行政院國家科學委員會補助專題研究計畫
□ 成 果 報 告
■ 期中進度報告
PIN1BPs 與 PIN1 蛋白之交互作用及對 PIN1 之功能調控(1/3)
計畫類別:■ 個別型計畫 □ 整合型計畫
計畫編號:NSC
97 – 2311 – B – 006 - 003 - MY3
執行期間: 97 年 8 月 1 日至 98 年 7 月 31 日
計畫主持人: 呂佩融
共同主持人:
計畫參與人員: 李建興、曾昱豪
成果報告類型(依經費核定清單規定繳交):■精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位: 國立成功大學
中 華 民 國 98 年 5 月 29 日
附件一中文摘要
本報告為計劃 NSC 97-2311-B-006 -003 -MY3 第一年計劃期中報告,計劃名稱:PIN1BPs 與 PIN1 蛋 白 之 交 互 作 用 及 對 PIN1 之 功 能 調 控 (1/3) ; Pin1 是 一 個 具 特 異 性 的 phosphorylation-dependent PPIase,可專一性地辨認 phosphorylated Ser/The-Pro motif,並是 第一個發現細胞生長所需的 PPIase。在人類及酵母菌細胞中,Pin1 同時具有負面調控細胞 進入 mitosis 以及正面調控離開 mitosis 的過程。之前研究指出,在 in vitro 的情況之下,Pin1 會結合及調控一系列 mitotic phosphoproteins,以結構與功能性區分,Pin1 可分為 N 端的 WW domain 和 C 端的 PPIase domain。我們先前的研究已經證明,Pin1 N 端的 WW domain 會辨認 phosphorylated Ser/Thr-Pro motif 且專一性地結合至磷酸化的受質蛋白,而 Pin1 C 端 的 PPIase domain 則被證明可專一性的催化 phosphorylated Ser/Thr-Pro peptide bond 間 cis/trans 之構型改變。過去十年的研究亦顯示,Pin1 除了改變 cis 及 trans 的蛋白質構型, 並可影響蛋白質功能,這些 Pin1 受質蛋白包括轉譯分子、訊息傳遞分子及細胞週期調控分 子。Pin1 於是被認為在磷酸化蛋白質之後修飾機制中,扮演著樞紐角色去除了調控細胞生 理反應及正常細胞的細胞分裂。目前研究發現,Pin1 之功能異常,與一些疾病形成相關, 包括阿茲海默症和癌症。在人類乳癌、攝護腺癌和肝癌中,Pin1 已被證明為過度表現且被 證實在致癌之訊息途徑中扮演重要角色,因此 Pin1 被認為可以作為癌症治療的一個標靶。 然而,對於細胞內 Pin1 的功能調控機制,目前仍然了解不多。在此為期三年的研究計畫中, 我們將系統性的探討 Pin1 結合蛋白(PIN1BPs)如何與 PIN1 WW domain 結合,並對 Pin1 結 構及功能產生調控及可能扮演的角色進行研究。我們已利用 yeast two-hybrid 以及 molecular cloning 等實驗,成功的發現並選殖到 6 個蛋白質能以非磷酸化的形式與 Pin1 WW domain 結合,我們的假說為藉由 PIN1BPs 與 Pin1 的交互作用,PIN1BPs 可負向的調控 Pin1 蛋白 功能。本計劃的主要目標是證明以上的假設。因此,本研究計劃將包括六個主要的研究目 標以探討 PIN1BPs 與 Pin1 蛋白之交互作用及 PIN1BP1 對 Pin1 之功能調控。第一年裡(目標 1 和目標 2),我們將利用生化及細胞分子生物學之方法來確認 Pin1 蛋白與 PIN1BP1 之 in
vitro 及 in vivo 之交互作用。第二年中(目標 3),我們將確認 PIN1BP1 與 Pin1 WW domain
之專一性結合部位並解出其 binding mode,在目標 4 與 5 的部分,我們則將研究 PIN1BP1 對於 Pin1 各項功能影響之評估。在第二年與第三年中(目標 6),我們將研究 PIN1 其他五種 結合蛋白與 Pin1 之交互作用及對其功能之調控。第一年期間,我們進行 Pin1 及 Pin1BP1 之蛋白質交互作用的生化分析,確定了 Pin1BP1 與 Pin1 WW domain 是以非磷酸化形式結 合,且明確指出 Pin1BP1 與 Pin1 WW domain 結合位置。此外,我們亦知道 Pin1BP1 具有 NLS 會將 BP1 帶入核內且與核仁共位,利用螢光染色我們也測到 Pin1 與 Pin1BP1 共位於 核內。 本計劃透過蛋白質與蛋白質的互相作用研究,來了解 Pin1 蛋白的調控機制。此調控機制的 缺失,或許可以解釋癌細胞中 Pin1 蛋白之功能異常及過度表現,此外,本研究結果將可提 供未來針對 PIN1 過度表現的癌症進行之治療方向。 關鍵字:脯胺酸異構酶,細胞週期,磷酸化,磷酸激酶,脯胺酸異構酶結合蛋白
Abstract
Pin1, a unique phosphorylation-dependent PPIase that specifically recognizes phosphorylated Ser/The-Pro motifs, has been demonstrated as the first PPIase that is essential for cell growth. Pin1 both negatively regulates entry into mitosis and positively regulates progression through mitosis in human and yeast cells (1). Moreover, it has been shown that Pin1 binds and regulates a subset of mitotic phosphor-proteins, MPM2 antigens in vitro (1). In addition, our preliminary results showed that Pin1 itself likely is phosphorylated in a cell cycle- regulated manner. Structurally and functionally, PIN1 can be divided into N-terminus WW domain and C-terminus PPIase domain. We have demonstrated that the WW domain works as a phosphoserine/ threonine-proline (pSer/Thr-Pro) binding module that targets to phosphoproteins. The PIN1 PPIase domain has shown to specifically isomerize pSer/Thr-Pro motifs in a defined subset of phosphoproteins. PIN1 therefore provides a novel post-phosphorylation regulatory mechanism that induces conformational changes following phosphorylation to control protein functions. This post-phosphorylation regulatory mechanism is pivotal for controlling many physiological processes and normal cell cycle progression. Deregulation of PIN1can lead to some human diseases including Alzheimer’s disease and cancer. Overexpression of PIN1 has been demonstrated in many human cancers including breast, prostate, and liver cancers, where it functions as a crucial catalyst for multiple oncogenic pathways suggesting that PIN1 may serve as a target for cancer treatment. However, very little functional regulation of PIN1 is known although we have demonstrated that phosphorylation of the WW domain at Ser16 can inhibit the ability of PIN1 to bind to its substrates and to control the localization and function of PIN1 [25]. We propose to systematically study the physical and functional interaction between novel PIN1 binding proteins (PIN1BPs) and WW domain in this three-year study. By yeast two-hybrid screen using PIN1WW domain as bait, we have successfully isolated and cloned six PIN1-binding proteins (PINBPs) that bind to WW domain in a phosphorylation independent manner indicating that PIN1BPs may negatively regulate the PIN1 functions through protein-protein interaction. The major goal of this proposal is to test this hypothesis. Our preliminary results strongly supported this hypothesis by showing that PIN1BP1 colocalizes and co-immunuoprecipitates with PIN1, PIN1BP1 can abolish the phosphoproteins binding to PIN1 WW domain, and inhibition of PIN1function by PIN1BP1 overexpression can cause cancer cell death. We attempt to study all six PIN1BPs in this three-year study and will start with PIN1BP1. Six specific aims will be carried out to study the physical and functional interaction between PIN1 and PIN1BPs. First, by biochemically characterizing the physical interaction between PIN1 and PIN1BP1 (Aim 1 and 2, year 1), we will be able to determine the binding motif of PIN1BP1 and the binding pocket within the WW domain. Next, by determine the binding affinity and specificity of the interaction between PIN1 and PIN1BP1, we hope to construct the binding mode of this interaction and answer whether PIN1BP1 specifically regulates PIN1 through binding to PIN1 WW domain or just acts as a common WW domain modulator (Aim3, year1 and 2). Then, by examining the functional effects of PIN1BP1 to PIN1 and vies versa (Aim 4 and 5, year 1and 2), we can explain how PIN1BP1 affects on phosphoproteins binding, prolyl isomerase acitivity, Ser16 phosphorylation by PKA, PIN1-mediated tumorigenesis and PIN1 protein stability. Finally, we will apply all the strategies and experimental designs described in specific aim1-5 to study other PIN1BPs (Aim6, year 3). In the first grant year, we focused on the interaction between Pin1
and a novel Pin1 binding protein 1, named Pin1BP1. The results form RT-PCR experiments suggested that Pin1BP1 was an ubiquitous protein that expressed in all tissues examined. By immunocytochemistry study, we have demonstrated that a nuclear localization signal (NLS) located at N-terminus, a regulatory domain was lined on next to the NLS, and a Zn2+ binding domain located at C-terminus. By Co-Immunoprecipitation and GST-pull down assay, we have demonstrated the interaction between Pin1BP1 and Pin1 was direct and in a non-phosphorylation dependent manner. The results of this study will shed light on the regulatory mechanism of PIN1 through protein-protein interaction. Moreover, the defect of this regulatory mechanism may explain why Pin1 is overexpressed in cancer and can provide strategies for treating Pin1-overexpressing cancer in the future.
Key words: Pin1,cell cycle,Phosphorylation,phosphatase,Pin1BP1
Introduction
Phsophoserine-binding modules help determine the specificity of signal transduction events. Indeed, Proline- directed protein kinases, such as cyclin- dependent kinases (Cdks), play important roles in regulating eukaryotic cell division. At the G2-to-M phase transition, a rapid activation of Cdc2, the mitotic Cdk, results in phosphorylation of many proteins on serine or threonine residues that are followed by praline (Ser/Thr-Pro sites). These phosphorylation events are thought to trigger many of the structural modifications that occur during mitosis. However, it is not clear what effects the phosphorylation of mitotic proteins actually and how these abrupt changes in phosphorylation state lead to an organized and programmed set of the mitotic events. Pin1, a unique phosphorylation-dependent PPIase that specifically recognizes phosphorylated Ser/ The-Pro motifs (2). It has been demonstrated that Pin1 is essential for mitotic progression in human and yeast cells (3), and is required for the DNA replication checkpoint in Xenopus extracts (4). Pin1 substrates are a defined subset of phosphorylated proteins, including many MPM-2 antigens (1,3,5). Pin1-catalyzed prolyl isomerization regulates the conformation and function of phosphoproteins (3-6) and also facilitates dephosphorylation because of the conformational specificity of some phosphatases (7). Thus, Pin1-dependent peptide bond isomerization is a critical post- phosphorylation regulatory mechanism, allowing cells to turn phosphoprotein function on or off with high efficiency and specificity (1-6).
The consistent level of Pin1 during the cell cycle suggested that Pin1 is likely regulated by a post-translational modification. Given the current interest in aberrant cell cycle regulation in carcinogenesis and the ability to kill cells specifically at mitosis by inhibiting Pin1 function, the results obtained in these studies will be important for understanding the detailed molecular mechanism of the cell cycle control, should shed light on mechanisms of carcinogenesis, and might be eventually used to develop new therapeutic reagents for cancer.
It becomes clear that PIN1 level is balanced by growth signaling and proteolytic degradation pathway. PIN1 expression is induced by growth signals through E2F transcription factors, with protein levels fluctuating during the cell cycle in normal, but not in cancerous cells (2,12,32). The ubiquitination-dependent proteosome has been demonstrated to involve in PIN1 degradation (34) that is inhibited by Polo-like kinase 1 mediated phosphorylation (35). However, it is not known
whether the function of PIN1 is regulated or how PIN1 is regulated. The only revealed functional regulation of PIN1 is by phosphorylation of the WW domain at Ser16 that inhibits the ability of PIN1 to bind to its substrates and controls the localization and function of PIN1 (18). We have demonstrated that PIN1 WW domain is phosphorylated on Ser16 both in vitro and in vivo and phosphorylation of the PIN1 WW domain on Ser16 regulates its ability to function as a pSer/Thr-binding module (18). Furthermore, phosphorylation regulates the ability of the WW domain to mediate PIN1 substrate interaction and cellular localization (2,18). Moreover, the biological significance of the phosphorylation is demonstrated by the findings that the mutant PIN1S16A or WW S16A, but not PIN1 S16E or WW S16E induces mitotic block and apoptosis and increases multinucleated cells with 8n DNA content. These results have suggested a new mechanism for regulation of the phosphorylation-specific isomerase PIN1 and constitute the first demonstration that a pSer/Thr-binding module is subjected to post-translational modification and resultant functional modification Fig.6 below. Since Ser16 of the PIN1 WW domain is highly conserved in many other WW domains (89 out of total about 200 WW domains), some of these latter WW domains may also be subjected to phosphorylation-mediated in vivo regulation. Significantly, SH2 domain Tyr phosphorylation also modulates its binding specificity (2,12). Therefore, our data suggested that phosphorylation may be a general mechanism for controlling the function of at least some pSer/Thr phosphoprotein-binding modules. In addition, we have demonstrated that the phosphorylation of PIN1 is regulated during the cell cycle and might be altered during oncogenesis (2,12). It is then important to determine all sites of PIN1 phosphorylation, their changes and to identify the enzymes or PIN1 binding proteins that modulate this phosphorylation during oncogenesis.
In this proposed study, we will systematically study the physical and functional interaction between novel PIN1 binding proteins (PIN1BPs) and WW domain in this three-year proposed study. Six specific aims will be carried out to study the physical and functional interaction between PIN1 and PIN1BPs. Since our team has generated most of reagents for this study and I have had extensive experience working with PIN1 since 1999, our team should be able to complete all specific aims of the proposal as expected.
EEPERIMENT PROCEDURES
Yeast two hybridization -
DNA constructions - The coding sequence of human Pin1BP1 from human testis cDNA library
were amplified by polymerase chain reaction (PCR) using Pin1BP1 primer. The PCR products were cloned into TA vectors (pGEM®-T Easy Vector, Promega) as pGEMT/Pin1BP1 and confirmed by sequencing. To generate HA-Pin1BP1, Flag-Pin1BP1 and GFP-Pin1BP1, cDNA encoding Pin1BP1 was amplified by PCR using Pin1BP1 primer from pGEMT/Pin1BP1 and then subcloned into pcDNA3.1B-HA-CPO, pCMV-Flag-CPO and pEGFP-C1-CPO vectors at RsrII sites. These constructs were confirmed by digestion with RsrII and sequencing.
Reverse transcription-polymerase chain reaction (RT-PCR) – Total RNAs were isolated from
quantified by spectrophotometer at 260/280 nm before cDNA synthesis. The cDNA was reversely transcribed from 1mg of total RNA using ImProm-IITM Reverse Transcription System (Promega). The RT reaction were performed in a reaction using oligo-dT as the primer at 70oC for 60 min, added nuclease-free water to bring up to 5µl, and incubated at 70oC for 5 min followed by chilling immediately on the ice. cDNA synthesis buffer mixture was then added and incubated at 25oC for 5 min, 42oC for 1 hr and then 70oC for 15 min. Finally, the cDNA concentration was quantified by spectrophotometer at 260/280 nm. Synthesized cDNA were amplified with Pin1BP1 primer by PCR. All PCR reaction were preformed in a final volume of 20µl with an initial 5 min for strand separating at 94oC, 25 cycle of 94oC 1min, 55oC 1min, 72oC 1min and a final 10min elongation at 72oC after the last cycle. PCR products were separated on an agarose gel and visualized with ethidium bromide under UV light. All primer pairs used are listed below.
Pin1BP1-F:5’-gtggatccatgaagaaggtcaagaag-3’ Pin1BP1-R:5’-cagaattctcagtgatgtagtaggc-3’ GAPDH-F:5’-tggtatcgtggaaggactca-3’ GAPDH-R:5’-agtgggtgtcgctgttgaag-3’
Expression and Purification of Recombinant Proteins – To generate an amino-terminally GST- or
His-PinBP1 fusion proteins, Pin1BP1 cDNA encoding PinBP1 was subcloned into a pGEX or pET28a vector both at BamHI (5’) and EcoRI (3’) sites, and the resulting fusion proteins were expressed and purified by glutathione or Ni2+-NTA agarose column, as described (5). Recombinant GST- or His-Pin1BP1 protein was eluted from the beads and concentrated to 5 mg/ml with a Centricon-10 (Amicon), followed by storing at -80oC. Both preparation were stored in a buffer containing 20mM HEPES (pH7.5), 50mM NaCl, and 1mM DTT. All proteins were quantified by the method of Bradford (Bio-Rad) using BSA as a standard. The same method is used to purify various GST-Pin1 or its mutant proteins.
Cell culture, Transfection and Immunofluorescence staining - Hela cells were grown in
Dulbecco’s modified Eage medium (DMEM, Invitrogen), supplemented with 10% fetal bovine serum (FBS, Hyclone) and incubated at 37oC with 5% CO2. Cells were plated on 12-well
chamber slides 1 day before transfection at 50% confluence. Transfections were carried out using Superfect reagent according to the manufacturer’s instructions (Qiagen). Briefly, 1.5 µg of GFP-PinBP1 or its mutant plasmid was complexed with 7.5 µl of the Superfect reagent in 75 µl opti-MEM (Invitrogen) for 15 min at 37 °C, combined with 400 µl DMEM and incubated for 2 hr at 37 °C. Two hours following transfection, medium containing the complexes was replaced with fresh medium and incubated for 16 hr. For immunofluorescence, the cells were fixed with 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.2% Triton X-100 in PBS for 15 min, blocked with 1% fetal bovine serum in PBS for 1 hr, incubated with a 1:100 diluted anti-nucleolin mouse monoclonal antibody (Santa Cruz) in blocking buffer for overnight, and stained with a 1:200 diluted rhodamine-conjugated goat anti-mouse antibody (Boehringer Mannheim). After washing, the slides were counterstained with 4,6-diamino-2-phenylindole (DAPI) and analyzed using a fluorescence microscope (Olympus).
Fluorescence Spectroscopy – Fluorescence spectra were recorded at 30oC with a Hitachi F-2000 spectrophotometer. Interactions between Pin1BP1 and various concentrations of Zinc were accessed by monitoring the tryptophan fluorescence with excitation wavelength at 292 nm. The buffer used for the fluorescence experiments consist of 50mM HEPES (pH 7.4) and 100mM NaCl. Various Zinc concentrations ranged from 4.5 µM to 100µM, were introduced into 1000µl of the buffer containing 2µg His-Pin1BP1.
GST pull-down and Co-Immunoprecipitation - Hela cells were transiently transfected with
GFP-Pin1BP1or its mutants for 16h. Cells were lysed with RIPA buffer (50mM Tris-HCl (pH 7.4), 150mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1mM EDTA, 1mM PMSF, 1mM NaF, 1mM Na3VO4, 1µg/ml aprotinin, 1µg/ml leupeptin and 1µg/ml pepstatin) and incubated with 30
µl of agarose beads containing GST-Pin1 or GST at 4°C for 2 hr. For in vitro phosphatase treatment, cell lysates were incubated with calf intestine phosphatase (0.2 U/µl) at 30oC for 30 min. The precipitated proteins were washed three times with wash buffer (50mM HEPES (pH 7.4), 100mM NaCl and 0.5% Tween-20) and subjected to SDS-PAGE. For co-immunoprecipitation, cells were harvested at 16 hr after transfection with Flag-Pin1BP1 and lysed with RIPA buffer. Cell lysates were precleared with 50µl anti-mouse IgG beads on ice for 30 min , and precipitated for 1 hr with 5µg anti-Flag antibody followed by another 1 hr with 50µl protein A/G agarose beads. After washing three times with RIPA buffer, pellets were analyzed on SDS-PAGE gels and subjected to immunoblotting analysis.
RESULTS
Yeast two hybridization and cDNA Cloning of Pin1BP1- Pin1BP1 contained an open reading
frame of 549 nucleotides and encoded 183 amino acids. Results of primary structure analysis showed that the homology between the human and the mouse Pin1BP1 gene has 55% identity.
Ubiquitous expression of Pin1BP1 - Human Pin1BP1 full-length was isolated cDNA clone and it
subsequently subjected for DNA sequencing. Expression of Pin1BP1 mRNA in normal human or rat tissues was semiquantified using RT-PCR. The cDNA were reversely transcribed from mRNA isolated from various normal human or rat tissues species described in detail in Experiment procedures. Human tissues including oral, breast, liver, stomach, small intestine, kidney, bladder, testis and ovary or rat tissues including brain, lung, liver, spleen, small intestine, bladder, testis and muscle were examined and the results showed that the 550-bp Pin1BP1 single transcript can be detected in all tissues examined (Fig. 1A and 1B). The bottom panels of Figure 2A and 2B showed the PCR results of Glyseraldehyde-3- phosphate dehydrogenase (GAPDH) gene as the control to demonstrate equal cDNA loading. These results suggested that Pin1BP1 mRNA was ubiquitously expressed in the human or rat tissues. We next targeted on the Pin1BP1 protein level and first raised anti-Pin1BP1 antibody using His-Pin1BP1 as the immunogen. In order to examine the specificity of anti-Pin1BP1 antibody, we generated GFP-, Flag- and HA-tagged Pin1BP1 by subcloning it into pEGFP, pCMV or pcDNA3.1 vector, respectively. Hela cell extracts were transfected with GFP-Pin1BP1 followed by immunoblotting analysis with anti-GFP or anti-Pin1BP1 antibodies, respectively. As shown in Figure 2C, GFP-Pin1BP1 was recognized
as a major 60kDa protein. In addition, we also generated the depleted anti-Pin1BP1 antibody to confirm the specificity by neutralizing antibody with overloaded recombinant His-Pin1BP1 protein. As we expected, Flag- and HA-Pin1BP1 fusion protein could be detected as a single 32 kDa protein by anti-Flag, anti-HA or anti-Pin1BP1 antibodies, but not depleted anti-Pin1BP1 antibody. The unidentified 60kDa band as indicated by arrowhead may be due to the Pin1BP1 postmodification. These results indicated that anti-Pin1BP1 antibody exhibited great specificity to recognize Pin1BP1. Since the specificity of anti-Pin1BP1 antibody is certain, we further examine the protein expression of Pin1BP1 by introducing human tissue specimens including oral, breast, liver, gall, stomach, intestine, bladder, testis or fallopian tube followed by immunoblotting using anti-Pin1BP1 antibody. Pin1BP1 could be detected in 9 human adult tissues examined and was around 30 kDa that nearly equals to molecular weight of 32 kDa HA-Pin1BP (data not shown). However, oral tissue for example, Pin1BP1 particularly existed in normal tissue and revealed obvious down regulation within tumor part of non-tumor/tumor pairs (Fig. 1D). The similar down regulation was also found in liver or bladder specimens. The bottom panel of Figure 1D showed the immunoblotting results of tubulin used as the internal control. These results indicated that human Pin1BP1 was also a ubiquitously expressing protein and it may be associated with tumorgenesis. To further confirm the ubiquitous existence of Pin1BP1 and its subcellular localization, Immunohistochemistry (IHC) was then applied by using tissue array which contains 20 locations of human tissue, such as oral, skin, adrenal, brain, breast, heart, large and small intestine, liver, lung, spleen, stomach, thyroid, tonsil, pancreas, placenta, prostate, uterus, testis, ovary etc. Pin1BP1 was detected in all tissues examined (data not shown) with the nuclear localization and the patterns appeared from the oral and skin tissues may particularly look like the nucleolus compartment localization (Fig. 1E). These results suggested that Pin1BP1 may locate in nucleolus compartment of the nucleus which was later demonstrated using immunocytochemistry study.
Identification of Pin1BP1 Nuclear Localization Sequence and its Zinc-binding ability – To study
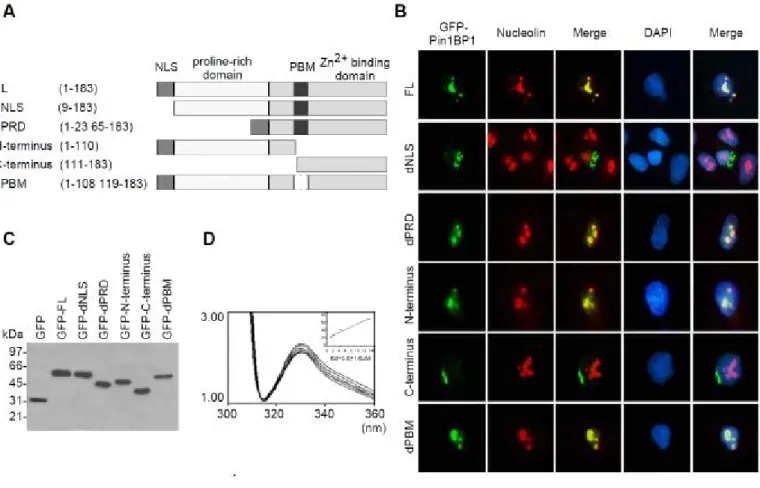
the structure and relationship of Pin1BP1, we divided Pin1BP1 into several domains according to the primary structure analysis and these domains were constructed as GFP fusion proteins for the following experiments as shown in Figure 2A. We first found that Pin1BP1 exhibited nuclear localization in the majority of Hela cells after 16 hr transient transfection with full length Pin1BP1/pEGFP. We found that full length Pin1BP1 colocalized with nucleolin but not SC35 and displayed the nucleolus location pattern. The subcellular localization of N-terminus of Pin1BP1 is exclusively nucleolus whereas C-terminus of Pin1PB1 locates outside of nucleus indicated that N-terminus carried nuclear localization sequence (NLS). Together with the NLS database prediction, these results suggested that the N-terminal first 8 amino acid may be the NLS of Pin1BP1. To further prove it, we constructed NLS deletion and chimerical NLS-C-terminus (dPRD) mutants as shown in Figure 3A. As we expected, the NLS deletion showed the localization outside the nucleus in contrary to nucleolus localization of dPRD mutant. Taken together, we identified first eight amino acid at the N-terminus, MKKVKKKR, as the NLS of Pin1BP1 (Fig. 2B). Furthermore, immunoblotting analysis revealed that molecular weight of GFP-Pin1BP1 was 60kDa and other GFP-Pin1BP1 mutants were correctly expressed as expected as shown in Figure 2C . The GFP-fusion proteins with no degradation confirmed that the
localizations showed in Figure 1B were not due to the degradation of Pin1BP1 (Fig. 2C). Since the molecular weight
of GFP shown in SDS-PAGE was 29kDa, it indicated that molecular weight of Pin1BP1 was around 30kDa. Primary sequence analysis suggested that beside NLS, Pin1BP1 also contains a Zn finger-like structure located at C-terminus of Pin1BP1.
To demonstrate that Pin1BP1 has Zinc-binding ability, tryptophan fluorescence emission spectra of Pin1BP1 was performed according to the method previously described. Human Pin1BP1 containing two C-terminal tryptophan residues (Try-114 and Try-133) in its C-terminus that allowed us to examine the Zinc-binding to C-terminus of Pin1BP1 by monitoring the tryptophan fluorescence with the excitation wavelength set to 292 nm (Raghunathan 1992). The emission maximum for Pin1BP1 was observed at 334 nm, which is consistent with the reported data and indicates a partially embedded microenvironment for these tryptophan residues (Metzler 1994). As shown in Figure 2D, Zinc at various concentrations quenched the fluorescence of Pin1BP1 in a dose dependent and saturable manner with no blue shift presented. A linear relationship was obtained by using the equation: 1/[1-(ΔF/ΔFmax)] and [Zinc]/ (ΔF/ΔFmax). Accordingly,
dissociation constants were calculated from the scatchard plot with a Kd of 3.6±1.6µM (n=3). The
Zinc-binding character of Pin1BP1 was further supported by inductively couple plasma mass spectrometry (ICP-MS). The recombinant His-Pin1BP1 purified from E coli. was subjected for ICP-MS analysis and the results showed that one mole of Zinc can be detected within one mole of Pin1BP1. These results demonstrated that Pin1BP1 was indeed a Zinc-binding protein.
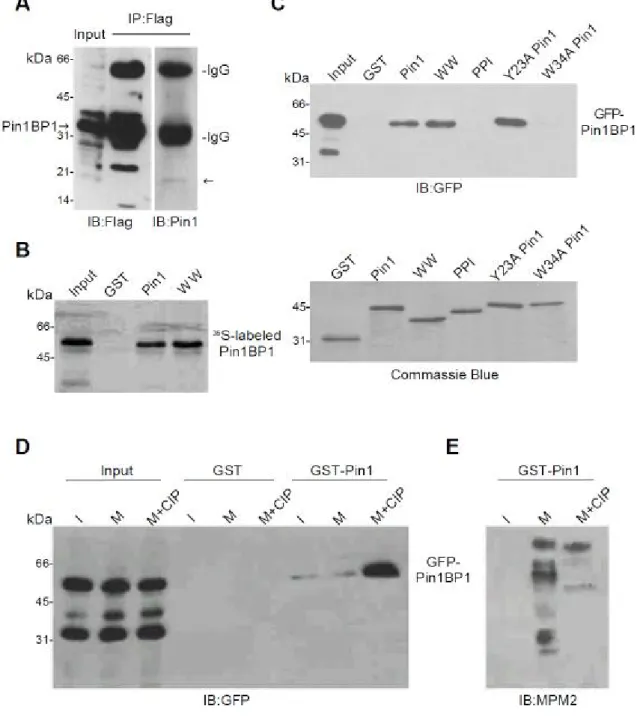
Pin1 WW domain interacted with Pin1BP1 in a phosphorylation and cell cycle independent manner – The yeast two hybrid screening showed that Pin1 can interact with Pin1BP1 in vivo. In
order to confirm the in vivo interaction between Pin1BP1 and Pin1, coimmunoprecipitation experiment was performed that Hela cells were transfected with Flag-Pin1BP1, and the lysate was prepared after 16 hr transfection for investigating whether Pin1BP1 interact with Pin1 in vivo. Figure 3A showed that Pin1BP1 overexpression in Hela cell was confirmed by immunoblot analysis using anti-Flag antibody and subsequently subjected for immunoprecipitation and immunoblotting assay using the same antibody. Using anti-Pin1 antibody, we have demonstrated Pin1 could be coimmunoprecipitated with Pin1BP1 that supported the interaction between Pin1 and Pin1BP1 in vivo. We next investigate whether the interaction between Pin1 and Pin1BP1 through direct or indirect binding. We first determined in vivo interaction using [35S]-labeled Pin1BP1 prepared by in vitro transcription and translation in GST-Pin1 pull down assay. Interestingly, [35S]-labeled Pin1BP1 was able to interact with Pin1 and the WW domain, but not with the PPI domain (Fig 3B). To confirm these finding, Hela cells expressing GFP-Pin1BP1 for 16 hr were also used and lysates were prepared followed by GST-Pin1 pull down assay. Consistent with our previous results, Pin1 and its WW domain, but not PPI domain, bound to Pin1BP1 (Fig. 3C, upper panel). The bottom panel showed equal amount of Pin1 were used in GST pull down assay (Fig. 3C). Together, these results showed that Pin1 is through direct binding to WW domain of Pin1BP1. In the previous study, it has been found that Pin1 bound to the phospho-Ser/Thr Pro motif with three important residues including Ser16, Tyr23 and Trp34 in WW domain. Thus, we introduced substitution of Tyr23 with alanine (Y23A) and Trp34 with alanine (W34A) Pin1 mutants that have been demonstrated to specifically affect phospho-binding
activity of Pin1. That allowed us to investigate whether Pin1BP1 binds to the same binding pocket that phosphoproteins bound in WW domain. In GST-Pin1 pull down assay, W34A Pin1 but not Y23A Pin1 in the WW domain showed no binding ability Pin1BP1 (Fig. 3C). These results indicated that Trp34 residue may play an important role in the Pin1 WW domain for Pin1BP1 binding and the binding may not follow the phosphorylation dependent manner. To further address whether Pin1 binds to Pin1BP1 in a phosphorylation or cell cycle dependent manners as many mitotic proteins do, we utilized cell lysate prepared from asynchronized cells and Nocodazole block mitotic cells as described previously. In addition, alkaline phosphatase (CIP) was used to dephosphorylate GFP-Pin1BP1 after cell lysate was prepared. Surprisingly, Pin1 showed Pin1BP1 association with no difference between interphase (I) and mitotic phase (M), but strong association after CIP treatment (M+CIP) indicated their interaction was in a phosphorylation and cell cycle independent manner (Fig. 3D). Efficient dephosphorylation of CIP treatment was verified by immunoblot of lysates with MPM-2 antibody which specifically recognized mitotic- and phosphoproteins (Fig 3E). These results demonstrated that Pin1 WW domain interacted with Pin1BP1 in a phosphorylation and cell cycle independent manner.
計畫成果自評
本研究報告利用生化及細胞分子生物學之方法來確認 Pin1 蛋白與 PIN1BP1 之 in vitro 及 in
vivo 之交互作用。此與三年計畫之第 1 年 specific Aim 相符合;具有學術價值及校稿中,將
待整理更多數據後,發表於國際生化期刊。
REFERENCES
1. Hanes, S.D., Shank, P.R. and Bostian, K.A. (1989). Yeast 5, 55-72. 2. Lu, K.P., Hanes, S.D., and Hunter, T. (1996). Nature 380, 544-547.
3. Winkler, K.E., Swenson, K.I., Kornbluth, S. and Means, A.R. (2000). Science 287, 1644-1647.
4. Yaffe, M.B. et al. (1997) Science 278, 1957-1960.
5. Shen, M., Stukenberg, P.T., Kirschner, M.W., and Lu, K.P. (1998). Genes Dev. 12, 706-720. 6. Lu, P.J., Zhou, X.Z., Shen, M., and Lu, K.P. (1999). Science 283, 1325-1328.
7. Rangannathan, R., Lu, K.P., Hunter, T. and Noel, J.P. (1997). Cell 89, 875-886. 8. Lu, P.J., Wulf, G., Zhou, X.Z., Davies, P., and Lu, K.P. (1999). Nature 399, 784-788. 9. Stukenberg, P.T. and Kirschner, M.W. (2001). Mol Cell 7, 1071-1083.
10. Wulf, G..M., Ryo, A., Wulf, G.G., Lee, S.W., Niu, T. and Lu, K.P. (2001). EMBO J 20, 3459-3472.
11. Ryo, A., Nakamura, M., Wulf, G., Liou, Y.C., and Lu, K.P. (2001). Nat. Cell Biol. 3, 793–801.
12. Ryo, A., Liou, Y.C., Wulf, G., Nakamura, N., Lee, S.W. and Lu, K.P. (2002). Mol Cell Bio 22, 5281-5295.
13. Wulf, G.M., Liou, Y.C., Ryo, A., Lee, S.W., and Lu, K.P. (2002). J. Biol. Chem. 277, 47976–47979.
14. Ryo, A., Suizu, F., Yoshida,Y., Perrem, K., Liou, Y.C., Wulf, G., Rottape, R., Yamaoka, S. and Lu, K.P. (2003). Mol Cell 12, 1413–1426.
Blandino, G., Schneider, C., and Del Sal, G. Nature 419, 853–857.
16. Raghunathan, V., Mowery, P., Rozycki, M., Lindberg, U. and Schutt, C. (1992). FEBS Lett. 297, 46-50
17. Metzler, W.J., Bell, A.J., Ernst, E., Lavoie, T.B. and Mueller, L. (1994). J. Biol. Chem. 296, 4620-4625
18. Wulf, G. M., Ryo, A., Wulf, G. G., Lee, S. W., Niu, T., and Lu, K. P. (2001). EMBO J. 20, 3459–3472
Figures
Fig. 1. Sequence analysis of Pin1BP1 cDNA and its ubiquitous existence.
(A) Nucleotide and deduced amino acid residues are numbered on the right. Detection of Pin1BP1 mRNA level in various human (B) or rat (C) tissues by RT-PCR (D) Pin1BP1 level in clinical specimen
Fig. 2. Functional characterization of Pin1BP1.
(A) Schematic representation of Pin1BP1 and its mutants. NLS, nuclear localization sequence; FL, full length; PRD, proline-rich domain; PBM, Pin1-binding motif.
(B) Nuclear localization sequence (NLS) in Pin1BP1 constructs. Compared to FL Pin1BP1 located in the nucleolus, GFP-constructs without NLS appeared outside the nuclei. Nucleolin was used as the nucleolus marker.
(C) Western blotting of the GFP-Pin1BP1 and its mutants.
(D) Tryptophan excitation spectra of recombinant Pin1BP1 by Zinc titration. Zinc binds to Pin1BP1 with a Kd of 3.6±1.6 M.
Fig. 3. Pin1BP1 interacts with Pin1 WW domain in a non-phosphorylation dependent manner. (A) In vivo interaction of endogenous Pin1 and Pin1BP1. After transfection with Flag-Pin1BP1, protein extracts were subjected to immunoprecipitation with anti-Flag antibody and checked by anti-Flag or anti-Pin1 antibody.
(B) In vitro interaction of Pin1 and Pin1BP1. In vitro translated [35S] His-tag Pin1BP1 can be pull down by WW domain of Pin1.
(C) Interaction of Pin1 and Pin1BP1 in vitro. Cells expressing GFP-Pin1BP1 can be pull down by wt Pin1 or WW domain of Pin1, but not PPI domain or W34A Pin1 mutant. The GST protein input were also checked by commassie blue staining.
(D) Non-phosphorylation dependent interaction between Pin1BP1 and Pin1. Cells were
transfected with GFP-Pin1BP1 and synchronized in interphase (I), mitotic extracts (M) or mitotic extracts followed by dephosphorylation with phosphatase (M+CIP), and then pulled down by GST or GST-Pin1 beads. Efficient dephosphorylation of CIP treatment was verified by immunoblot of lysates with pS/pT-P-specific MPM-2 antibody.