ces of the pr When evalu her 989 tem find a tempormance
sets descri ee the impro
structures ein-DNA co
od is com h annotated
ture-based m
ng data of
uences cont method. 96 o0.001, so 3 employing 632, after r
omplexes
3.1.3, we hahey were a rotein-DNA uating the p mplate seque plate with e
e
ibed in 4.2 ovement aft by the trai omplex str mpared with
PFMs, and methods pre
f SABINE
tained in the of these pr 357 proteinthe DNA se efining
with anno
ave 26 diffe also used to A complex s proposed me ences and fie-value < 0
were used ter the refin ining data ructure to h structure
it turns out edict.
E
e training d rotein seque sequences equence in by the
otated PFM
ferent proteio evaluate structures w ethod, the q
nds a templ .001, so we
d to evaluat ning the PFM
of SABINE perform pr e-based me t that the pr
data of SAB ences canno
were tested the templa knowledge
Ms
in-DNA com the propos were used a query protei
late. Howev e finally use
te the propo Ms built by E. Structure rediction, s ethods usin roposed me
INE were u ot find a te
d. As show ate ( ) ebase, an av
mplex struc sed method as an input q
in sequence ver, there ar e 24 protein
osed metho DNA sequ e-based me so in 4.3.2 ng protein- ethod can pr
used as quer emplate stru wn in Figure have an av verage simi
ctures . The query e runs re two ns for
od. In ences ethods 2, the
-DNA redict
ry for ucture e 4-1, verage
ilarity
of 0 infer In Ta refin proba proba anno of an becau temp resul impr infor
.682 is ach rring the PF
able 4-1, it nement. For ability = ability to C otated PFM.
nnotated PF use there is plate structu
lt. It can roving the p rmation give
hieved. Th M of a quer
is shown t r example, 1 to Thym Cytosine, s . Similar sit FM. On the s no any co ure, so pred
be observe performanc en by the D
his shows t ry protein se that PFM b
, at the 1 mine, after
o the PFM tuations can e other han ontact resid dictions at th
ed that trim e, because DNA sequen
that refinem equence.
becomes mo 11th positio refinemen M after refin
n be observ nd, PFM af due at 1st ~
hese positio mming the
trimming t nce in the tem
ment by th
ore similar on of anno nt, the kno
nement bec ed at 7th, 8t fter refinem
4th positio ons were tri ese unimpo these positi mplate struc
he knowled
to the anno otated PFM owledgebase
comes more
th, 9th, 10th a ment is shor n of DNA immed befo ortant posit
ions filters cture.
dgebase do
otated PFM M,
e divides re similar t and 13th po rter than
sequence i ore reportin tions also the unimpo
help
after gives some to the
sition , in the ng the helps ortant
F
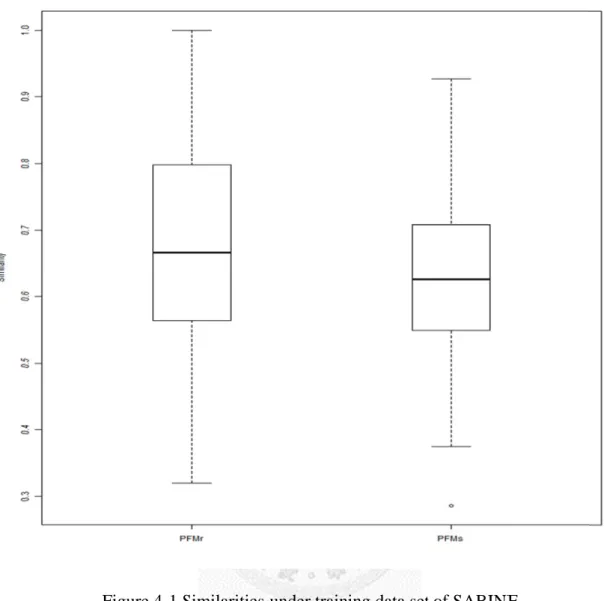
Figure 4-1 SSimilarities under trainning data sett of SABINEE
An
DN o
re k
4.3.2
Next For t an av by kn know obser PFM afterTable 4
nnotated PFM
NA sequenc of template
PFM after efinement by
knowledge
2 Protein
t, 24 protein the PFMs b verage simi nowledgeba wledgebaserved that m Ms built by e
refinement
4-2 PFM be
M
ce
y
n-DNA co
n sequencesuilt by emp ilarity of 0.6
ase, an aver we built do most of all th employing t t)
ecome more B
omplexes
in protein-D ploying the D 642 over thrage simila o help refini he cases hav
the DNA se
e similar wit Binding Pro
with anno
DNA comp DNA seque he 24 protei arity of 0.71ing the pred ve improve equences of
th annotatio ofile
otated PFM
plex structurence in the t ns. After in 11 is achiev diction of P ement after r
f templates,
on after refin
Ms
res were use template ( ncorporating ved. It show PFM. In Tab refinement.
, and
nement Similari
0.498
0.796
ed for valid ), they g the PFMs ws again tha
ble 4-2, it c . ( de denotes P
ity
ation.
y have s built at the can be enotes PFMs
PDB
1MD 1GT 1MN
3CO 2NN 3BP 1HL 1A0 1NK 2UZ 3G7 2PI 6PA 1YS 1B7 1MH
1TS 1D6 1ZM 1KB 1MN 1AK 2HA 1CD
Aver
Table 4-3
B ID
DMA TWA NNA O7C NYA PYA LOA
AA KPA ZKA 73A 0A AXA
AC 72B HDA
RB 66A MEC
B2A NMA KHA
APC WA rage
Difference
0.498 0.618 0.650 0.706 0.507 0.723 0.662 0.876 0.879 0.671 0.473 0.678 0.440 0.405 0.600 0.509 0.850 0.596 0.454 0.544 0.751 0.962 0.657 0.716 0.642
before and after PFM r
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
refinement b
0.535 0.660 0.689 0.904 0.639 0.853 0.751 0.862 0.957 0.835 0.556 0.659 0.547 0.375 0.669 0.695 0.960 0.994 0.611 0.587 0.433 0.962 0.622 0.716 0.716
by the know D
wledgebase Difference
0.037 0.042 0.039 0.198 0.132 0.130 0.089 -0.014 0.078 0.164 0.083 -0.019 0.107 -0.03 0.069 0.186 0.110 0.398 0.157 0.043 -0.318
0 -0.035
0 0.074
The
‘all-a make struc
The by T trans to ge predi propo all-at struc predi mode
In T all-at deno predi
Com meth (prot the p
contact mo atom model e comparis ctures of the
superimpos TM-align [1 sformed coo enerate a su
iction. The osed metho tom model ctures of the
iction. As a el has an av
Table 4-4, s tom model ote using na iction, respe
mpared with hod achieve tein structur proposed m
del propose l’ in the foll son with th ese 24 prote
sed structure 4]. The ori ordinates of
uperimpose templates od. As a re
has an av ese 24 prot a result, con verage simil
similarities are the alg ative structu
ectively.
h structure-b s a better p res without method with
ed in [5, 6]
owing cont he propose ins were us
es were con iginal prote f the query s ed protein-D
selected he esult, contac
verage simi teins were ntact model larity of 0.6
of differen gorithms de
ure and sup
based meth erformance DNA), the h the prote
and the all text) propos d method.
sed for PFM
nstructed by ein chains i
structure wa DNA comp ere are the ct model ha ilarity of 0
also used a l has an ave
89.
nt algorithm escribed in t
perimposed
hods with s e. For users ey could hav
ein sequenc
-atom know sed in [7] w
Superimpo M prediction
y applying t in the temp
as appended lex structur same as the as an avera 0.679. On t as input of erage simila
ms are inc this section structure a
superimpose have unbou ve better pr
ce of the
wledgebase ere implem osed structu
respectivel
the rotation late were r d into the te re for struc e templates age similari the other h
the two m arity of 0.7
luded. Con n. Native an as query for
ed structure und query p redicted PFM
unbound st
potential (c mented in ord tures and n
ly.
n matrix rep removed an emplate stru cture-based s selected b ity of 0.692 hand, the n methods for 16, and all-
ntact mode nd superimp
r structure-b
es, the prop protein struc Ms if they tructure. A
called der to native
ported nd the ucture PFM by the
2 and native
PFM -atom
l and posed based
posed ctures apply As for
struc same accom struc
cture-based e (or better,
mplishment cture-based
method wit compared t that the method.
th native st with all-ato
proposed,
tructures, se om model) , sequence
equence-bas performanc -based me
sed method ce as it does thod can
d can achiev s. This is a do as we
ve the great ell as