新型聚苯并咪唑之開發與特性及在 高溫質子交換膜燃料電池之應用
實務專題報告書
指導教授:陳志堅 研究生:蕭智中
系所:材料科學與工程系
學號:B10004110
1
目錄
目錄 ... 1
一、摘要 ... 2
二、前言 ... 2
三、研究動機與目的 ... 5
四、實驗方法 ... 5
4.1 新型四胺單體之合成 ... 5
4.2 新型聚苯并咪唑合成 ... 6
4.3 聚苯并咪唑薄膜之製備及磷酸摻雜方法 ... 7
4.4 膜電極組(Membrance electrode assembly, MEA)之製備 ... 7
五、結果與討論 ... 8
5.1 新型四胺單體之合成與性質表徵 ... 8
5.2 新型聚苯并咪唑之合成與性質表徵 ... 11
5.3 熱穩定性測試結果 ... 12
5.4 氧化穩定性測試 ... 13
5.5 聚苯并咪唑磷酸摻雜結果 ... 14
5.6 質子傳導率測試 ... 16
5.7 質子交換膜燃料電池測試 ... 17
六、結論 ... 19
七、參考文獻 ... 19
2
一、摘要
本 研 究 已 成 功 合 成 新 型 四 胺 單 體 2,2'-(3,5-Bis(trifluoromethyl)benzyl) - 4,4',5,5'-tetraaminodiphenyl ether(6)。其中反應涉及醯胺化、硝化、鈴木偶合(Suzuki Coupling)、水解以及還原反應。單體(6)之氫譜在化學位移為 4.50、5.04、6.24、
6.79、7.86 及 7.93 (ppm)處為單重峰。質譜測得其分子離子峰為 654.3 [M]+。熔點 介於 96~98 ℃。單體(6)與 4, 4’-oxybis(benzoic acid)單體(7)以 Eaton’s reagent(10 wt%)為溶劑可成功聚合出新型聚苯并咪唑 P8,其黏度為 1.11 dL/g。並且具有良 好之熱性質,10 wt%重量損失之熱裂解溫度為 445 ℃,及玻璃轉移溫度大於 450
℃。此高分子在 60 ℃下可溶於 DMAc, NMP 等有機溶劑並塗佈成可撓性之薄膜。
此薄膜利用Fenton’s test 進行氧化穩定性測試,經過 216 小時,薄膜重量損失為 4.4 %,呈現良好氧化穩定性。薄膜於常溫下浸泡於不同濃度磷酸水溶液進行磷 酸摻雜,所得之飽和磷酸摻雜量介於 70 %~200 %間。磷酸摻雜量為 199 %之 P8 薄膜在 140 及 160 ℃下質子傳導率分別為 4.33×10-2及 5.34×10-2 S cm-1。在單電 池測試方面,160 ℃下測得最高電功率密度為 475 mW/cm2。
二、前言
近年來,由於石油價格波動頻繁,為了降低對於石油的依賴,替代能源的開 發與相關研究也日益漸增,例如:太陽能電池(Solar cell)與燃料電池(Fuel cell)等。
其中,質子交換膜燃料電池(Proton Exchange Membrane Fuel Cell, PEMFC)除了具 有低汙染、能量轉換效率高等一般燃料電池的特點外,還具備可體積微小化、接 近常溫操作與啟動快速的特性,具有可應用於運輸工具及可移動電子設備的潛力。
Figure 1. 質子交換膜燃料電池示意圖
Figure 1 為 PEMFC 示意圖。中間為質子導電聚合物膜作為電解質分隔陽 極和陰極。氫氣由陽極流入經由氧化反應分離成質子和電子,電子與質子分別 經由外電路與質子交換膜導入陰極,並於陰極使氧氣經還原反應成水,形成完 整電路,多餘反應物與水則由電極出口排放出。
3
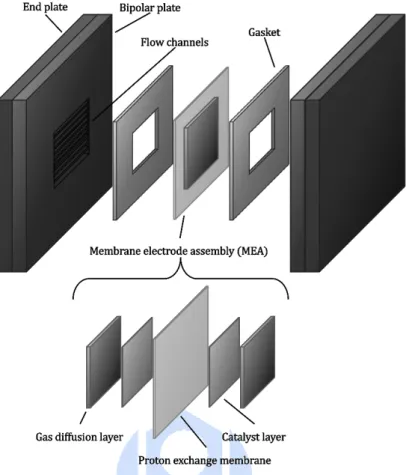
Figure 2. PEMFC 之單電池結構圖
Figure 2 為 PEMFC 之單電池結構圖。其基本結構以一個核心膜電極組向外 堆疊成,由內向外依序是膜電極組(Membrane electrode assembly, MEA)、密封墊 片、雙極板與端板,其中電池效能主要受膜電極組影響。質子交換膜燃料電池各 個結構功能如下所述。
(1) 膜電極組由質子交換膜、觸媒層以及氣體擴散層所組成:質子交換膜為可傳 遞質子之聚合物製備而成,作為 PEMFC 之固態電解質,可以將質子從陽極 傳遞到陰極,同時阻絕兩端電極之氣體。觸媒層則為含有附於碳微粒上之鉑 金屬及電解質,同時作為催化劑以及電極,催化氣體進行氧化、還原反應。
氣體擴散層為多孔材料如碳布,使氣體能夠滲透至觸媒層進行作用。
(2) 雙極板:其中一側置有使反應氣體均勻分佈的流道,確保電池堆的溫度均勻 分佈和達到散熱效果,亦作為導線用途串連電池形成電池堆。在單電池測試 中雙極板僅具有疏導氣體作用,因此又稱為流場板。
(3) 密封墊片:密封住膜電極組與雙極板之間隙以防止氣體外洩。
(4) 端板:作為電池支架固定整體電池結構。
PEMFC 根據操作溫度又可細分為低溫型(≦80 ℃)與高溫型(100 ℃~200 ℃) 兩種。目前應用於低溫型 PEMFC 中商業化的質子交換膜是以美國杜邦(Du pont) 公司開發之聚全氟磺酸膜(Polyperfluorosulfonicacid)又稱 Nafion 為主。然而,
Nafion 與其它聚全氟磺酸膜有五個主要缺陷:(1)造價昂貴,(2)含氟材料對環境
4
衝擊較大,(3)應用於直接甲醇燃料電池(DMFC)時,於較高的操作溫度下會出現 嚴重的甲醇穿透現象(Methanol crossover),(4)操作溫度必須在 80 ℃以下這些缺 陷大大限制了 Nafion 的發展性。許多研究皆指出更高的工作溫度(100 ℃~200 ℃) 將更有利於 PEMFC 的實際操作。
高溫型PEMFC(HT-PEMFC)則是以磷酸當作質子傳導的媒介。聚并咪唑 (Polybenzimidazoles, PBI)本身因具有優良的熱穩定性、化學穩定性與機械性質,
且與磷酸摻雜後在高溫下具有高質子傳導率,而被廣泛使用在燃料電池上。
由於PBI具有剛硬的全芳香族結構與分子鏈間氫鍵,因此在室溫下是相當難 溶於一般的極性非質子溶劑。有三種常見的方式可達到提升PBI溶解度之目的。
(1) 高分子改質1:以不同的官能基置換imidazole雜環氮上的氫原子,藉此降低高 分子鏈間氫鍵,如Scheme 1所示。
Scheme 1. Structure modification of PBI.
(2) 導入巨大側基:Kim等人2在p-PBI側鏈上導入巨大的benz imidazole基團,如 Scheme 2所示。相較於m-PBI,由於分子鏈間的自由體積增加,使BIpPBI可在室 溫下溶於一般極性非質子溶劑中。
Scheme 2. Synthetic route for BIpPBI.
(3) 導入柔軟鏈段3,4:高分子主鏈上導入不同的柔軟鏈段,如Figure 1所示,藉 此增加分子鏈柔性及減少分子鏈段的規則排列,藉此提升PBI之溶解度。
Figure 3. Various structure of PBI with flexible linkage.
PBI 薄膜之質子傳導率(Acid conductivity)主要受到磷酸摻雜率(Acid doping level, ADL)影響5, 6,ADL 定義為平均每莫耳重複單元上磷酸分子莫耳數含量。
ADL 越高 PBI 薄膜之質子傳導率越佳,但高磷酸摻雜量也降低分子鏈間作用力 使機械強度明顯下降。根據研究指出,聚并咪唑摻雜磷酸後,磷酸分子會以兩種 型式留存在聚并咪唑結構中:苯并咪唑雜環上三級氮原子的孤電子對會攻擊磷酸 (H3PO4)上的質子成為亞磷酸根(H2PO4-)7 離子,這些亞磷酸根離子固定於苯并咪 唑雜環形成為(H2PO4--NH+)共同離子對,而未形成離子對之磷酸分子則以氫鍵
5
與亞磷酸根及磷酸相結合,儲存於聚并咪唑分子鏈間自由體積中。Ma 等人以 m- PBI 摻雜磷酸實驗,提出兩種質子傳導機制8,如 Figure 4 所示。當磷酸摻雜率 (ADL)≦2 時,質子傳導主要透過分子鏈間質子化及非質子化之氮上(H2PO4-- NH+),而當(ADL)≧4 時,高分子自由體積間存在許多自由磷酸,質子交換主要 發 生 在 磷 酸 - 亞 磷 酸 根 (H3PO4…...H2PO4-) 以 及 磷 酸 - 磷 酸 分 子 (H3PO4…...
H2PO4-)之間進行跳躍傳導。此結果證實高質子傳導率主要藉由磷酸間的質子遷 移所致。
Figure 4. 質子傳導機制示意圖
三、研究動機與目的
目前已經有許多新型結構之PBI被合成出來,但大部分的新型PBI都是以聯 苯四胺(TAB)與不同結構之二酸單體進行聚縮合反應合成而得,極少關於新型四 胺單體的合成及其新型PBI之報導。由於四胺單體之合成路徑較為複雜,且須將 四胺單體儲放在鈍氣環境甚至低溫才可維持其安定性。因此,在四胺單體之選擇 性不多的前提下,我們認為開發新型四胺單體將成為深具價值的研究方向,對於 新型PBI之開發將有極大幫助。
四、實驗方法
4.1 新型四胺單體之合成
4,4'-Diacetamido-2,2'-dibromodiphenyl ether(2)
將醋酸酐 200 ml 加入三頸瓶,以磁石攪拌均勻再加入粉狀 2,2'-dibromo-4,4'- dioxydianiline(1)9 5.00 g(13.97 mmol),室溫下反應 24 小時。直接過濾並水洗至中 性,收集沉澱物並以 100 ℃真空烘乾。得到 5.99 g 化合物(2) (產率 97 %)。mp:
243~246 ℃, 1H NMR (500 MHz, DMSO-d6, δ, ppm) 2.04 (s, 6H), 6.86 (d, J=7.35 Hz, 2H), 7.44 (dd, J1=7.35 Hz, J2=2.05 Hz, 2H), 8.07 (d, J=2.05 Hz, 2H), 10.08 (s,2H).
EIMS (m/z):calcd for C16H14Br2N2O3, 439.9; found, 439.9 [M]+ 4,4'-Diacetamido-2,2'-dibromo-5,5'-dinitrodiphenyl ether(3)
將化合物(2) 7.04 g(15.95 mmol)、濃硫酸 22 ml 加入三頸瓶,室溫下攪拌至 全溶,接著冰浴 30 分鐘後,以加藥漏斗緩慢加入硝酸 2ml,於 0~5 ℃下反應 2
6
小時後再於室溫下反應 2 小時。最後將溶液倒入冰水中攪拌,過濾並水洗至中 性,收集沉澱物並以 100 ℃真空烘乾。得到 7.89 g 化合物(3) (產率 93 %)。mp:
260~265 ℃, 1H NMR (500 MHz, DMSO-d6, δ, ppm):2.08 (s, 6H), 7.66 (s, 2H), 8.09 (s, 2H), 10.31 (s, 2H). EIMS (m/z):calcd for C16H12Br2N4O7, 529.9; found, 529.9 [M]+. 4,4'-Diacetamido-2,2'-(3,5-bis(trifluoromethyl)benzyl)-5,5'-dinitrodiphenyl ether (4)
將化合物(3) 4.00 g(15.03 mmol)、甲苯 200 ml 加入有氮氣導入/出之三頸瓶,
攪拌均勻並加熱至 60 ℃,加入碳酸鈉水溶液(碳酸鈉 3.50 g(33.02 mmol)/水 35 ml)、Tetrakis(triphenylphosphine)palladium(0)0.55 g,升溫至 80 ℃反應 1 小時,
接 著 加 入 硼 酸 乙 醇 溶 液 ( 硼 酸 3,5-Bis(trifluoromethyl)benzeneboronic acid 5.00 g(19.39 mmol)/乙醇 40 ml),反應三天。溶液降至室溫後以矽藻土過濾催化劑,收 集濾液並以 500 ml 飽和食鹽水水洗,收集有機層以無水硫酸鎂除水,利用減壓 濃縮機抽乾溶劑,以二氯甲烷作為沖提液進行液相層析分離法純化,得到 2.28 g 化合物(4)(產率 38 %)。mp:234~235 ℃,1H NMR (500 MHz, DMSO-d6, δ, ppm):
2.10 (s, 6H), 7.81 (s, 2H), 7.89 (s, 2H), 8.09 (s, 4H), 8.17 (s, 2H), 10.36(s, 2H). EIMS (m/z):calcd for C32H18F12N4O7, 798.1; found, 798.0 [M]+
2,2'-(3,5-Bis(trifluoromethyl)benzyl)-5,5'-dinitro-4,4'-diaminodiphenyl ether(5) 將化合物(4)9.10 g(11.42 mmol)、甲醇 40 ml、氫氧化鉀溶液(氫氧化鉀 2.16 g/
甲醇 10 ml)、氫氧化鉀 1.20 g 加入錐形瓶,加熱至 85 ℃迴流 6 小時。溶液降至 室溫後倒入水中攪拌,收集沉澱物並水洗至中性,以 100 ℃真空烘乾。將粗產物 以乙酸乙酯:己烷=3:5 作為沖提液進行液相層析分離法純化,得到 7.74 g 化合 物(5)(產率 95 %)。mp:110~112 ℃, 1H NMR (500 MHz, DMSO-d6, δ, ppm):7.26 (s, 2H), 7.46 (s, 4H), 7.74 (s, 2H), 8.00 (s, 4H), 8.16 (s, 2H). EIMS (m/z):calcd for C28H14F12N4O5, 714.1; found, 714.1[M]+
2,2'-(3,5-Bis(trifluoromethyl)benzyl)-4,4',5,5'-tetraaminodiphenyl ether(6)
將化合物(5) 3.23 g(4.52mmol)、乙酸乙酯 24 ml、Ammonium formate 3.40 g、
乙醇 24 ml、10 wt% Pd/C 0.4 g 依序加入錐形瓶,反應 24 小時。以矽藻土過濾催 化劑,水洗濾液後收集有機層,以無水硫酸鎂除水,利用減壓濃縮機抽乾溶劑,
以 100 ℃真空烘乾。得到 2.80 g 單體(6)(產率 95 %)。mp:96~98 ℃, 1H NMR (500 MHz, DMSO-d6, δ, ppm):4.50 (s, 4H),5.04 (s, 4H), 6.24 (s, 2H), 6.79 (s, 2H), 7.86(s, 2H), 7.93 (s, 4H). EIMS (m/z) : calcd for C28H18F12N4O, 654.1; found, 654.3[M]+.
4.2 新型聚苯并咪唑合成
將單體(6)0.5616 g(0.8581 mmol)及 Eaton’s reagent(10 wt%) 7.87 ml 加入有氮 氣導入/出之三頸瓶,機械攪拌均勻並加熱至 90 ℃。待單體(6)全溶後將單體(7) 0.2216 g(0.8581 mmol)加入,升溫至 110 ℃。待單體(7)全溶後升溫至 120 ℃,反 應 10 小時。接著 15 分鐘內升溫至 130 ℃,反應 3 小時。同樣 15 分鐘內升溫至 140 ℃,反應 6 小時。當高分子黏度有明顯提升後倒入水中沉澱成絲,收集沉澱
7
物過濾並水洗至中性,以乙醇索氏萃取 24 小時後再以 100 ℃真空烘乾。得到得 到 P8 0.6996 g(產率 93 %)。
4.3 聚苯并咪唑薄膜之製備及磷酸摻雜方法
將 P8 與 NMP 配置成 3~5 %(w/v)之聚苯并咪唑溶液,經離心機以轉速 6000 rpm 離心 1 小時後,收集上層澄清聚苯并咪唑溶液,並均勻塗佈於玻璃板上,並 在氮氣環境下以 60 ℃烘烤 6 小時後降溫。接著將降至室溫之玻璃板浸泡於水中 以 80 ℃水煮二小時,最後將薄膜在 200 ℃下重壓並真空烘乾得到平整薄膜。
將薄膜裁切成適當尺寸後先秤其乾膜重(Wdry),接著將薄膜分別浸泡於 85
%、80 %及 75 %磷酸水溶液中,經過固定時間之磷酸摻雜後將薄膜取出,以拭 淨紙擦拭掉薄膜表面磷酸,再以 100 ℃下真空烘乾後秤其濕膜重(Wwet)。磷酸 摻雜量(Phosphoric acid content, PA content)定義為摻雜磷酸重量與乾膜重量之百 分比,以下列公式表示:
PA content=(Wwet-Wdry)/Wdry×100%-Eq.(1)
4.4 膜電極組(Membrance electrode assembly, MEA)之製備
以 Pt/C(40 % on carbon black, Johnson Matthey Company)、聚苯并咪唑與 DMAc 之比例為 20:1:150 (w/w/μl)10配置成觸媒與聚苯咪唑之混合溶液,攪拌 均勻後,將溶液以刷毛均勻塗佈在碳布上,再以 70 ℃下真空烘乾。計算 Pt 在陰 極及陽極之含量分別為 0.8 mg cm-2以及 0.6 mg cm-2。最後將碳布浸泡於 10 %磷 酸水溶液中即完成電極之製作。
在進行燃料電池測試前,將電極從磷酸水溶液中取出,以拭淨紙擦拭掉表面 大部分之磷酸後,再依流場板/密封墊片/陽極/質子交換模/陰極/密封墊片/流場板 之順序堆疊成單電池,接著將完成知單電池至於端板間,以氣壓式單電池治具 (ASCT-5, Asia Pacific Fuel Cell Technologies, Ltd)將電池固定即完成膜電極之製 作。成品如 Figure 5 所示。
Figure 5. 膜電極(MEA)成品示意圖
8
五、結果與討論
5.1 新型四胺單體之合成與性質表徵
本研究參考合成 3,3',4,4'-tetraaminodiphenyl ether(TADE)之相關文獻11,並以 2,2'-Dibromo-4,4'-dioxydianiline (1)9 為起始物,經過五個步驟製備成 2,2'-(3,5- Bis(trifluoromethyl)benzyl)-4,4',5,5'-tetraaminodiphenyl ether(6)。反應包括醯胺化反 應、硝化、鈴木偶合(Suzuki Coupling)、水解以及還原反應,成功合成新型四胺 單體(6)如 Scheme 3 所示。
MeOH KOH
O Br H2N
Br
NH2 Acetic anhydride
O Br NH
Br
HN C CH3 O C
H3C O
O Br NH
O2N Br NO2
HN C CH3 O C
H3C O O
NH O2N
NO2 HN
F3C
CF3
C CH3 O C
H3C O
HNO3 H2SO4
Suzuki coupling Toluene F3C
CF3
1 2
4 3
O H2N
O2N
NO2 NH2
F3C
CF3 F3C
H2N O H2N
NH2 NH2
F3C
CF3 CF3 F3C
Pd/c Ammonium formate
CF3
5 6
yield:97 %
yield:38 % yield:93 %
yield:95 % yield:95 %
Scheme 3. 四胺單體合成流程圖
將化合物(1)與醋酸酐在室溫下反應得到化合物(2),其氫核磁共振光譜如 Figure 6 所示,在化學位移 δ 6.86、7.44、8.07 及 10.08 ppm 分別為 Hd、Hc、Hb
及 Ha。Hd與 Hc偶合,其偶合常數 J=7.35 Hz,Hc與 Hb偶合,其偶合常數 J=2.05 Hz。由積分值 3 可判斷 δ 2.04 ppm 處之單重峰為 He。經由氫核磁共振光譜判斷 此結構與目標化合物相符。同時以質譜測得此化合物分子離子峰為 439.9 [M]+, 與預期相符。化合物(2)之熔點為 243~246 ℃。
此反應將胺基(-NH2)替換成醯胺基(-NHCOCH3)使得 5,5'處較 6,6'處陰電性更
9
強,使得後續之硝化親電子取代反應傾向取代 5,5'位置之氫,可減少副反應發生。
Figure 6. 化合物(2)氫核磁共振光譜
化合物(2)經硝化反應得到化合物(3),其氫核磁共振光譜如 Figure 7 所示,
在δ 7.66、8.09 及 10.31 ppm 分別為 Hc, Hb與 Ha。由積分值 3 可判斷 δ 2.08 ppm 處之單重峰為 Hd。經由質譜測得此化合物分子離子峰為 529.9 [M]+,與預期相 符。化合物(3)之熔點為 260~265 ℃。
Figure 7. 化合物(3)氫核磁共振光譜
化合物(3)經鈴木偶合反應得到化合物(4)初產物,經過液相層析法純化後以 氫核磁共振光譜檢測,其氫核磁共振光譜如 Figure 8 所示,在 δ 7.81、8.09 及 10.36 ppm 分別為 He, Hc與 Ha。由積分值 2 可判斷 δ 7.89 及 8.17 ppm 處之單重 峰為 Hd與 Hb。經由質譜測得此化合物分子離子峰為 798.0 [M]+,與預期相符。
化合物(4)之熔點為 234~235 ℃。
10
Figure 8. 化合物(4)氫核磁共振光譜
化合物(4)經水解反應得到化合物(5),其氫核磁共振光譜如 Figure 9 所示,
在δ 7.46、7.74 及 8.00 ppm 分別為 Hd, Hc與 Hb。由積分值 2 可判斷 δ 7.26 及 8.16 ppm 處之單重峰為 He與 Ha。經由質譜測得此化合物分子離子峰為 714.1 [M]+, 與預期相符。化合物(5)之熔點為 110~112 ℃。
Figure 9. 化合物(5)氫核磁共振光譜
化合物(5)經還原反應得到單體(6)。其氫核磁共振光譜如 Figure 10 所示,在 δ6.24、6.79 及 7.86 ppm 分別為, Hd、Hc與 Hb。由積分值 2 可判斷 4.50、5.04 及 7.93 ppm 處之單重峰為 Hf、He與 Ha。經由質譜測得此化合物分子離子峰為 654.3 [M]+,與預期相符。經由以上數據判斷單體(6)已經被成功合成。
11
Figure 10. 單體(6)氫核磁共振光譜
5.2 新型聚苯并咪唑之合成與性質表徵
本研究使用Eaton’s reagent (10 wt%)做為聚合溶劑,在 140 ℃下完成 P8 之 聚合,另外我們也以聚磷酸作為溶劑成功聚合聚苯并咪唑 P9,P8 及 P9 之結構 如 Scheme 4 所示。由於單體(6)在 170 ℃以上會有昇華現象,因此無法以聚磷 酸作為聚合溶劑,我們選用Eaton’s reagent 除了此溶劑流動性較佳,主要是因為 此溶劑對於單體及聚合物溶解度較佳。聚合之單體濃度控制約 0.3 mmol/ml。待 單體完全溶解後,先在 120 ℃下反應一小時,此目的為使高分子鏈能均勻成長,
否則在 140 ℃下反應造成局部分子鏈成長太快以致於短時間內溶液黏度迅速提 升,造成凝膠現象(Gelation)。在較低溫進行聚合可延緩反應速度之現象則可由 Ueda 等人針對 Eaton’sreagent 聚合苯并咪唑之研究證實12。
Eaton's reagent
O
F3C
CF3 CF3 F3C
N HN
O N
NH
n O
H2N
H2N
NH2
NH2
F3C
CF3 CF3 F3C
6
O
COOH COOH
7 P8
PPA
O N
H
N O
N NH
n O
H2N
H2N
NH2
NH2 O
COOH COOH
P9
Scheme 4 高分子合成流程圖
12
Table1. 高分子固有黏度(ŋinh)a與溶解度b測試
Polymer ŋinh(dL g-1) Solventc
DMAc DMF NMP DMSO H2SO4 MSA
P8 1.11 ++d + ++ - ++ ++
P9 1.13 +- +- +- +- ++ ++
a. Measured at a polymer concentration of 0.2 g/dL in methanesulfonic acid at 35
℃.
b. The solubility was determined by using 30 mg of sample in 1 mL of stirred solvent.
c. DMAc : N,N-dimethylacetamide; NMP : N-methyl-2-pyrrolidone; DMSO : dimethyl sulfoxide; DMF:N,N-dimethylformamide; MSA:Methanesulfonic acid.
d. ++ : soluble at 60 ℃; + : soluble at 80 ℃; +- : soluble at 100 ℃; - : partial soluble or insoluble at 100 ℃.
Table2. 高分子之固有黏度及分子量
Polymer ŋinha(dL g-1) Mnb×10-4(Da) Mwb×10-4(Da) PDIc
P8 1.11 8.81 22.47 2.55
P9 1.13 11.13 25.11 2.26
The solubility was determined by using 30 mg of sample in 1 mL of stirred solvent.
a. Measured at a polymer concentration of 0.2 g/dL in methanesulfonic acid at 35 ℃.
b. By GPC in DMAc (relative to polystyrene standards).
c. Mw/Mn
由 Table 1 可得知 P8 與 P9 在相近之固有黏度下,P8 於 DMAc、DMF 及 NMP 具有較佳之溶解度。因此可證實在導入 3,5-Bis(trifluoromethyl)benzyl 基團可以增 加高分子鏈間之自由體積,分子鏈段不易排列,進而有效提升 PBI 之溶解度。
Table 2 則為以凝膠滲透層析儀(GPC)測得分子量之結果。新型聚苯并咪唑 P8 之數目平均分子量(Mn)為 88093 Da,此分子量之 PBI 已足夠塗佈成具可撓性之 薄膜。
5.3 熱穩定性測試結果
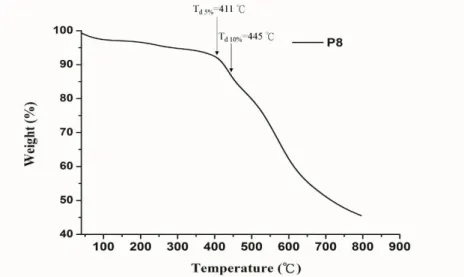
HT-PEMFC 之工作溫度介於 100 ℃至 200 ℃間,因此薄膜必須有良好熱穩 定性質,包含熱裂解溫度(Td)以及玻璃軟化溫度(Tg)。熱穩定性測試方式如下,將 烘乾之高分子置於熱重分析儀(TA TGA Q500)後維持在氮氣環境,以每分鐘 10
℃升溫至 800 ℃為止,所得到的熱重分析圖如 Figure 11 所示。從圖中我們可以 觀察到 100 ℃以前有些微重量損失,推論是水分所致,由於苯并咪唑為吸水性 官能基團,即使經過真空烘乾仍會吸收空氣中水氣。而在 100 ℃介於 350 ℃以 前則無明顯重量損失,顯示出良好熱穩定性,P8 其 5 %即 10 %重量損失溫度分 別為 411 ℃及 445 ℃。另一方面,我們以熱機械分析法(Thermo-mechanical
13
analysis, TMA)測量 P8 之 Tg,然而在 450 ℃以前均無法測得其值,推測由於巨 大側基造成立體障礙較高所致。
Figure 11. 熱重分析圖 Thermogravimetric analysis, TGA
5.4 氧化穩定性測試
在 HT-PEMFC 電化學反應過程中,氧氣在陰極若反應不完全成,將形成過 氧化氫(H2O2)分子13,而過氧化氫在觸媒作用下會分解成.OH 與.OOH 自由基
14-16,自由基可能攻擊高分子鏈上氫原子造成分子鏈降解,許多研究已經將
Fenton’s test 視為檢視質子交換膜氧化穩定性之測試方法。Fenton’s test 測試方法 如下:配置出含 4 ppm Fe2+之 3 %過氧化氫水溶液,將裁切好之薄膜先秤得薄膜 重(Wi),接著浸泡於過氧化氫水溶液中,升溫至 68 ℃,每間隔 24 小時將薄膜取 出並以去離子水洗滌數次後,再以 100 ℃真空乾燥,秤得薄膜重(Wf),重複此流 程達到 216 小時以上。重量損失百分比(Wloss)以 Eq.(2)表示:
Wloss=(Wi -Wf)/ Wi×100%-Eq.(2)
實驗結果如 Figure 12 所示,可得知在超過 200 小時氧化穩定性測試後,P8 薄膜僅有 4.4 wt%的重量損失,而數目平均分子量為 33000 Da 之 m-PBI 的重量 損失則為 23 wt%。此外根據文獻值17指出,固有黏度為 1.51 及 0.63 dLg-1之 m- PBI 在相同條件測試下,200 小時後重量損失為 19 wt %及 27 wt %,以上數據皆 顯示出 P8 相較 m-PBI 有較良好的氧化穩定性。
14
Figure 12. P8 與 m-PBI 薄膜氧化穩定性
5.5 聚苯并咪唑磷酸摻雜結果
將 P8 薄膜依照 4.3 提及之方法進行磷酸摻雜,分別於 7、15、39、63、87 小 時候,取出薄膜並以 Eq.(1)求得不同時間下之磷酸摻雜量。結果如 Table 3 及 Figure 13 所示。從 Figure 13 中我們可以觀察到,在 0~7 以及 7~15 小時這兩個 區段內,浸泡較高濃度磷酸可以得到較高之磷酸摻雜。在 15 小時之後磷酸摻雜 速率減慢,甚至在 39 小時候幾乎無明顯增加,說明已經接近飽和點。
Table 3.新型聚苯并咪唑之磷酸摻結果。
Time (hr)
PA Contenta (%)
85 % b 80 % 75 % 60 %
7 110a 97 75 61
15 160 135 97 63
39 185 147 123 65
63 195 159 131 69
87 199 161 136 70
a. PA content was caucalated from Eq. (1).
b. Concentration of phosphoric acid solution.
15
Figure 13. P8 薄膜磷酸摻雜結果
為了將新 P8 薄膜應用在燃料電池上,我們將不同磷酸摻雜薄膜進行機械強 度測試,結果如 Figure 14 所示。P8 薄膜斷裂強度介於 8~11 MPa,並隨著磷酸 摻雜量增加其機械強度降低,但即使酸摻雜量為 199 %之薄膜仍能維持 8.1 MPa 之斷裂強度以及 175 %之體積膨脹率。此結果證實磷酸摻雜量為 199 %之 P8 薄 膜具備足夠機械強度及尺寸安定性,可做為高溫 PEMFC 之質子交換膜,我們更 進一步將此薄膜作後續單電池測試。
Figure 14. P8 薄膜之機械強度之機械強度測試
16
Table 4. P8 薄膜之機械強度a及尺寸安定性。
PA conc.
(%)
P8 Strength
(MPa)
Strain (%)
S.V.
(%)
0 80.3 23.7 0
75 11.2 29.3 142
80 10.3 33.8 154
85 8.1 39.6 175
a. Measurements were performed with a constant cross-head speed of 5 mm min-1 under ambient atmosphere.
b. The tensile strength at break of membranes.
c. The tensile strain at break of membranes.
d. Swelling volume ratio of membranes after PA doping.
5.6 質子傳導率測試
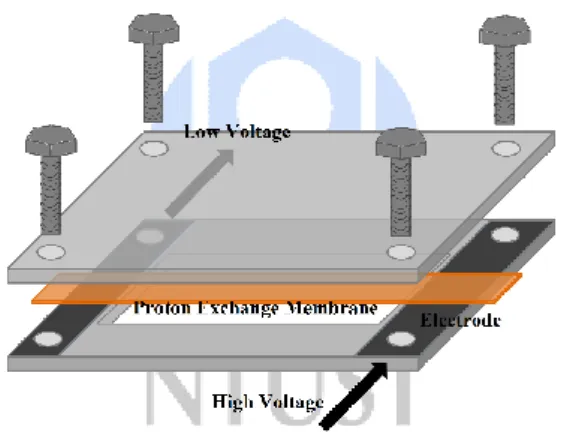
Figure 15. 兩點式電化學阻抗量測示意圖
P8 薄膜在完成磷酸摻雜後即可進行質子傳導率測試。以兩點式電化學阻抗 儀(Electrochemical impedance spectroscopy, EIS)量測薄膜之質子傳導阻抗,將薄 膜固定於電極上,如 Figure 15 所示。首先,為除去薄膜內之水分,升溫至 120
℃維持 30 分鐘,接著降至 60 ℃以後即可進行阻抗量測。兩端電極分別接上高 電位與低電位,並以交流電導通。藉由變化電流頻率量測質子傳導阻抗,每提 高 20 ℃時測得一次阻抗值,並於 160 ℃時完成測量,最後得到不同磷酸摻雜 量的薄膜其質子傳導率與溫度之對應關係如 Figure16 所示。質子傳導率則以 Eq.(3)計算而得,σ 為質子傳導率,L 為電極之間距離並固定為 1.1 cm,A 為質 子交換膜之截面積,R 為 EIS 測得之阻抗值。
σ(S cm-1)=L (cm) / R(Ω)A(cm2)-Eq.(3)
17
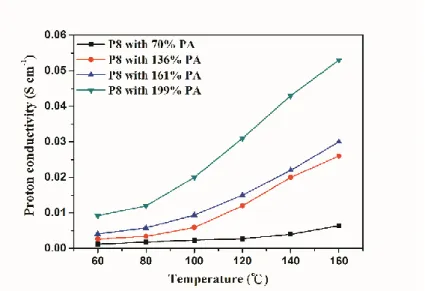
Figure 16. P8 薄膜質子傳導率
由 Figure 16 我們觀察到質子傳導率與溫度呈現正相關,而磷酸摻雜量高之 薄膜其質子傳導率隨溫度變化較明顯,進一步以 Arrhenius equation, Eq.(4)計算 質子傳導率之活化能(Activation energy, Ea),數值列於 Table 5。磷酸摻雜量 136、
161 及 199 %對應之活化能分別為 30.0、24.7 及 22.4 kJ/mol。此結果證明薄膜隨 磷酸摻雜量增加,質子傳導所需克服的活化能越低。
σ=σ0exp (-Ea/ RT)-Eq.(4)
Table 5. P8 薄膜之質子傳導率及其活化能 PA content.
(%)
P8 薄膜 Ea
(kJ/mol) 60 ℃ 80 ℃ 100 ℃ 120 ℃ 140 ℃ 160 ℃
70 0.12 0.18 0.23 0.27 0.40 0.64 18.6 136 0.26 0.34 0.59 1.21 2.21 2.62 30.0 161 0.41 0.58 0.94 1.50 2.22 3.00 24.7 199 0.92 1.23 2.01 3.11 4.33 5.34 22.4
5.7 質子交換膜燃料電池測試
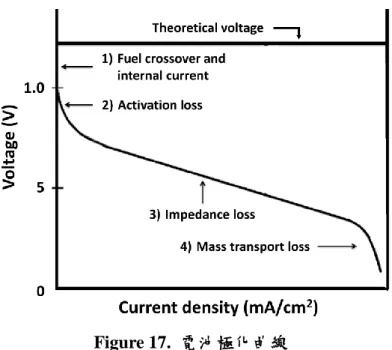
將完成之單電池接上單電池測試儀器(FCED-P200),在陽極導入氫氣,陰極 導入氧氣,流速分別為 0.2 以及 0.5 L/min,並保持其內部背壓為 2 大氣壓。固定 單電池之導入電流,量測其輸出電壓,得到電流密度與電壓關係為一極化曲線如 Figure 17 所示。
18
Figure 17. 電池極化曲線
一般而言,電壓隨電流密度變化可分為四個階段:(1)由於些微燃料穿透(Fuel cell crossover)過薄膜或微小電流流經薄膜(Internal current)導致開路電壓(Open circuit voltage, VOC)無法達到理論值。(2)氧化還原反應所需活化能造成電壓下降,
(3)在薄膜內進行質子傳導,其阻抗值造成電壓下降。(4)在高電流密度下,燃料擴 散速率大於氧化還原反應速率,使電壓快速衰退18。
Figure 18. P8 薄膜電池極化曲線
Figure 18 為 P8 薄膜進行單電池測試之極化曲線,由於分子支鏈上含氟原子 造成分子鏈吸引力較強,薄膜安定性佳且燃料不易穿透,使得燃料電池於 140 ℃ 及 160 ℃之開路電壓分別保有 0.973 V 及 0.968V。在 160 ℃測得之電功率密度 可達 475 mW/cm2。一方面,在 140 ℃下測試時,於階段(3)之電壓衰退較 160 ℃
19
下較快速則歸咎於低溫下質子傳導率較差之緣故。
Table 6. P8 薄膜燃料電池測試結果。
a. The temperature of fuel cells testing.
b. VOC:open circuit voltages.
c. The maximum performance offuel cells.
六、結論
本 研 究 已 成 功 開 發 新 型 四 胺 單 體 2,2'-(3,5-Bis(trifluoromethyl)benzyl) - 4,4',5,5'-tetraaminodiphenyl ether(6)。此單體(6)可與 4, 4’-oxybis(benzoic acid)單體 (7)以 Eaton’s reagent (10 wt%)為溶劑聚合成新型聚苯并咪唑 P8,其黏度為 1.11 dL/g。P8 具有良好之熱性質,10 wt%重量損失之熱裂解溫度為 445 ℃,及玻璃 轉移溫度大於 450 ℃。P8 在 60 ℃下可溶於 DMAc, NMP 等有機溶劑並塗佈成 可撓性之薄膜。P8 薄膜利用 Fenton’s test 進行氧化穩定性測試,經過 216 小時,
薄膜重量損失為 4.4 %,具備良好氧化穩定性。薄膜於常溫下浸泡於不同濃度磷 酸水溶液進行磷酸摻雜,所得之磷酸摻雜量介於 70 %~200 %間。磷酸摻雜量為 199 %之 P8 薄在 140 及 160 ℃下質子傳導率分別為 4.33×10-2及 5.34×10-2S cm-
1。在單電池測試方面,160 ℃下測得最高電功率密度為 475 mW/cm2。以上數 據證明 P8 具備優良質子交換膜特性。
七、參考文獻
1. J. A. Asensio, S. Borrós and P. GómezJ-Romero, J. Polym. Sci. Part A: Polym Chem. 2002, 40, 3703.
2. S. K. Kim, T.H. Kim, J. W. Jung and J. C. Lee, Polymer. 2009, 50, 3495.
3. Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci. 2009, 34, 449.
4. A. Carollo, E. Quartarone, C. Tomasi, P. Mustarelli, F. Belotti, A. Magistris, F.
Maestroni, M. Parachini, L. Garlaschelli and P. P. Righetti, J. Power Sources. 2006, 160, 175.
5. A. Schechter, R. F. Savinell, Solid State Ionics. 2002, 247, 181.
6. C. E. Hughes, S. Haufe, B. Angerstein, R. Kalim, U. Mähr, A. Reiche, M. Baldus, Polymer Temperaturea
(℃)
PA content (%)
Proton Conductivity (×10-2 S cm-1)
VOC(V)b
Power densityc (mW/cm2)
P8 140 199 4.33 0.973 361
160 199 5.34 0.968 475
20
J. Phys. Chem. B. 2004, 108, 13626.
7. R. Bouchet, E. Siebert, Solid of State Ionics. 1999, 118, 287.
8. J. A. Asensio, S. Borrós, P. Gómez-Romero, J. Electrochem. Soc. 2004, 151, A304.
9. J. Lobato, P. Cañizares, M. A. Rodrigo, J. J. Linares, F. J. Pinar, Int. J. Hydrogen Energy. 2010, 35, 1347.
10. J. C. Chen, Y. T. Liu, C. M. Leu, H. Y. Liao, W. C. Lee, J. Appt, polym. Sci. 2010, 117, 1144.
11. R. T. Foster, C. S. Marvel, J. Polym. Sci., Part A:Polym. Chem. 1965, 3, 417.
12. M. Ueda, M. Sato, A. Mochizuki, Macromolecules. 1985, 18, 2723.
13. K. Teranishi, K. Kawata, S. Tsushima, S. Hirai,Electrochem. Solid-State Lett. 2006, 9, A475.
14. F. N. Buchi, B. Gupta, O. Haas, G. G. Scherer, Electrochim. Acta. 1995, 40, 345.
15. G. Hubner, E. Roduner, J. Mater. Chem. 1999, 9, 409.
16. A. Pancheko, H. Dilger, E. Moller, T. Sixt, E. Roduner, J. Power Sources. 2004, 127, 325.
17. J. S. Yang, L. N. Cleemann, T. Steenberg, C. Terkeksen, Q. F. Li, J. O. Jensen, H.
A. Hjuler, N. J. Bjerrum, R. H. He. Fuel Cells. 2014, 14, 7.
18. J. Laminie, A. Dicks, “Fuel Cell Systems Explained”, 2 edition, John Wiley &
Son Ltd, England. 2003.