行政院國家科學委員會專題研究計畫 成果報告
利用生物資訊方法預測癌症相關之 microRNA 並建立生物實 驗平台
研究成果報告(精簡版)
計 畫 類 別 : 個別型
計 畫 編 號 : NSC 98-2221-E-468-013-
執 行 期 間 : 98 年 08 月 01 日至 99 年 07 月 31 日 執 行 單 位 : 亞洲大學生物資訊學系
計 畫 主 持 人 : 吳家樂 共 同 主 持 人 : 李尚熾
計畫參與人員: 碩士班研究生-兼任助理人員:林晏安 碩士班研究生-兼任助理人員:張哲豪 碩士班研究生-兼任助理人員:江文欽 碩士班研究生-兼任助理人員:王傑瑋 碩士班研究生-兼任助理人員:謝蕙霞
報 告 附 件 : 出席國際會議研究心得報告及發表論文
處 理 方 式 : 本計畫可公開查詢
中 華 民 國 99 年 10 月 26 日
精簡報告 目錄
報 告 內 容 - In silico study of expression profiles correlation between microRNAs and cancerous genes
參考文獻 計畫成果自評
In silico study of expression profiles correlation between microRNAs and cancerous genes
Keywords: microRNA, oncogene, tumor suppressor gene, gene expression profile, correlation coefficient
Abstract: We investigate the possibility that microRNA can act as an oncogene or tumor suppressor gene.
Experimentally verified microRNA target genes information (TarBase) are integrated with microRNA and mRNA expression data (NCI-60) to study this hypothesis, in which the Pearson correlation and Spearman rank coefficients are used to quantify these relations for nine cancer types. Correlation coefficients with negative values are used to filter out microRNA targets. Biological annotations of the targets are supplied by using the TAG, GO and KEGG records. The above information are utilized to provide a platform in identifying potential cancer related microRNAs. A web based interface is set up for information query and data display.
1 INTRODUCTION
MicroRNAs (miRNAs) are a class of small non- coding RNAs that bind to its target mRNA sequence in the 3′-untranslated region (3′UTR), and induce either translation repression or mRNA degradation.
Recent studies indicated that microRNA could possibly play an important role in human cancer where microRNA targets oncogene (OCG) or tumor suppressor gene (TSG) to regulate the gene expression (Zhang et al., 2007, He and Cao, 2007, Wu and Hu, 2006, Garzon et al., 2006). When microRNA plays an oncogenic role, it targets TSG and leads to tumor formation. On the other hand, if microRNA plays the tumor suppressor role, it would target OCG and suppress tumor formation.
This work utilized the following databases; the TarBase (Sethupathy et al., 2005), miRBase (Griffiths-Jones et al., 2006) and NCI-60 (Shankavaram et al., 2007, Blower et al., 2007), Online Mendelian Inheritance in Man (OMIM), Tumor Associate Gene (TAG) (Chan, 2006) Gene Ontology (Gene Ontology Consortiium, 2006), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2008) databases to set up a platform for predicting human microRNA targeting cancerous genes information. Table 1 states the general information provided by the databases used in the current study.
The platform mainly provides the following two functionalities; (i) human microRNA target information, and (ii) nine cancer types’ Pearson
correlation and Spearman rank coefficients of microRNA and its target expression level for three Affymetrix chips.
Table 1. General information provided by the databases used in the current study.
Database General information provided TarBase experimentally tested miRNA target genes
miRBase
information for precursor microRNAs, mature microRNAs, FASTA sequences, and their target genes.
NCI-60 human cancer cell lines mRNA and miRNA expression data
OMIM human diseases genetic data TAG OCG, TCG and cancer related genes
GO
Three types of gene annotations; i.e.
molecular function, biological process and subcellular localization
KEGG Metabolic pathways, disease pathways
2 MATERIALS AND METHODS
It is known that microRNA binds with mRNA and can induce mRNA cleavage or inhibit translation. In order to investigate the regulatory role of microRNA in cancer diseases, we study the expression profiles correlation between microRNA and its target genes, in particular the OCG and TSG targets.
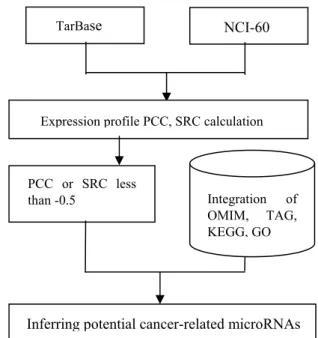
Figure 1 depicted the process flowchart for identifying potential cancer related microRNAs. The microRNA-target pairs information is obtained from TarBase, whereas the expression profiles for microRNAs and mRNAs are retrieved from the NCI-60 dataset. Then, the expression profiles correlation between microRNA and its target genes are quantified by computing the correlation coefficients. If the microRNA and its target gene are direct interact, the correlation coefficient results should reveal significant negative values.
MicroRNA target pairs with the correlation coefficients below a given threshold are filtered for further investigation. These pairs suggest a regulatory relationship between the microRNAs and their targets. The TAG dataset is used in order to sort out the microRNA-OCG and microRNA-TSG pairs.
These pairs are further annotated by using the OMIM, GO and KEGG biomedical terms. With multiple biological annotations, i.e. disease type, relevant biological function and pathway, for the negative correlated pairs, this platform should
provide helpful guidance for investigating the role of micorRNAs in tumor formation.
Figure 1. Process flowchart to identify potential cancer related microRNAs based on quantifying the expression profile correlation between microRNA and its target genes.
2.1 TarBase and miRBase datasets
TarBase is a manually curated collection of experimentally tested microRNA target genes. Each experimentally validated target site is extracted from the literatures. TarBase includes several species, such as human, mouse, fruit fly, and worm, microRNA target gene records.
There are many tools available for microRNA target genes prediction, such as miRanda (Enright et al., 2003), RNAhybrid (Kruger and Rehmsmeier,
2006), and TargetScans (http://genes.mit.edu/tscan/targetscanS2005.html). A
major problem of microRNA target genes prediction is that the prediction accuracy remains rather high, there were reports indicated that the false positive rate could be as high as 50% for human (John et al., 2004), 24-39% and 22-31% when using miRanda (Bentwich, 2005), and TargetScan (Bentwich, 2005) respectively.
The primary goal of this work is to develop a bioinformatics tool to investigate the possibility that microRNA can act as an OCG or TSG. The main advantage of using TarBase in constructing the microRNA targeting information is that all the target genes recorded by TarBase are experiment verified, and TarBase provides their PubMed ID. From a biologist point of view, experimental verified targets imply higher confidence. If the miRNA:mRNA targeting part is uncertain, then any further results derived are doubtful. The TarBase version 5 dataset
TarBase NCI-60
Expression profile PCC, SRC calculation
PCC or SRC less
than -0.5 Integration of
OMIM, TAG, KEGG, GO
Inferring potential cancer-related microRNAs
from DIANA Lab. website is employed in the present study. The miRBase database collects information for precursor microRNAs, mature microRNAs, FASTA sequences, and their target genes. Currently the latest version of the miRBase sequence database is 13.0, which includes microRNA information across 103 species. In version 13, a total of 706 Homo Sapiens mature micorRNA entries are recorded.
2.2 Expression datasets
In this study, we made use of the NCI-60 cancer cell line mutation data to investigate the possibility that microRNA can act as an OCG or TSG. This can be achieved by calculating the correlation coefficient between the expression levels of microRNAs and their experimentally validated target genes.
The NCI-60 is a set of 60 human cancer cell lines derived from diverse tissues. These cell lines include nine tissues’ microRNA and mRNA expression information, that is, breast cancer, central neural system (CNS) cancer, colon cancer, leukemia, melanoma, non-small cell lung cancer, ovarian cancer, prostate cancer, and renal cancer,.
Four publicly available datasets of gene expression profiles are selected in this study; including the microRNA expression, and the Affymetrix U95(A- E), U133A and U133B mRNA expression datasets.
Affymetrix mRNA expression datasets use three types of normalization methods, that is, GCRMA, MAS5 and RMA. Therefore, a total of ten datasets are used, including one microRNA dataset and nine Affymetrix RNA expression datasets. The NCI-60 website provides a tool, called CellMiner (Shankavaram et al, 2009), to query those chip datasets.
2.3 Tumor Associated Gene Database
The Tumor Associated Gene (TAG) database presents information about cancer related genes. In TAG, cancer related genes are classified into OCGs, TSGs and tumor-associated genes. All genes in the TAG are retrieved through text-mining approach from the PubMed database. Currently, TAG documented 519 genes, including 198 OCGs, 170 TSGs, and 151 genes related to oncogenesis. In addition, more cancer related microRNA gene information are obtained by using Pipeline PilotTM, which is a commercial bioinformatics text mining package to do keywords search against PubMed. At present, a total of 111 microRNAs are retrieved that are related to certain types of cancers. These microRNAs and their target records are stored in our platform for further analysis.
2.4 OMIM, GO and KEGG Databases
Online Mendelian Inheritance in Man (OMIM) is a compendium of human genes and genetic phenotypes. It contains information on all known mendelian disorders.
The GO database includes three structured controlled vocabularies (ontology) that describe gene products in terms of their associated biological processes, cellular components and molecular functions in a species-independent manner.
KEGG is short for Kyoto Encyclopedia of Genes and Genomes which is a collection of manually drawn pathway maps. In our study, we focused on cancer related pathways information.
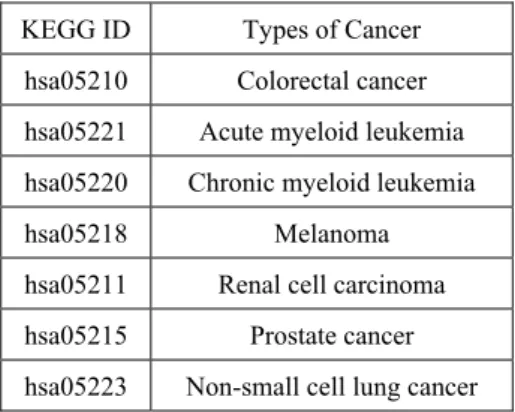
Investigation of cancer related pathway in KEGG can help us determine the biological functions of the target genes. We investigate which cancer related pathways consist of the microRNA target genes. By going through biological function keywords search, a list of microRNA target genes which participated in certain KEGG pathways are obtained. Table 2 shows the cancer related pathways which are processed in this study.
Table 2. Cancer related pathways which are processed in this study.
KEGG ID Types of Cancer hsa05210 Colorectal cancer hsa05221 Acute myeloid leukemia hsa05220 Chronic myeloid leukemia
hsa05218 Melanoma
hsa05211 Renal cell carcinoma hsa05215 Prostate cancer hsa05223 Non-small cell lung cancer
2.5 Preprocessing
The TarBase and NCI-60 datasets used different ID formats for microRNAs, therefore, both sets of ID are standardized using the miRBase IDs. We also converted the mRNA gene IDs in TarBase and NCI- 60 Affymetrix RNA expression datasets IDs to NCBI official symbols by using Gene Name Service (Lin et al., 2007). Gene Name Service website provides query services for 26 types of gene identifiers of Homo sapiens genes. We obtained 44855, 20169 and 16441 entries for the U95(A-E), U133A and U133B mRNA datasets after pre- processing.
3. Methods
3.1. Expression profiles correlation
For a given cancer tissue type, we calculated both the Pearson correlation coefficient (PCC) and Spearman rank coefficient (SRC), ρ, between the expression level of a microRNA and it's target genes.
PCC or SRC is given by
∑ ∑
∑
= =
=
−
−
−
= −
n i
n
i i
i n
i i i
y y x x
y y x x
1 1
2 2
1
) ( ) (
) )(
ρ ( (1)
where xi and yi denote the expression intensity of microRNA and the microRNA's target gene respectively;
x
andy
denote the mean expression intensity of microRNA and the target gene respectively; and n is the total number of the expression data entries. In case of SRC, the expression intensity values of xi, yi,x
andy
are replaced by their ranks.The PCC for each microRNA and Affymetrix RNA expression profile are computed for nine tissue types. PCC takes a value between -1 and +1. One of the troubles with quantifying the strength of correlation by PCC is that it is susceptible to being skewed by outliers. Outliers that is a single data point can result in two genes appearing to be correlated, even when all the other data points not.
SRC is a non-parametric statistical method that is robust to outliers. It can ignore the magnitude of the changes. The idea of SRC is to transform the original values into ranks, and then to compute the correlation between the series of ranks.
Gene expression values of microRNA and mRNA in the same tissue type are ordered in ascending order, the lowest value is assigned to rank one. In case of ties mid-rank is assigned, as for example, when both values are ranked five, a rank of 5.5 is assigned. After ranking the expression profiles of microRNA and mRNA for a particular tissue, SRC can be calculated by Eq. (1), with the ranks and the average value of ranks of microRNA and mRNA are used instead of the expression intensity and average expression intensity. The SRC also takes a value between -1 and +1.
4. Result
Both the PCC and SRC of microRNA expression levels and their targeting mRNA expression levels for nine types of cancer tissues are computed.
For example, microRNA hsa-miR-16 targets the breast cancer gene, BCL2, have PCC less than -0.7 for the three Affymetrix datasets with three different
normalization methods for each chip. The results are reported in Table 3. We can understand negative PCC (SRC) based on the following reasoning. It is known that microRNA is able to repress and/or cleavage mRNA by incomplete or complete complementary binding with the mRNA. If a microRNA and its target gene is directly interacting, the result of PCC (SRC) of their expression profiles should reveal negative correlation. Table 3 list the U95 results which suggested that microRNA, hsa- miR-16, can possibly play a role in regulating the cancer gene BCL2.
Table 3. PCC of the expression profile for hsa-miR-16 and BCL2 in the breast tissue.
Dataset Normalization PCC U95(A-E) GCRMA -0.918
MAS5 -0.949 RMA -0.718 Downstream cancer targets are easily obtained by cross-referencing the target gene results with the TAG dataset. The results of cancerous genes found in both TarBase and TAG are listed in Table 4.
Table 4. MicroRNA target genes which are corresponded with TAG entries.
Cancer Gene Gene Symbols
OCG
AXL, BCL2, CCND1, CDK4, CTGF, ESR1, FGF20, KIT, MAP3K8, MYB, MYBL1, MYCN, RYK, TEAD1
TSG
CAV1, CDKN1A, CDKN1C, CDKN2A, HTATIP2, MXI1, NF2, PTEN, PTPN12, RB1, SERPINB5, TGFBR2
4.1 Evaluation of results by OMIM
Disease disorder keywords provided by the OMIM, is compared with the negative (under or equal to -0.5) PCC results. It is found that 82 entries involved in disease disorder. Among these 82 entries, 19 are cancer genes. In these cancer genes, only 17 entries belong to the nine cancer types. Table 5 presents five of the 17 OMIM entries.
Table 5. The 17 OMIM entries with negative PCC (≦ -0.5) and their corresponding cancerous types.
OMIM ID Gene Symbol Cancerous Types
605882 BACH1 Breast 151430 BCL21 Leukemia 168461 CCND1 Colon 123829 CDK41 Melanoma
600160 CDKN2A CNS||Melanoma For a given Affymetrix chip, it is found that the PCC score is independent of the normalization methods. Similarly, for a given normalization method, the PCC score is also rather independent of the chip type. Furthermore, it is also found that target results (microRNA:mRNA) obtained by PCC versus PCC and SRC are rather consistent. Table 4 reports the results for the U95(A-E) chip with the GCRMA normalization method.
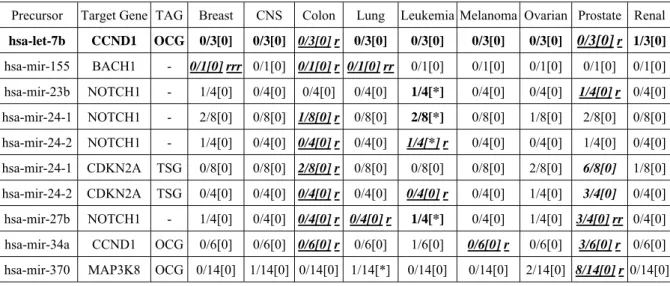
According to Table 6, four pairs of negative microRNA:mRNA are found belonging to leukemia, and one pair belongs to lung cancer. Among these five pairs, only hsa-mir-370 targets an OCG, i.e.
MAP3K8. Besides the OMIM recorded data in Table 4, we also identified five (in Italic) microRNA targets in which half of the probes’ PCC and SRC are under or equal to -0.5 in prostate cancer. Among these pairs, one of the target is a TSG, i.e. CDKN2A, and two targets are OCGs, i.e. CCND1 and MAP3K8.
Table 6. OMIM evaluation results of both PCC and SRC under or equal to -0.5 for GCRMA-U95(A-E) dataset.
Precursor Target Gene TAG Breast CNS Colon Lung Leukemia Melanoma Ovarian Prostate Renal hsa-let-7b CCND1 OCG 0/3[0] 0/3[0] 0/3[0] r 0/3[0] 0/3[0] 0/3[0] 0/3[0] 0/3[0] r 1/3[0]
hsa-mir-155 BACH1 - 0/1[0] rrr 0/1[0] 0/1[0] r 0/1[0] rr 0/1[0] 0/1[0] 0/1[0] 0/1[0] 0/1[0]
hsa-mir-23b NOTCH1 - 1/4[0] 0/4[0] 0/4[0] 0/4[0] 1/4[*] 0/4[0] 0/4[0] 1/4[0] r 0/4[0]
hsa-mir-24-1 NOTCH1 - 2/8[0] 0/8[0] 1/8[0] r 0/8[0] 2/8[*] 0/8[0] 1/8[0] 2/8[0] 0/8[0]
hsa-mir-24-2 NOTCH1 - 1/4[0] 0/4[0] 0/4[0] r 0/4[0] 1/4[*] r 0/4[0] 0/4[0] 1/4[0] 0/4[0]
hsa-mir-24-1 CDKN2A TSG 0/8[0] 0/8[0] 2/8[0] r 0/8[0] 0/8[0] 0/8[0] 2/8[0] 6/8[0] 1/8[0]
hsa-mir-24-2 CDKN2A TSG 0/4[0] 0/4[0] 0/4[0] r 0/4[0] 0/4[0] r 0/4[0] 1/4[0] 3/4[0] 0/4[0]
hsa-mir-27b NOTCH1 - 1/4[0] 0/4[0] 0/4[0] r 0/4[0] r 1/4[*] 0/4[0] 1/4[0] 3/4[0] rr 0/4[0]
hsa-mir-34a CCND1 OCG 0/6[0] 0/6[0] 0/6[0] r 0/6[0] 1/6[0] 0/6[0] r 0/6[0] 3/6[0] r 0/6[0]
hsa-mir-370 MAP3K8 OCG 0/14[0] 1/14[0] 0/14[0] 1/14[*] 0/14[0] 0/14[0] 2/14[0] 8/14[0] r 0/14[0]
Bold font denotes the OMIM matching data.
Italic and bold font denotes that half of the microRNA probes’
PCC and SRC are ≦-0.5. Inside the box, the numerator of the fraction denotes the number of times where both of PCC and SRC are less than -0.5.
The denominator represents the total number of PCC values calculated (i.e. the number of microRNA probes times the number of mRNA probes). Inside the square bracket, zero ([0]) implies no matching with the OMIM data and [*] denotes matching with the OMIM data.
4.2 Evaluation of results by KEGG
The KEGG pathways are compared with the PCC results. A total of 26 pairs (with an asterisk) of negative correlated microRNA and its target genes matched with KEGG data for four cancer types, which are colon, leukemia, melanoma and prostate.
Among these KEGG matched pairs, twelve pairs (with an asterisk) have both the PCC and SRC under or equal to -0.5 in more than half of the probes. In these twelve pairs, two target genes are TSG, i.e.
CDKN1A and PTEN, and one gene is an OCG, i.e.
CCND1.
In OMIM and KEGG evaluation results, two pairs of microRNA:mRNA, i.e. hsa-mir-24- 1:CDKN2A and hsa-mir-24-2:CDKN2A, both
present significant negative correlation in prostate cancer, with the correlation coefficients under or equal to -0.5 in more than half of the probes. At present, it is still unclear whether the hsa-mir-24-2 and CDKN2A has any regulatory relationship in prostate cancer. These predicted negative correlated microRNA:mRNA pairs maybe subjected to further investigation in order to identify the exact regulatory situations in prostate cancer.
According to OMIM (Table 6) and KEGG evaluation results, one pair presents negative correlation in leukemia which is hsa-mir- 34a:CCND1, but the PCC and SRC scores don’t present in more than half of the probes are under or equal to -0.5. Although the evidence is not strong, but it suggests that hsa-mir-34a may also be a potential regulator of CCND1 in leukemia.
Final, we set up a web based service to provide the computed results. The web site is availabled at http://ppi.bioinfo.asia.edu.tw/mirna_target/index.html.
5. CONCLUSION
Recent studies indicate that microRNA could possibly play an important role in human cancer, where microRNA targets TSG or doesn’t target OCG. Experimentally verified microRNA targeted genes information (TarBase) are integrated with microRNA and mRNA expression data (NCI-60) to study this hypothesis, in which two correlation coefficients, PCC and SRC, are used to quantify the correlation between microRNA and its targets expression profiles. The predicted results are evaluated with reference to the OMIM and KEGG data. It is found that the obtained results are rather independent of the chip types and the normalization methods too.
In the OMIM evaluation with both PCC and SRC less than or equals to -0.5, five pairs of negative correlated microRNA and its target genes matched with OMIM records, in which four of them belong to leukemia and the rest one is lung cancer. In these five pairs, only one of them is an OCG, i.e.
MAP3K8. Besides, we also got five pairs of significantly negative correlated microRNA and its target in prostate cancer in which both of PCC and SRC are under or equals to -0.5. Among these five pairs, only one gene is a TSG, i.e. CDKN2A, and only two genes are OCGs, i.e. CCND1 and MAP3K8. These five pairs can be browsed in Table 5 in which they are denoted with italic and bold font.
Similar conclusions are obtained for the KEGG evaluation.
Given that more than half of the probes’
correlation coefficients are negative correlated, we identified certain putative pairs of microRNA and its cancer related targets in different cancer types, such as, hsa-mir-24-1:CDKN2A and hsa-mir-24- 2:CDKN2A in prostate cancer and hsa-mir- 19a:PTEN in both leukemia and prostate cancer.
It is suggested that those negative correlated pairs of microRNA and target can be subjected to further investigation, such as performing in vivo experiments to valid the hypothesis that microRNA could possibly play an important role in human cancer.
ACKNOWLEDGEMENTS
K-L Ng work is supported by the National Science Council of R.O.C. under the grant of NSC 98-2221- E-468-013
REFERENCES
Bentwich I. 2005. Prediction and validation of microRNAs and their targets. FEBS Lett., 579, 5904.
Blower P.E., Verducci JS, Lin S, Zhou J, Chung JH, Dai Z, Liu CG, Reinhold W, Lorenzi PL, Kaldjian EP, Croce CM, Weinstein JN, Sadee W., 2007. MicroRNA expression profiles for the NCI-60 cancer cell panel, Mol. Cancer Ther., 6, 1483-1491.
Chan Hsiang-Han 2006. Identification of novel tumor- associated gene (TAG) by bioinformatics analysis.
MSc. Thesis, Institute of Molecular Medicine, National Cheng Kung University, Taiwan.
Enright, A.J., John, B., Gaul, U., Tuschl, T., Sander, C., Marks, D.S., 2003. MicroRNA targets in Drosophila.
Genome Biology, 5(1):R1.
Garzon Ramiro, Fabbri Muller, Cimmino Amelia, Calin George A. and Croce Carlo M., 2006. MicroRNA expression and function in cancer. Trends in Molecular Medicine, 12, 580-588.
Gene Ontology Consortium 2006. The Gene Ontology (GO) project in 2006. Nucl. Acids Res, 34, D322–326.
Griffiths-Jones S., Grocock Russell J., van Dongen Stijn, Bateman Alex, Enright Anton J., 2006. miRBase:
microRNA sequences, targets and gene nomenclature.
Nucl. Acids Research, 34, 140-144.
He Xiaoting, Cao Xiufeng 2007. MicroRNA and esophageal carcinoma. J.N.M.U., 21, 201-206.
John B., Enright A.J., Aravin A., Tuschl T., Sander C., Marks D.S., 2004. Human MicroRNA targets. PLoS Biol. 2(11), e363.
Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., Katayama T., Kawashima S., Okuda S., Tokimatsu T., and Yamanishi Y., 2008. KEGG for linking genomes to life and the environment. Nucl.
Acids Res., 36, 480-484
Kruger Jan and Rehmsmeier Marc 2006. RNAhybrid:
miRNA target prediction easy, fast and flexible, Nucleic Acids Research, 34, 451-454.
Lin Kuan-Ting, Liu Chia-Hung, Chiou Jen-Jie, Tseng Wen-Hsien, Lin Kuang-Lung, Hsu Chun-Nan 2007.
Gene Name Service: No-Nonsense Alias Resolution Service for Homo Sapiens Genes. Proceedings of the 2007 IEEE/WIC/ACM International Conferences on Web Intelligence and Intelligent Agent Technology- Workshops, 185-188.
Sethupathy P., Corda B., Hatzigeorgiou A. 2005 TarBase:
A comprehensive database of experimentally supported animal microRNA targets. RNA, 12, 192- 197.
Shankavaram U.T., Uma T., Reinhold William C., Nishizuka Satoshi, Major Sylvia, Morita Daisaku, Chary Krishna K., Reimers Mark A., Scherf Uwe, Kahn Ari, Dolginow Douglas, Cossman Jeffrey,
Kaldjian Eric P., Scudiero Dominic A., Petricoin Emanuel, Liotta Lance, Lee Jae K., Weinsteinu John N, 2007. Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Molecular Cancer Therapeutics, 6, 820-832.
Shankavaram U.T., Varma S, Kane D, Sunshine M, Chary KK, Reinhold WC, Pommier Y, Weinstein JN., 2009.
CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. BMC Genomics,10, 277..
Wu Dan and Hu Lan 2006. Micro-RNA: A New Kind of Gene Regulators. Agricultural Sci. in China, 5, 77-80.
Zhang Baohong, Pan Xiaoping, Cobb George P., Anderson Todd A., 2007. microRNAs as oncogenes and tumor suppressors. Develop. Biol, 302, 1-12.
計畫成果自評
請就研究內容與原計畫相符程度、達成預期目標情況、研究成果之學術或應用價值、
是否適合在學術期刊發表或申請專利、主要發現或其他有關價值等,作一綜合評估。
研究達成預期目標,本計畫主要是探討微型核糖核酸(miRNA)扮演致癌及抑癌基
因的角式。透過整合不同類型之資料,即 miRNA 標靶資料(TarBase)、miRNA 及
mRNA 表達資料(NCI60)、癌症基因微晶片資料(ArrayExpress)、基因功能註解資料庫 (Gene Ontology)及文獻資料(PubMed)等,探討 miRNA 扮演此調控的角式。
研究成果在學術期刊及學術研討會發表。對於參與之學生獲得訓練,如生物資
料 庫 之 使 用 、 程 式 撰 寫(Perl),將計算之結果建立成資料庫(MySQL),架設網站
(Dreamweaver)等。
對於學術研究之貢獻為探討miRNA 標靶致癌及抑癌基因之可能性。
行政院國家科學委員會補助國內專家學者出席國際學術會議報告
99 年 1 月 27 日 報告人姓名
吳家樂
服務機構 及職稱
亞洲大學 生物資訊系 教授
時間 會議 地點
20-23 Jan. 2010 Valencia, Spain
本會核定
補助文號 NSC 98-2221-E-468-013 會議
名稱
(中文) 2010 年 第一屆生物資訊國際研討會
(英文) First International Conference on Bioinformatics 2010 發表
論文 題目
(中文) 微型核糖核酸與癌症基因表達量之相關性分析
( 英 文 ) In silico study of expression profiles correlation between microRNAs and cancerous genes
附件三
表 Y04
2
報告內容應包括下列各項:
一、參加會議經過
Jan. 20
Below is the listed of talks I attended;
Interdisciplinary Synergies in Biomedical Engineering
Keynote Speech
Session 1
Jan. 21
Session 4
Keynote Speech
Session 5
Session 6
Session 7
Jan. 22
Session 8
Keynote Speech
Poster Session
Session 9 – no bioinformatics paper
Session 10 – no bioinformatics paper
Session 11– no bioinformatics paper
Jan. 23
Session 12
Keynote Speech 二、與會心得
The First International Conference Bioinformatics 2010 (ICB10) is one of the four co-located conferences of the Third International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2010). The First International Conference Bioinformatics intends to proportionate a discussion place to researchers and practitioners interested in the application of computational systems and information technologies to the field of molecular biology, including for examples the use of statistics and algorithms to understanding biological processes and systems, with a focus on new development in genome bioinformatics and computational biology. The conference is held at the Hotel Sidi Saler, Valencia, Spain, Jan. 20 to 23, 2010.
The scientific program of ICB10 included 4 keynote talks, and 113 oral presentations.
The joint conference, BIOSTEC received a total of 410 paper submissions from more than 55 countries, and 46 full papers and 113 position papers were accepted for presentation.
The program committee accepts approximately 11% and closed to 39% of full and position papers respectively, showing the intention of preserving a high quality forum for this conference. A variety of bioinformatics papers were presented at this conference and the topics include TFBS prediction, protein domain phylogenetic study, microarray data analysis, gene regulation, RNA non-standard pseudo-knot structure prediction, protein structures study, and biological pathways layout studies.
I had an oral presentation on Jan. 21 (Thur.) 12:25 p.m. Title of my presentation is “In
silico study of expression profiles correlation between microRNAs and cancerous genes”.
On the other hand, I had attended most of the talks during the four days conferences.
There are several good talks presented in this conference. Attended talks with their titles and brief contents are listed in below.
表 Y04
3
Jan. 20, 2010
Interdisciplinary Synergies in Biomedical Engineering
This session had all of the four invited speakers to give 10 minutes briefly summary of the keynote. After the presentation is a one hour opening discussion.
z Interoperate, support paradigm shift, privacy
z Too many parameters, how to interpret the results and make decision is difficult
Keynote Speech
HEALTH AND MEDICAL INFORMATION SYSTEMS - A Demanding Perspective Prof. Rui M.C. Ferreira
z Medical informatics is complicate; it involved different environments, and different staffs; such as physicians, nurses, patients, technicians, and administrators.
z Hospital department organization – from local department to central system
z Medical practice: a process of making decisions
z The usage of IT systems has not been installed as a routine in hospital to make decision, so it implies a need for a deeper changes in the personnel health system (PHS)
z Concept: (1) data collection, data integration, knowledge acquisition, (2) health/medical information system (HIS/MIS)
z Data from individual data Æ population-cohot level data Æ regional/national data
z Avoid “text free” problem by standardizing the options (list of choices)
z Similar user interface
z HIS availability – everywhere, bed side, emergence room, operation room, iphone, NB
Session 1
SEGMENTATION OF SES FOR PROTEIN STRUCTURE ANALYSIS
Virginio Cantoni, Riccardo Gatti and Luca Lombardi
z Concepts of Van de Waal surface, solvent excluded surface (SES, a concavity volume), solvent accessible surface (SAS), convex Hall (CH)
z SAS - Dilation operation from the mathematics morphology
z SES - Erosion operation from the mathematics morphology of SAS
z A propagation algorithm – joint connected areas
z To find regions of interests : tunnels and pockets by back propagation
z Practical case: first pocket is the deepest pocket
PROTEIN DOMAIN PHYLOGENIES - Information Theory and Evolutionary Dynamics
K. Hamacher
z Study protein domain superfamily (SF)
z Define information based distance
z Signal to noise is better for domain than rRNA, PNAS (2005), 102, 373
z Study selection on domain not DNA seq.
z Start from genome data Æ HMM domains Æ feature vectors
z House keeping genes evolve slower
z KL distance for two domain which is a new measure of the evolution distance
z Compute the distance of SF folds
z Compute the Jansen-Shannon distance
z Construct the NJ tree, then do a bootstrapping step
z One is able to study the evolution dynamics of every domain, somewhat similar to the Poisson process
表 Y04
4
PROTEIN FOLDING, MOLECULAR DOCKING, DRUG DESIGN - The Role of the Derivative “Drift” in Complex Systems Dynamics
Corrado Giannantoni
z Unsolvable problem – three body problem
z Intractable problem – NP hard problem
z Thermodynamics – the 4th law, Maximum Em-power principle
z The 1st and 2nd law defined the concept of exergy by Z. Rant (1955)
z The 3rd law by Nernst (1906)
z Em = Tr * Ex = transformity * (energy quantify)
z Non-conservative algebra, co-production ( 1+1 = 1+ a), interaction ( 1*1 = 1 + b), (3) feedback ( 1*1 = 1 + c)
z Ordinary differential is a functional relationship
z Incipient differential is co-instantaneous, an ordinal relationship (1) generative causality, (2) adherent logic, (3) ordinal relationship
Session 2
ReHap: AN INTEGRATED SYSTEM FOR THE HAPLOTYPE ASSEMBLY PROBLEM FROM SHOTGUN SEQUENCING DATA
Filippo Geraci and Marco Pellegrini
z Paternal and maternal type of SNP
z Better donar/recipient matching ((HLA one marrow transplant)
z Genetic association in population
z Personal genomics
z Aim: cataloguing individual SNP variation
z Pipeline: (1) sample perparatin, (2) seq. and imaging, (3) genome aligment
z 2007 Levy et al., found 3.2 M SNP
z Sequencing technology – (1) automatic Sanger sequencing, (2) Single nucleotide addition (pyroseq), 93) cycle reversible termination
z Given a set of reads determine the SNP position and the wo parenatal haplotype, (1) read length and SNP density, (2) ratio of heteroqygate/homozygoate, (3) sequencing error
z Construct a platform with 5 assemble algorithms tools available for the users, speedHap seems to perform the best
z The author presented Re-Hap, an easy-to-use AJAX based web tool that provides a complete experimental environment for comparing five different assembly algorithms under a variety of parameters setting, taking as input user generated data and/or providing several fragment-generation simulation tools.
z
http://bioalgo.iit.cnr.it/rehap/
LINEAR–TIME MATCHING OF POSITION WEIGHT MATRICES
Nikola Stojanovic
z Building a PSSM, it is an O(n*n) calculation. PSSM is not inefficient on shorter sequences, it is too expensive for whole–genome searches.
z Knuth-Morris-Pratt (KMP) algorithm
z Aho-Corasick algorithm – build a finite state automata (FSA) from a given PSSM, FSA can track several pieces of information, such as position, score and suffix.
z After the initial pre–processing of the matrix it performs in time linear to the size of the genomic segment.
z Limitation: FSA extend to multiple PSSM, reduce the time complexity of pre-processing. http://bioinformatics.uta.edu/toolkit/motifs/
Session 3
表 Y04
5
BUILDING VERY LARGE NEIGHBOUR-JOINING TREES
Martin Simonsen, Thomas Mailund and Christian N. S. Pedersen
z developed a simple branch and bound heuristic, RapidNJ, which significantly reduces the average running time
z The author presented two extensions of RapidNJ which reduce memory requirements and enable RapidNJ to infer very large phylogenetic trees efficiently.
z also present an improved search heuristic for RapidNJ which improves RapidNJ’s performance on many data sets of all sizes
Jan. 21, 2010
Session 4
STRUCTURE PREDICTION OF SIMPLE NON-STANDARD PSEUDOKNOT
Thomas K. F. Wong and S. M. Yiu
z Finding pseudoknot is an NP hard problem
z Simple standard pseudoknot join in adjacent regions
z Many classes of pseudoknot – consider a new class of pseudoknot structures, the simple non-standard pseudoknots (NSPK), cover more complicated secondary structures
z No three bps cross each other
z None of the previous algorithms can handle this class of pseudoknots. Only two of them, which run in O(m6) and O(m5)
z Perform dynamics programming, provide a prediction algorithm that runs in O(m4) time for simple non-standard pseudoknots of degree 4 which already covers all known secondary structures of RNAs in this class
z Need an energy model
z In Rfam only 7 families out of ~ 1000 families have such NSPK
REACTION KERNELS - Structured Output Prediction Approaches for Novel Enzyme Function
Katja Astikainen, Esa Pitkänen, Juho Rousu, Liisa Holm and Sándor Szedmák
z explore a structured output learning approach, where enzyme function—an enzymatic reaction—is described in fine-grained fashion with so called reaction kernels which allow interpolation and extrapolation in the output (reaction) space.
z Two structured output models are learned via Kernel Density Estimation and Maximum Margin Regression to predict enzymatic reactions from sequence motifs
PARALLEL CALCULATION OF SUBGRAPH CENSUS IN BIOLOGICAL NETWORKS
Pedro Ribeiro, Fernando Silva and Luís Lopes
z calculating the frequency of all subgraphs of a certain size, also known as the subgraph
census problem.
z The subgraph census problem is computationally hard and the author present several parallel strategies to solve this problem.
z The initial strategies were refined towards achieving an efficient and scalable adaptive parallel algorithm. This algorithm achieves almost linear speedups up to 128 cores
z MAVISTO, FANMOD, MFINDERS
Keynote Speech
MOLECULES TO DEVICES - The Role of Engineering in Next Generation Point of Care Tests
Tony Cass
z Point of Care Tests (PoCT) offer many advantages in the management of disease
z They require the discovery of new prognostic biomarkers, the engineering of molecular
表 Y04
6
receptors that recognize these markers in complex samples, the design of new, minimally invasive sampling tools and the exploitation of novel sensing modalities.
z It is their integration and overlay with informatics for decision support that will determine their ultimate adoption.
Session 5
PMSGA: A FAST DNA FRAGMENT ASSEMBLER
Juho Mäkinen, Jorma Tarhio and Sami Khuri
z DNA sequencers output fragments, the original genome must be reconstructed from these small reads
z a new fragment assembly algorithm, Pattern Matching based String Graph Assembler (PMSGA), is presented.
z The algorithm uses multipattern matching to detect overlaps and a minimum cost flow algorithm to detect repeats.
z The algorithm is faster than other assemblers. PMSGA
z The algorithm produced high quality assemblies with prokaryotic data sets
z The results for eukaryotic data are comparable with other assemblers.
A SUBSPACE METHOD FOR THE DETECTION OF TRANSCRIPTION FACTOR BINDING SITES
Erola Pairo, Santiago Marco and Alexandre Perera
z Algorithms to search binding sites are usually based on PSSM, where each position is treated independently.
z Mapping symbolical DNA to numerical sequences, a detector has been built with a PCA of the numerical sequences, taking into account covariances between positions.
z The performance on the detector depends on the estimation of missing values and the percentage of missing values considered in the model.
IN SILICO STUDY OF EXPRESSION PROFILES CORRELATION BETWEEN MICRORNAS AND CANCEROUS GENES
Ka-Lok Ng and Chia-Wei Weng
z My presentation
Session 6
CELLMICROCOSMOS 4.1 - An Interactive Approach to Integrating Spatially Localized Metabolic Networks into a Virtual 3D Cell Environment
Björn Sommer, Jörn Künsemöller, Norbert Sand, Arne Husemann,Madis Rumming and Benjamin Kormeier
z an approach which allows the creation and exploration of an abstract compartmented cell environment,
z can be used for (semi-)automatic, species- and organelle specific mapping and the comparison of metabolic data
HOLY-II: IMPROVED HIERARCHICALLY ORGANIZED LAYOUT FOR VISUALIZATION OF BIOCHEMICAL COMPLEX PATHWAYS
Jyh-Jong Tsay, Bo-Liang Wu and Guo-Gen Huang
z pathways are described as hierarchical structures in which a pathway is recursively partitioned into several sub-pathways, and organized hierarchically as a tree.
z a hierarchically organized layout algorithm HOLY which takes the advantages of the hierarchical structures inherent in complex pathways has been proposed.
z The author presented a new layout algorithm HOLY-II which follows the basic
表 Y04
7
principle of HOLY, but improves HOLY by introducing a new algorithm for joining layouts
z gives better visualization for many examples from MetaCyc, CADLIVE and HOLY.
Session 7
CONCEPTUAL MODELING OF HUMAN GENOME MUTATIONS - A Dichotomy Between what we Have and What we Should Have
M. Ángeles Pastor, Verónica Burriel Coll and Óscar Pastor
z the millions of mutations and polymorphisms that occur in human populations are potential predictors of disease
z Finding sound strategies for going from the Genotype to the Phenotype
z A systematic approach to large-scale analysis of Genotype-Phenotype correlations can be developed
z stores the current information about genome mutations, Human Gene Mutation Database
Jan. 22, 2010 Session 8
ON THE GRADIENT-BASED ALGORITHM FOR MATRIX FACTORIZATION APPLIED TO DIMENSIONALITY REDUCTION
Vladimir Nikulin and Geoffrey J. McLachlan
z better to describe the data in terms of a small number of metagenes, derived as a result of matrix factorization
z based on decomposition by parts that can reduce the dimension of expression data from thousands of genes to several factors.
z Unlike classification and regression, matrix decomposition requires no response variable and thus falls into category of unsupervised learning methods
z demonstrate the effectiveness of this approach to the supervised classification of gene expression data, i.e. the colon cancer, leukemia, lymphoma
Keynote Speech
DATABASES AND ALGORITHMS FOR PATHWAY BIOINFORMATICS
Peter D. Karp
z A family of hundreds of Pathway/Genome Databases (PGDBs) now exists for organisms with sequenced genomes
z The BioCyc PGDB collection developed by SRI contains PGDBs for more than 500 organisms
z the Pathway Tools software, which contains a large suite of algorithms for manipulating biological networks and genome data.
z Pathway Tools includes inference modules for inferring the metabolic pathways of an organism, and for predicting which genes fill missing reactions in the predicted pathways
z extensive visualization tools for individual metabolic pathways, for complete metabolic networks, and for complete regulatory networks.
z These visualization tools can also be used for analysis of omics datasets.
z Pathway Tools also contains algorithms for systems biology analyses of metabolic networks, including detection of dead-end metabolites and reachability analysis
Poster session
RECURSIVE BAYESIAN NETS FOR PREDICTION, EXPLANATION AND CONTROL IN CANCER SCIENCE - A Position Paper
Lorenzo Casini, Phyllis McKay Illari, Federica Russo and Jon Williamson
z Recursive Bayesian Net (RBN) formalism was originally developed for modelling
表 Y04
8
nested causal relationships.
z It is argue that the formalism can also be applied to modelling the hierarchical structure of physical mechanisms.
z The resulting network contains quantitative information about probabilities, as well as qualitative information about mechanistic structure and causal relations.
z RBN can be applied to all these tasks.
z RBN can be used to model mechanisms in cancer science.
z The highest level of the proposed model will contain variables at the clinical level, while a middle level will map the structure of the DNA damage response mechanism and the lowest level will contain information about gene expression
Jan. 23, 2010 Session 12
CODING BIOLOGICAL SYSTEMS IN A STOCHASTIC FRAMEWORK - The Case Study of Budding Yeast Cell Cycle
Alida Palmisano
z the process algebra language called BlenX is used to model ODEs
z characterized biological network: the cell cycle of the budding yeast.
z It is interesting to note that the experimental phenotypic characterization of some mutants cannot be explained by the deterministic solution
z so BlenX is proposed to perform a translation of the model in the stochastic framework in order to be able to verify if the inconsistencies are due to the noise that is affecting the system
z This method cannot handle the parameter estimation problem, such as the problem of reaction rate constants which have to be given as the inputs
STRUCTURAL MOTIF ENUMERATION IN TRANSCRIPTIONAL REGULATION NETWORKS
Claire Luciano and Chun-Hsi Huang
z The transcriptional regulation network of E. coli has been represented as a network and evaluated using both full enumeration and an edge sampling algorithm.
z Several significant network motifs were identified, including feedforward loops and bipartite graphs.
z both full enumeration and a different sampling algorithm, randomized enumeration are applied to the E. coli network using the newer software tool FANMOD
z Sampling identified fewer and less significant motifs than full enumeration Keynote Speech
PERSONAL HEALTH - The New Paradigm to make Sustainable the Health Care System
Vicente Traver
z focused on the chronic conditions and how they are currently managed,
z the raising costs to cope with such diseases
z the need of a new paradigm where information communication technologies and patient empowerment will be the tools to drive towards a new sustainable health system based on a patient centered approach.
三、考察參觀活動(無是項活動者省略) 無
表 Y04
9
四、建議
I had discussions with the following speakers during the ICB 2010 conference.
z K. Hamacher, Bioinformatics & Theoretical Biology Group, Technische Universit¨at
Darmstadt, Germany. [email protected]
z Phyllis McKay Illari, Philosophy, University of Kent, Canterbury, U.K.
[email protected],
z Erola Pairo, Institut de Bioenginyeria de Catalunya, Baldiri i Reixac 13, 08028,
Barcelona, Spain. Departament d’electronica, Universitat de Barcelona, Mart´ı i Franqu`es 1, 08028, Barcelona, Spain. [email protected], [email protected]
z Pedro Ribeiro, CRACS & INESC-Porto LA, Faculdade de Ciˆencias, Universidade do
Porto Rua do Campo Alegre 1021, 4169-007 Porto, Portugal. [email protected]
In summary, I had several nice discussions with some of the speakers in the conference. The level and quality of the talks are in general good.
五、攜回資料名稱及內容 資料名稱:
(1) CD for ICB 2010
(2) BIOSTEC Final Program and Book of Abstracts.
六、其他 無
無研發成果推廣資料
98 年度專題研究計畫研究成果彙整表
計畫主持人:吳家
樂
計畫編號:98-2221-E-468-013-
計畫名稱:利用生物資訊方法預測癌症相關之 microRNA 並建立生物實驗平台
量化成果項目
實際 已達 成數
(被 接受 或已 發 表)
預期 總達 成數 (含 實際 已達 成 數)
本 計 畫 實 際 貢 獻 百 分 比
單位
備註(質 化 說 明 : 如 數 個 計 畫 共 同 成 果 、 成 果 列 為 該 期 刊 之 封 面 故 事 ...等)
期刊論文 0 0 10 0%
研 究 報 告 /
技術報告 0 0 10 0%
研 討 會 論
文 3 3 10
0%
篇
1.江文欽,'利用蛋白質交互作用探討癌症相關蛋白之交互作用研 究',第十八屆細胞及分子生物新知研討會。墾丁,2010/1/20~22 2.王傑瑋,'題目:探討由選擇性剪接造成蛋白質域的變化與肝癌 之 關 係 ' , 第 十 八 屆 細 胞 及 分 子 生 物 新 知 研 討 會 。 墾 丁 , 2010/1/20~22
3. 薛一勤,'辨識人類、黑猩猩、老鼠及大鼠的 MicroRNA 核心 啟動子',ITIA 2010 資訊技術與產業應用研討會。北台灣科學技 術學院資訊管理系,2010/6/4
論文 著作
專書 0 0 10
0%
申 請 中 件
數 0 0 10
專利 0%
已 獲 得 件
數 0 0 10
0%
件
件數 0 0 10
0% 件 技術
移轉 權利金 0 0 10 0%
千 元
碩士生 5 5 10
0%
博士生 0 0 10
0%
博 士 後 研
究員 0 0 10
0%
國 內
參與 計畫 人力
(本 國籍)
專任助理 0 0 10 0%
人 次
期刊論文 3 2 10 0%
1. K.L. Ng; C.H. Huang, M.C. Tsai (2010). Vertebrate microRNA genes and CpG-islands. WSEAS Transactions on Biology and BioMedicine 3(7), 73-81
http://www.worldses.org/journals/biology/biology-2010.htm 2. H.J. Shiu; K.L. Ng; J.F. Fang; R. C.T. Lee; C.H. Huang (2010). Information Sciences , 180(11), 2196-2208.
IF =3.095, Rank: Computer Science, Information systems, 6/116=5.2%.
3. Ka-Lok Ng, Jin-Shuei Ciou, Chien-Hung Huang (2010).
Computers in Biology and Medicine, 40, 300-305.
研 究 報 告 /
技術報告 0 0 10 0%
研 討 會 論
文 3 3 10
0%
篇
Ka-Lok Ng and Chia-Wei Weng (2010). In silico study of expression profiles correlation between microRNAs and cancerous genes. Bioinformatics 2010, Jan. 20-23, 2010, Valencia, Spain (oral presentation.
謝蕙霞 (2010). Ganoderma tsugae extracts induce cell cycle arrest and apoptosis in lung cancer cells. 35th FEBS congress molecules of life, Gothenburg, Sweden, June 26 – July 1, 2010. B2. 37
謝蕙霞 (2010). A whole genome microarray approach to signal patheway of growth inhibition effects of Ganoderma tsugae extracts on lung cancer cells. 35th FEBS congress molecules of life, Gothenburg, Sweden, June 26 – July 1, 2010. B2.38 論文
著作
專書 0 0 10
0%
章 / 本 申 請 中 件
數 0 0 10
專利 0%
已 獲 得 件
數 0 0 10
0%
件
件數 0 0 10
0% 件 技術
移轉 權利金 0 0 10 0%
千 元
碩士生 0 0 10
0%
博士生 0 0 10
0%
博 士 後 研
究員 0 0 10
0%
國 外
參與 計畫 人力
(外 國籍)
專任助理 0 0 10 0%
人 次
其他成 果 (無 法 以 量 化 表 達 之 成 果 如 辦 理 學 術 活動、獲
得 獎
項、重要 國 際 合 作、研究 成 果 國 際 影 響 力 及 其 他 協 助 產 業 技 術 發 展 之 具 體 效 益 事 項等,請 以 文 字 敘 述 填 列。)
本計畫共發表三篇論文,其中兩篇為 SCI 論文,另一篇為具有審查機制之國外期刊。
參予本計畫之學生共五位,均在國內外之研討會發表論文。
成果項目 量化 名稱或內容性質簡述
測驗工具(含質性與
量性) 0
課程/模組 0 電腦及網路系統或
工具 0
教材 0
舉辦之活動/競賽 0 研討會/工作坊 0 電子報、網站 0 科
教 處 計 畫 加 填 項 目
計畫成果推廣之參 與(閱聽)人數 0
國科會補助專題研究計畫成果報告自評表
請就研究內容與原計畫相符程度、達成預期目標情況、研究成果之學術或應用價 值(簡要敘述成果所代表之意義、價值、影響或進一步發展之可能性)、是否適 合在學術期刊發表或申請專利、主要發現或其他有關價值等,作一綜合評估。
1. 請就研究內容與原計畫相符程度、達成預期目標情況作一綜合評估
■達成目標
□未達成目標(請說明,以 100 字為限)
□實驗失敗
□因故實驗中斷
□其他原因 說明:
2. 研究成果在學術期刊發表或申請專利等情形:
論文:■已發表 □未發表之文稿 □撰寫中 □無 專利:□已獲得 □申請中 ■無
技轉:□已技轉 □洽談中 ■無 其他:(以 100 字為限)
三篇論文
Ng et al (2010) WSEAS Tran on Biol. and BioMed 3(7),73 Shiu et al (2010) Info Sci. 180, 2196
Ng et al (2010) Comput. Biol and Med, 40,300
3. 請依學術成就、技術創新、社會影響等方面,評估研究成果之學術或應用價 值(簡要敘述成果所代表之意義、價值、影響或進一步發展之可能性)(以 500 字為限)
對於學術研究之貢獻為探討 miRNA 標靶致癌及抑癌基因之可能性,