行政院國家科學委員會專題研究計畫 成果報告
各類批式蒸餾系統之操作及控制(3/3) 研究成果報告(完整版)
計 畫 類 別 : 個別型
計 畫 編 號 : NSC 96-2221-E-011-039-
執 行 期 間 : 96 年 08 月 01 日至 97 年 07 月 31 日 執 行 單 位 : 國立臺灣科技大學化學工程系
計 畫 主 持 人 : 錢義隆
計畫參與人員: 碩士班研究生-兼任助理人員:魏民忠 碩士班研究生-兼任助理人員:柯喬鐙 碩士班研究生-兼任助理人員:林義皓 碩士班研究生-兼任助理人員:Gunawan
報 告 附 件 : 出席國際會議研究心得報告及發表論文
處 理 方 式 : 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢
中 華 民 國 97 年 09 月 15 日
行政院國家科學委員會補助專題研究計畫 √ 成 果 報 告
□ 期中進度報告 各類批式蒸餾系統之操作及控制(3/3)
計畫類別: √ 個別型計畫 □ 整合型計畫 計畫編號:NSC 96-2221-E-011-039
執行期間: 96 年 08 月 01 日至 97 年 07 月 31 日
計畫主持人:錢 義 隆 共同主持人:
計畫參與人員: 魏民忠、柯喬鐙、林義皓、Gunawan
成果報告類型(依經費核定清單規定繳交):□ 精簡報告 √ 完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
√ 出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:國立臺灣科技大學化工系
中 華 民 國 97 年 09 月 15 日
行政院國家科學委員會專題研究計畫期中進度報告
各類批式蒸餾系統之操作及控制(3/3)
Operation and control of various batch distillation systems 計畫編號:NSC 96-2221-E-011-039
執行期限:96/08/01-97/07/31
主持人:錢義隆 國立台灣科技大學化工系教授 計畫參與人員:魏民忠、柯喬鐙、林義皓、Gunawan
一、 中文摘要
本研究針對各類批式蒸餾系統之操作及控制 進行共三年一系列完整的探討。第一年針對不含共 沸點混合物的批式蒸餾進行探討,此類系統的操作 方 式 計 有 以 下 四 種 : 一 般 批 式 蒸 餾 (Regular column)、逆轉式批式蒸餾(Inverted column)、中槽 式批式蒸餾(Middle vessel batch distillation)與多槽 式批式蒸餾(Multivessel batch distillation),各有其 特色與優點。本研究針對二成份、三成份與四成份 等非理想系統以不同的操作方式進行純化分離,並 加以比較。第二年的研究主要針對批次萃取蒸餾進 行最適化操作的模擬,探討的系統包括分離丙酮和 甲醇以水為萃取劑,以及分離乙酸甲酯和環己烷以 四氯化碳為萃取劑,利用改變再沸器熱量並以夾帶 劑連續進料流量、產物流的回流比、不純物的回流 比為變數,並以不同的目標函數可以探討批次萃取 蒸餾的最佳化操作。除此之外,研究中並探討利用 丙酮-甲醇-水系統應用在中槽式批次萃取蒸餾來 操作,期望藉由塔底、塔頂與中間儲槽回收三個產 物,以改進因為在一般批次萃取蒸餾當中的不純物 而導致產物回收率不高與操作時間過長的缺點。本 研究第三年則針對非均勻相批式共沸蒸餾系統進 行操作步驟與控制方面的探討,其中第一部分為分 離最低共沸點混合物(1,4-二氧陸圜和水)使用苯為 共沸劑,第二部分為分離近沸點混合物(醋酸和水) 使用幾種酯類為共沸劑,研究中也利用中槽式批式 蒸餾塔的架構應用在非均勻相共沸蒸餾系統,其望 藉由塔底、塔頂與中間儲槽回收產物,以提升產物 回收率及操作時間,探討使用不同夾帶劑以及改變 控制器設定點、改變夾帶劑含量的模擬結果,並比 較一般式批式蒸餾塔與中槽式批式蒸餾塔的優缺
獲得。計算使用的是一套精準的模擬軟體“Aspen plus®”與“Aspen dynamics™”。
關鍵詞:批式蒸餾、批式萃取蒸餾、非均勻相批式 共沸蒸餾、中槽式批式蒸餾、操作與控制
Abstract
This three-year’s research studied the operation and control of various batch distillation systems.
The first-year’s research studied the separation of mixtures without azeotrope. The suitable batch distillation systems used in this situation can be categorized into four types, as shown in the following:
regular column, inverted column, middle vessel column and multi-vessel column. The operating strategies include fixing reflux ratio strategy, fixing composition strategy and total reflux operation for the separation of binary system, ternary system and quaternary non-ideal system. The second-year’s research was focused on batch extractive distillation systems for the separation of mixtures containing azeotrope. The studied systems include: separation of acetone and methanol using water as extractive agent and separation of methyl acetate and cyclohexane using carbon tetrachloride as extractive agent. By changing reboiler duty, flow rate of entrainer, reflux ratio, and with different object functions, the study on the operation of batch extractive distillation was carried out. The operation of batch extractive distillation with middle vessel was also investigated and applied to Acetone-Methanol-Water system. It was hope that this kind of operation with a middle vessel can
operation time for the regular batch extractive distillation. The third-year’s research was focused on heteroazeotropic batch distillation system. Both mixture containing minimum-boiling azeotrope and also a close-boiling system were studied. The operation of heteroazeotropic batch distillation with middle vessel was also investigated. It was hoped that this kind of operation with a middle vessel can improve the rate of recovery and operating time.
All process simulations in this three-year’s research were carried out with rigorous simulation softwares
“Aspen plus®”and “Aspen dynamics™”.
Keywords: batch distillation; batch extractive distillation; heteroazeotropic batch distillation; batch distillation with middle vessel; operation and control.
二、 研究動機與目的
批次蒸餾廣泛使用於精密與特用化學品、生醫 及食品等工業。其製程可以僅利用單一的蒸餾塔即 能簡單、經濟、又高效率的進行多成份系統之分離 純化,將有價值、有用途的成分回收而且提高純 度,故所需使用之硬體設備成本較連續式蒸餾節省 許多。另外,批式蒸餾可以處理不同進料組成的比 例、同時分離化合物之物理及化學性質甚為不同的 物種及可以指定獲得不同純度規格的產物,因此操 作非常具有彈性,故批式蒸餾廣泛使用於高價值、
低產量之精細化學品等製程中。此類批式蒸餾之操 作及產物規格之控制幾乎無穩態可言,操作由起始 至終了,各變數均會隨著時間不斷改變,因為穩態 操作點不易達到,故而系統具有非線性的現象,造 成操作及控制上的困難。因此若能有效的掌握程序 的動態,而具備良好的控制效果,則除了可以使產 能增加與提高產品品質之外,且可降低成本與增強 安全性。
在目前有關無共沸物批式蒸餾的文獻中(如參 考文獻中 1 至 5 篇)出料的迴流比與再沸比皆是由 線上量測塔頂及塔底之純度來進行控制,真實工業 界線上量測之塔頂及塔底純度多未可得,且以上各 文獻中的系統亦均為理想之汽液平衡系統,使用固
定之相對揮發度或採用理想化勞特定律與蒸汽壓 的方式來描述,而本研究第一年所探討的化學系統 包括多成分的非理想系統,系統之汽液平衡描述是 利用 Aspen Tech 公司強大完整之熱力學模式資料 庫進行精確真實的模擬,採用較準確之液相活性係 數模式(如 NRTL 或 Wlison 模式)的方式來描述真 實系統。研究重點將在假設產物純度線上量測器不 存在之前提下,探討利用可線上量測之塔板溫度間 接的進行產物純度線上控制,使得產物純度在規格 之內,並且探討各類製程之最適化操作步驟,包 括:一般批式蒸餾塔、逆轉式批式蒸餾塔、結合上 兩形式之中槽批式蒸餾塔以及多槽批式蒸餾塔 等。研究中亦將比較以上各類批式蒸餾形式之優劣 點,比較之性能指標將以 Luyben [6]所提出對於多 成 分 系 統 批 式 分 離 之 指 標 : Capacity Factor (CAP),及以總批次操作的時間 (Total Batch time) 為準。Capacity Factor (CAP)定義如下:
5 . +0
= ∑
f i
t CAP P
(1) 其中 Pi (kmol)代表符合產物純度規格之 i 成分的 產量,tf (hr)代表總批次操作的時間(包括全回流操 作之時間),而 0.5 hr 則假設塔淨空及下批次注入 進料的時間定為三十分鐘。
若要分離含最低共沸點之混合物,工業界時 常另外加入一高沸點或中間沸點的萃取劑以利分 離出純成分,本研究第二年則針對批式萃取蒸餾進 行探討。有關此類批式蒸餾文獻中大部份的論文 (如參考文獻中 7-21 篇)僅探討批次萃取蒸餾操作 步驟的第一部份,即分離出混合物中的最輕成分,
對操作步驟中的其他部份則鮮少探討。本研究將以 兩個實施例子探討批次萃取蒸餾由空塔至批次終 了完整之操作步驟。第一個例子廣為文獻中論文所 採用,為以高沸點的水為萃取劑來分離丙酮及甲 醇。第二個例子為使用中間沸點的四氯化碳為萃取 劑來分離乙酸甲酯及環己烷。研究中會探討各操作 變數的最佳值,如預先與混合物共同置入塔底之萃 取劑量、操作步驟第一部份萃取劑連續注入的流 率、各階段操作回流比、以及各階段再沸器熱量 等。模擬中將會探討固定回流比及固定萃取劑流率 的操作,並會探討若使得此兩個操作變數變動時可 否進一步改善批次操作的效果。此兩個操作變數的
變動可由兩個溫度控制環路來決定。本研究也探討 三成份共沸系統使用中槽式批式蒸餾的應用可 能,並與一般批次萃取蒸餾相比較,期望能夠解決 一般批次萃取蒸餾因為不純物而使產率降低的缺 點。
本研究第三年則針對非均勻相批式共沸蒸餾 進行探討。此類操作在文獻中所敘述的操作步驟與 現實中運作有所差異,而在最後完成分離及回收產 物的步驟也未完整的描述。本研究將操作步驟更貼 近現實且完整化,另外,亦會探討各控制變數對模 擬結果造成的影響。在醋酸去水系統中亦將比較不 同的夾帶劑、不同的操作策略以及不同的蒸餾塔系 統,討論操作步驟以及過程之動態,並針對醋酸去 水系統對一般式均勻相批式共沸蒸餾與中槽式的 操作進行比較及討論。此部分所探討的化學系統之 汽液平衡描述亦是利用 Aspen Tech 公司強大完整 之熱力學模式資料庫進行精確真實的模擬,採用較 準確之液相活性系數模式(NRTL)的方式來描述真 實系統,探討具共沸物之多成份系統的批式蒸餾。
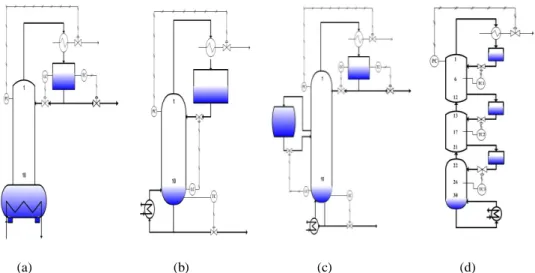
三、 各類無共沸物批式蒸餾的操作 3.1 固定回流比出料之操作
各類批式蒸餾之裝置圖如圖一所示,其中的一 般、逆轉式與中槽式批式蒸餾皆可進行固定回流比 出料之操作,而固定回流比出料之控制架構執行較 為簡單,當系統開始出料時,便控制塔頂回流槽出 料的液體流量,並保持固定回流比,導致塔頂產物 的組成持續隨著時間而改變,產品的濃度會由高濃 度慢慢降低,將高濃度與低濃度之所得產品混合成 所需產品之純度規格,再依序收集餾出的產物,隨 著蒸餾的時間加長,可以收集到沸點較高的產品,
然後依序更換收集的容器。而出料回流比的選擇是 以能夠獲得最大的 CAP 作為準則,因此以獲得最 大 CAP 作為目標函數去求得最適化的回流比。以 一般批式蒸餾二成份系統為例,如圖二所示,在回 流比為 2.3 時,可得最高的 CAP 值,而在逆轉式 批式蒸餾是以再沸比為 11.8 時,可得最高的 CAP 值。收集出料的產物以混合的方式來獲得輕成份產 品純度為 0.97,重成份產品純度為 0.95,但是會在 蒸餾的過程中產生不合規格的不純物,對於 N 成 份系統而言,將有最多 N-1 種不純物,每一種不純 物均接近於二成份混合物,將所有 N-1 種不純物混
合新鮮的原料於下次批次合併蒸餾,可以使原本成 為廢料的不純物資源再回收,以減去處理廢料的問 題與原料成本上的損失,回收不純物所影響僅為進 料組成上的改變。固定回流比出料所獲得組成與溫 度的動態分佈如圖三、四所示。
(a) (b)
(c) (d)
圖一 各類批式蒸餾之裝置圖(a)一般批式蒸餾(b) 逆轉式批式蒸餾(c)中槽式批式蒸餾(d)多槽式批式 蒸餾
0.286 0.288 0.290 0.292 0.294 0.296 0.298 0.300
1.5 2.0 2.5 3.0 3.5
Reflux ratio
CAP
圖二 一般批式蒸餾之 CAP-回流比關係圖
Time (hr)
0 5 10 15 20 25 30
Propylene oxide mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Z=0.665 Z=0.700 Z=0.735
Time (hr)
0 5 10 15 20 25 30
Temperature (oC)
20 25 30 35 40 45 50 55 60
Z=0.665 Z=0.700 Z=0.735
圖三 一般批式蒸餾固定回流比之組成與溫度之動 態分佈
Time (hr)
0 10 20 30 40
Propylene oxide mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Z=0.665 Z=0.700 Z=0.735
Time (hr)
0 10 20 30 40
Temperature (oC)
20 30 40 50 60 70
Z=0.665 Z=0.700 Z=0.735
圖四 逆轉批式蒸餾固定回流比之組成與溫度之動 態分佈
3.2 固定出料組成之操作
對於固定回流比出料的操作,產品是以混合的 方式來獲得,其操作較麻煩且不切實際,而且不易 得知儲槽混合後的組成。一般批式蒸餾塔在固定出 料組成的操作下,批式蒸餾若是想要不斷的取得符 合規格濃度的產品,在理論板數固定時,較可行的 方法就是不斷的調整回流比,這便是改變回流比之 操作的批式蒸餾。蒸餾的初期,加熱器內的液體混 合物會含有較多輕成分,因此只需要低回流比即可 達到產品純度的要求;隨著蒸餾的進行,因輕成分 變少,故要不斷提高回流比去控制塔頂回流槽的組 成在特定規格。
但是對出料進行直接組成控制的方式並不實 際,因為工業界並沒有即時組成量測的儀器,同時 對組成改變具有相依的物理性質並不多,由組成控 制的情況得知,若塔頂組成被控制而穏定時,其塔 頂的溫度也會維持穏定,所以若能控制此時的溫 度,就能夠間接的控制塔頂回流槽的組成。由於在 汽液平衡的前提下,若固定壓力則溫度與組成的關 係可由熱力學 T-xy 相圖得知,就可以得知在特定 組成的溫度為多少。因此可以溫度作為系統之控制 變數,作動塔頂出料流量來進行控制,由塔頂組成 的改變而造成溫度改變,則隨著時間改變塔頂出料 流量再收集產物。固定出料組成之操作所獲得組成 與溫度的動態分佈如圖五所示。
3.3 全回流之操作
批式蒸餾的操作除了起動期的全回流之外,
還必須進行生產期的出料,因為是批式的操作,所 以會不斷的重複起動期的全回流,整體而言其操作 並不單純,若能只進行全回流就可以來獲得產品,
不用再進行出料,則可大大的化簡操作的程序。一 般批式蒸餾塔本身具有一個回流槽與塔底容器,若 儲槽體積夠大,在持續全回流的操作下,輕成份不 斷的被往上蒸餾,則輕成份被收集在塔頂回流槽 中,而重成份會被收集在塔底中。以二成份糸統為 例,恰好可以分別被收集到兩個儲槽中,持續全回 流操作時,改變塔頂回流槽的液體回流量,控制的 方式有二,第一為控制儲槽液位以作動塔頂回流槽 的液體回流量,第二是以溫度控制來作動液體回流 量,控制蒸餾塔段中點溫度以改變塔頂回流槽液體 回流量。由於是全回流的操作,系統是會達到穏定 的狀態,可在塔頂獲得輕成份產品、塔底獲得重成
(b) 份產品,所以若整個系統達到穏定的標準,即可停
止蒸餾,可將塔頂回流槽與塔底的產物排出。
(a) (b)
Time (hr)
0 5 10 15 20 25 30
Propylene oxide mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Z=0.665 Z=0.700 Z=0.735
Time (hr)
0 5 10 15 20 25 30
Temperature (oC)
20 25 30 35 40 45 50 55 60
Z=0.665 Z=0.700 Z=0.735
Time (hr)
0 10 20 30 40
Propylene oxide mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Z=0.665 Z=0.700 Z=0.735
Time (hr)
0 10 20 30 40
Temperature (oC)
20 30 40 50 60
Z=0.665 Z=0.700 Z=0.735
(c) (d)
Time (hr)
0 5 10 15 20 25 30
Propylene oxide mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Z=0.665 Z=0.700 Z=0.735
Time (hr)
0 5 10 15 20 25 30
Temperature (oC)
20 30 40 50 60
Z=0.665 Z=0.700 Z=0.735
Time (hr)
0 5 10 15 20 25 30 35
Mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Z=0.18 Z=0.20 Z=0.22
Time (hr)
0 5 10 15 20 25 30 35
Temperature (oC)
20 40 60 80 100
Z=0.18 Z=0.20 Z=0.22
圖五 固定出料組成之溫度控制之動態分佈(a)一般批式蒸餾(b)逆轉批式蒸餾(c)中槽式批式蒸餾二成 份系統(d)中槽式批式蒸餾三成份系統
全回流的液位控制策略是由 Hasebe et al.[22]
所提出,液位控制較為簡單、容易,利用進料組成 與進料量來計算每個儲槽的最終滯留量,再將滯留 量換算成液位高度,然後將這些結果當作液位控制 器的設定值,利用簡單的回饋環路來控制各個貯槽 的液位高度。改變液體回流量使每個儲槽內的液體 回流到塔中,來控制每個儲槽的液位高度,若液位 高度大於設定值時,則加大液體回流量,反之,若 液位高度小於設定值時,則減少液體回流量。中槽 式批式蒸餾總共具有三個塔槽,在全回流的操作 下,可對三成份系統進行分離,分別收集至三個儲 槽中。但若應用在二成份系統的分離,則會多出一 個多餘的儲槽,所以 Barolo and Botteon[23]提出將 中間塔槽當作不純物的儲存槽,而可在進料組成不 確定的情況下,可以分別在塔頂與塔底收集到高純 度的產品。先決定所要容忍組成不確定的上限為±
0.05 莫耳組成,再分別假設進料的誤差全來自輕 成份或是全來自於重成份,分別求得所造成的過量 滯留量之量,此量即為中間塔槽所應收集的量。全 回流操作之液位控制所獲得組成與溫度的動態分 佈如圖六(a)(b)(c)所示。
全回流的溫度控制策略是由 Wittgens et al.[24]
所提出,以兩個槽的中間板之溫度為控制變數,而 去作動塔頂回流槽回流入塔內的液體流量,因為在 全回流的操作下會達到一個近似穏態,所以會有固 定的組成分佈,若在板數夠多的情況下,則會在蒸 餾塔的上下兩端獲得高純度的產品。且在汽液平衡 的情況下,也具有固定的溫度分佈,所以可應用控 制溫度來對組成進行控制,進而改變液體回流量使 儲槽內的液體回流到塔中,以達到特定塔板所要求 的溫度,若溫度高於設定值時,則加大液體回流 量,若溫度小於設定值時,則減少液體回流量。而 其溫度設定點的獲得是因為在理想分離的情況 下,可在各槽獲得純物質,因此在各槽此時的飽和 溫度即等於其各成份的沸點,所以可以假設在兩個 槽的中間的那一板的飽和溫度,即等於兩個槽其各 成份的沸點相加除以二,以環氧丙烷及丙酮之二成 份糸統為例,塔頂輕成份純物質沸點為 34.48℃,
塔底重成份純物質沸點為 56.14℃,則塔頂回流槽 與 塔 底 之 間 的 正 中 間 塔 板 的 溫 度 為 (34.48+56.14)÷2=45.31℃。則將此輕成分與重成份 沸點的平均值作為溫度控制器的設定點,以塔頂回 流槽與塔底之間的正中間塔板,作為溫度控制的塔 板,來作動液體回流量,塔頂回流槽的液位並未直
接控制,但是由於控制液體回流量的關係,故各儲 槽的滯留量會自行調整,間接調整儲槽內產品的組 成。全回流操作之溫度控制所獲得組成與溫度的動 態分佈如圖六(d)所示。
3.4 各類無共沸物批式蒸餾的比較 一般與逆轉批式蒸餾之比較
二成份系統之模擬棌用之輕成份為環氧丙 烷 , 沸 點 為 34.48℃ 與 重 成 份 丙 酮 , 沸 點 為 56.14℃,此二成份之選用與 Monroy-Loperena et al.[25]相同,所選用的熱力學模式為 UNIQUAC。
一開始將全部的進料量 10 kmol 充入塔底,基本的 進料組成為 Z = [0.7, 0.3],固定熱量為 0.03 GJ/hr,
板數為 10 板。一般批式蒸餾與逆轉批式蒸餾的硬 體裝置恰好相反,一個是將全部的進料加入塔底,
所以塔底的體積較大,另一個是將全部的進料加入 塔頂回流槽,所以塔頂回流槽的體積較大。這兩個 系統在裝置的成本上相差不大,所以需了解它們在 純化分離的應用上誰優誰劣,才能決定誰具有較高 的效能。因此考慮適用的組成範圍或所要求之產品 純度的高低,決定何者較為適用,是否可以最少的 時間而獲得最大的產率,則是本節中所要來加以探 討。理論上若是進料組成中輕成份含量較少時,在 所要求的純度不高的情況,使用一般批式蒸餾的效 能較高[26]。因為輕成份從塔頂出料的量較少,塔 底重成份佔大部份的量,則所需的蒸餾時間較短。
反之,若是進料組成中輕成份含量較多時,在所要 求的純度不高的情況,使用逆轉式批式蒸餾的效能 較高。因為重成份從塔底出料的量較少,塔頂輕成 份佔大部份的量,則所需的蒸餾時間較短。若是要 求獲得高純度產品的情況,所需的總蒸餾時間較 長,當進料組成中輕成份含量較多時,因為在蒸餾 的過程中,塔頂輕成份組成持續高純度的時間較 長,所以可以較小的回流比進行出料,出料流率較 大而節省前段出料的時間,則使用一般批式蒸餾的 效能較高。相反的,當進料組成中輕成份含量較少 時,因為在蒸餾的過程中,塔頂輕成份組成持續高 純度的時間較少,所以須以較大的回流比進行出 料,出料流率較小使前段出料的時間較長,則使用 一般批式蒸餾的效能較低。
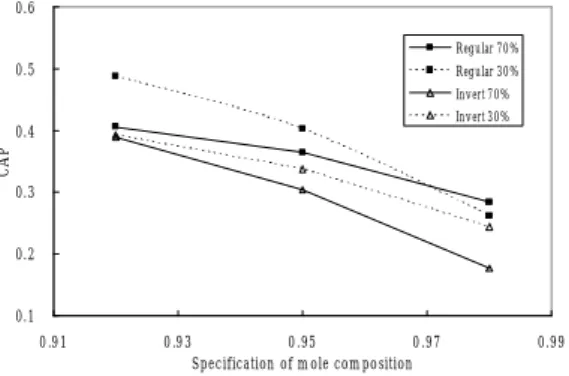
由模擬的結果得知如下,當進料組成中輕成份 含量較多時 (環氧丙烷 70%),如圖七中實線之連
(b)
(d) (a)
(c)
線即為輕成份含量較多時之情況,無論在何種產品 純度規格下,一般批式蒸餾的 CAP 值皆大於逆轉 式批式蒸餾。當進料組成中輕成份含量較少時 (環 氧丙烷 30%),如圖七中虛線之連線即為輕成份含 量較少時之情況,無論在何種產品純度規格下,一 般批式蒸餾的 CAP 值也是大於逆轉式批式蒸餾。
逆轉式批式蒸餾在理論上應該贏過一般批式蒸餾 的 地 方 都 沒 有 贏 , 如 在 低 產 品 純 度 規 格 (Xsepc=0.92),進料組成中輕成份含量較多時,應該 是逆轉式批式蒸餾具有較高的效能,但是只是與一
般批式蒸餾的效能相當而已。另外,在高產品純度 規格(Xsepc=0.98),進料組成中輕成份含量較少時,
也是同樣的情況。造成的原因應該為塔底的壓力較 大,而使相對揮發度變小,導致分離的難度提升,
所需的蒸餾時間增加,如圖八相對揮發度對壓力之 關係圖所示,所以由結果得知一般批式蒸餾可謂大 獲全勝,其無論在任何進料組成範圍,效能皆大於 逆轉式批式蒸餾,所以在純化分離的應用上可優先 考慮使用之。
(a) (b)
T im e ( h r)
0 1 0 2 0 3 0 4 0
Propylene oxide mole composition
0 . 0 0 . 2 0 . 4 0 . 6 0 . 8 1 . 0
Z = 0 . 6 6 5 Z = 0 . 7 0 0 Z = 0 . 7 3 5
T im e (h r )
0 1 0 2 0 3 0 4 0
Temperature (oC)
2 0 3 0 4 0 5 0 6 0 7 0
Z = 0 . 6 6 5 Z = 0 . 7 0 0 Z = 0 . 7 3 5
T im e ( h r)
0 5 1 0 1 5 2 0 2 5 3 0
Propylene oxide mole composition
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0
Z = 0 .6 6 5 Z = 0 .7 0 0 Z = 0 .7 3 5
T im e ( h r )
0 5 1 0 1 5 2 0 2 5 3 0
Temperature (oC)
2 0 3 0 4 0 5 0 6 0
Z = 0 .6 6 5 Z = 0 .7 0 0 Z = 0 .7 3 5
(c) (d)
T im e (h r)
0 5 1 0 1 5 2 0 2 5 3 0
Mole composition
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0
Z = 0 .1 8 Z = 0 .2 0 Z = 0 .2 2
T im e (h r)
0 5 1 0 1 5 2 0 2 5 3 0
Temperature (oC)
0 2 0 4 0 6 0 8 0 1 0 0
Z = 0 .1 8 Z = 0 .2 0 Z = 0 .2 2
T im e (h r)
0 5 1 0 1 5 2 0 2 5 3 0 3 5
Mole composition
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0
Z =0 .1 8 Z =0 .2 0 Z =0 .2 2
T im e (h r )
0 5 1 0 1 5 2 0 2 5 3 0 3 5
Temperature (oC)
2 0 4 0 6 0 8 0 1 0 0
Z = 0 .1 8 Z = 0 .2 0 Z = 0 .2 2
圖六 全回流操作之組成與溫度之動態分佈(a)一般批式蒸餾之液位控制(b)中槽式批式蒸餾二成份系統之 液位控制(c)中槽式批式蒸餾三成份系統之液位控制(d)中槽式批式蒸餾三成份系統之溫度控制
0.1 0.2 0.3 0.4 0.5 0.6
0.91 0.93 0.95 0.97 0.99
Specification of mole composition
CAP
Regular 70%
Regular 30%
Invert 70%
Invert 30%
圖七 一般與逆轉批式蒸餾之 CAP 比較圖
2.06 2.08 2.1 2.12 2.14 2.16 2.18
1.00 1.04 1.08 1.12 1.16
Pressure (bar)
Relative volatility
X1=0.95 X1=0.05
圖八 相對揮發度對壓力之關係圖
二成份系統之比較
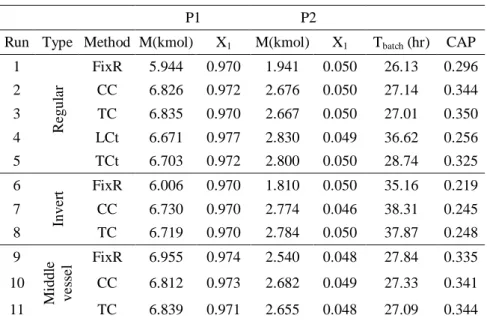
為了使二成份系統之比較能夠在公平的條件 下進行,在不同的操作與控制方法是以在獲得相同 的產品純度之前提下,比較 CAP 值的高低,所以 輕成份的產品組成為 0.97,重成份的產品組成為 0.95 來進行批式蒸餾,所比較的結果如圖九如示。
固定回流比出料即是沒有固定出料組成的操作對 於二成份系統的蒸餾效能,整體而言因為有不純物 的產生,所以並不佳,除了中槽式批式蒸餾是從塔 頂與塔底同時進行出料,而使出料的時間縮短、
CAP 值因而增加,而在一般批式蒸餾與逆轉式批 式蒸餾中所進行蒸餾的結果卻不盡理想。尤其是在 逆轉式批式蒸餾之固定再沸比出料的操作,因為全 回流的時間較長與塔頂所要求的產品組成較高的 情況下,其 CAP 值因而相當低。在全回流操作的 部份,一般批式蒸餾的液位控制之蒸餾效能較差,
因為需從塔底蒸上塔頂的量較多的情況下,液位達 到設定點的時間較長,而使液體從塔頂回流槽回流 入塔內的時間較晚,造成分離的效率較差,為此方 法的盲點。而溫度控制是以控制溫度去作動液體回
流量,溫度上升的速度較快,所以液體回流入塔內 的時間較早,致使分離的效果較佳。而中槽式批式 蒸餾之液位控制,因為中間塔槽也會收集產物,所 以縮短了把輕成份蒸上塔頂的時間,因而使 CAP 增加,雖然產生了不純物,但是可經由回收的方式 減少損失。固定出料組成的操作的部份,中槽式批 式蒸餾之溫度控制並沒有預料中的可以從塔頂與 塔底同時進行出料,而使出料的時間縮短,所以使 CAP 增加。主要是其進行全回流的時間過長的緣 故所造成,因為重成份的含量較少,所以塔底達到 高純度的時間較長,因而會使總蒸餾時間變長。反 觀一般批式蒸餾的溫度控制,因為輕成份的含量較 多,所以塔頂達到高純度的時間較短,雖然出料的 時間較長,但塔底需達到的產品組成相對較低,因 此總蒸餾時間略快於中槽式批式蒸餾。所以對於本 次模擬的二成份系統而言,一般批式蒸餾的溫度控 制是為最好的選擇。
0.0 0.1 0.2 0.3 0.4
R-TC R-CC Mv-TC Mv-CC Mv-FixR R-TCt Mv-LC R-FixR R-LC I-TC I-CC I-FixR
CAP
圖九 二成份系統之 CAP 比較圖
三成份系統之比較
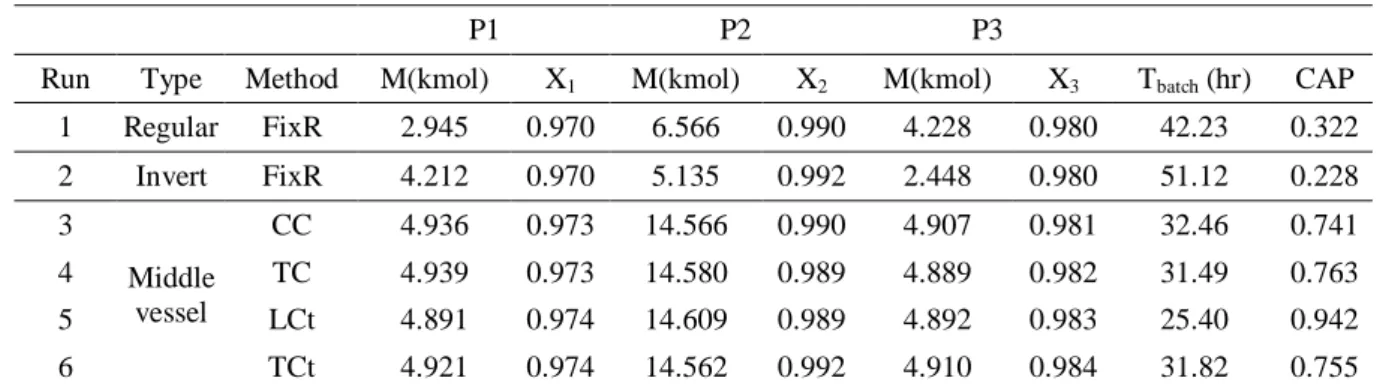
三成份系統之模擬棌用之三成份分別為輕成 份正戊烷,沸點為 36.05℃、中成份正己烷,沸點 為 68.73℃與重成份正庚烷,沸點為 98.40℃,此三 成份之選用與 Monroy-Loperena et al.[25]相同,應 用的熱力學模式為 NRTL。一開始將全部的進料量 25 kmol 充入塔底,基本的進料組成為 Z = [0.2, 0.6, 0.2],固定熱量為 0.1 GJ/hr,板數為 20 板。各類批 式蒸餾對於相同的三成份系統進行分離,達到輕成 份的產品組成為 0.97,中成份的產品組成為 0.99 與重成份的產品組成為 0.98 來進行批式蒸餾,所 比較的結果如圖十如示。一般與逆轉式批式蒸餾是 使用固定回流比出料的操作方式進行,因為會有不 純物產生,且需進行出料的量較多,因此所需的蒸
餾時間也較長,所以 CAP 值較低。在中槽式批式 蒸餾對於三成份系統進行分離卻有不錯的表現,全 回流操作之液位控制所獲得的 CAP 值最高,理應 為最佳之選,但是對於進料組成不確性的容忍性不 佳,是其最大的缺點。退而求其次,以第二名的固 定出料組成之溫度控制來說,具有可以容忍進料組 成不確定性的效果,也可在塔頂與塔底同時進行出 料,節省了許多蒸餾的時間,但是因為是上、下兩 端同時進行出料,所以需同時對塔頂與塔底進行溫 度控制,才能維持出料組成的固定,因此其控制的 架構較為複雜。全回流操作之溫度控制也是有水準 之上的表現,其操作方式與控制架構皆簡單,就能 在產品儲槽獲得高純度的組成,而且對於進料組成 的改變也具有容忍性,其蒸餾的效能很高。所以以 本次模擬的三成份系統而言,固定出料組成之溫度 控制與全回流操作之溫度控制是最值得去使用的 蒸餾方法,可以在最少的蒸餾時間,獲得最大的產 量。
0.0 0.2 0.4 0.6 0.8 1.0
Regular FixR Invert FixR Mv CC Mv TC Mv LC Mv TCt Type
CAP
圖十 三成份系統之 CAP 比較圖
*FixR:固定回流比出料,CC:固定出料組成之組成控制,TC:固定出
料組成之溫度控制,LC:全回流操作之液位控制,TCt:全回流操作之
溫度控制
四成份系統之比較
對於混合物含有的成份數在三成份以上的情 況下,若是用出料的方式進行操作,不論是用固定 迴流比出料的操作或是固定組成出料的方式進行 混合物的分離純化,其操作的步驟皆變成相當繁瑣 與複雜,可想而知,光是產品的依序出料就會花費 許多的時間,例如以一般批式蒸餾塔固定迴流比出 料的操作來分離四成份的混合物,則需在進行全迴 流之後,由塔頂出料依序獲得第一輕成份、第一不
純物、第二中成份、第二不純物、第三中成份與第 三不純物,最後才由塔底獲得第四重成份。所以若 是能只進行全迴流的操作,而不進行出料的操作,
則可以使整個分離程序化簡許多。只需在批式蒸餾 塔的塔身中間插入數個貯槽,即可構成多槽式批式 蒸餾塔,其槽的數目與組成數目有關,即組成數目 為 N 時,則槽的數目為 N-2,在整個蒸餾過程中,
系統保持在全回流的操作下,亦即完全沒有出料,
可依序由沸點高低從各個塔槽獲得高純度產品,如 低沸點產物在上方的儲槽,高沸點產物在下方之儲 槽。這種批式蒸餾具有兩項優點:(1)操作簡單只 需全迴流操作,產物會依沸點各自收集所以不需要 轉換收集槽;(2)控制架構簡單,因為全回流的操 作單純易行。
分離的系統以 Wittgens et al. [27]所提出的四 成份系統來進行模擬,分別是甲醇、乙醇、丙醇與 丁醇,進料組成為 Z=[0.26, 0.12, 0.18, 0.44],沸點 分別為 Tb=[64.53℃,78.31℃, 97.19℃, 117.68℃],
板數為 30 板。第一個中間儲槽置於 12 與 13 板之 間,第二個中間儲槽置於 21 與 22 板之間,將整個 蒸餾塔劃分成三段,第一段有 12 板,第二、三段 皆有 9 板,因為其相對揮發度分別為 α=[7.8, 4.5, 2.3, 1],甲醇與乙醇的相對揮發度較小,所以第一段較 第二、三段為多。三個溫度控制器分別所控制的板 之位置為 Nc=[6, 17, 26],其溫度設定點分別應為 Tsp=[71.42℃, 87.75℃, 107.44℃],溫度控制所獲得 組成與溫度的動態分佈如圖十一(a)所示。全回流 操作之液位控制所獲得組成與溫度的動態分佈如 圖十一(b)所示,由圖得知對於醇類的混合系統具 有如此非理想的現象,其四成份系統的分離效果是 出乎意料的不佳,由此說明了光靠液位控制,就算 是控制在所要求的液位上,也不一定可以獲得高純 度的產品,所以若能在每批出料取樣分析來修正設 定值,輔助液位控制所不足,則可提高產品的純 度。相對於溫度控制卻能夠有不錯的分離效果,所 以溫度與組成的關係的確較為正確,控制塔內的溫 度分佈,就能使塔內的組成分佈固定,而使產品儲 槽內的組成獲得較高的純度,就算是產品儲槽內的 組成未能達到所要求的規格,同樣可以經由在每批 出料的產品取樣分析來修正溫度設定值,由其中所 含不純物的比率,而可以作一個彈性的調整。
(a)
Time (hr)
0 2 4 6 8 10
Mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Vessel 1 (X1) Vessel 2 (X2) Vessel 3 (X3) Vessel 4 (X4)
Time (hr)
0 2 4 6 8 10
Temperature (oC)
20 40 60 80 100 120 140
Vessel 1 Vessel 2 Vessel 3 Vessel 4
Time (hr)
0 1 2 3 4 5 6
Mole composition
0.0 0.2 0.4 0.6 0.8 1.0
Vessel 1 (X1) Vessel 2 (X2) Vessel 3 (X3) Vessel 4 (X4)
Time (hr)
0 1 2 3 4 5 6
Temperature (oC)
20 40 60 80 100 120 140
Vessel 1 Vessel 2 Vessel 3 Vessel 4
圖十一 多槽式批式蒸餾全回流操作之動態分佈(a) 溫度控制(b)液位控制
四、 批式萃取蒸餾系統的操作與控制
4.1 丙酮-甲醇-水系統在一般批次萃取蒸餾之最適 化操作
圖十二為丙酮-甲醇-水系統之 RCM 圖,可知 丙酮與甲醇共沸物為系統最輕的成分,因此若用一 般批次蒸餾做分離,則塔頂會得到的是最輕的共沸 物組成,而無法得到高純度的丙酮;但是利用在塔 的中間板連續加入夾帶劑的方式,就可以在塔頂得 到高純度的丙酮。原理為在連續加入夾帶劑(水)
後,水在萃取段將甲醇吸收後帶至塔底,進入精餾 段的物流為丙酮、水與極少量的甲醇,而水與丙酮 就在精餾段中進行分離,在塔頂得到高純度的丙 酮。圖十三為批次萃取蒸餾系統固定回流比之控制 圖,固定回流比進料之控制架構執行較為簡單,當 系統開始出料時,便控制塔頂回流槽出料的液體流 量,並保持固定回流比,導致塔頂產物的組成持續 隨著時間而改變,產品的濃度會由高濃度慢慢降 低,將高濃度與低濃度之所得產品混合成所需產品 之純度規格,再依序收集餾出的產物,隨著蒸餾的 時間加長,可以收集到沸點較高的產品,然後依序 更換收集的容器。而夾帶劑的進料是從蒸餾程序一 開始就以連續方式加入,持續至收集最輕成份(P1) 的步驟完成。由於剛開始加熱時,塔內各板的壓力 變化幅度很大,所以夾帶劑進料流的壓力與閥的大 小需要格外注意。圖十三中之回流比及萃取劑流率 亦可採用串級控制的方式,由兩個溫度控制環路來 決定。本研究亦比較了固定此兩變數及使其變動的 不同結果。
圖十二、丙酮、甲醇和水在一大氣壓下的 RCM 圖
圖十三、批次萃取蒸餾系統固定回流比之控制圖 (b)
動態模擬結果
本系統第一階段所使用的最佳化目標函數為:
*
=丙酮產量 丙酮價格-夾帶劑花費-再沸器熱量花費P1
獲利 結束時間
此目標函數的物理意義為每單位時間的獲利,用此 目標函數搜尋的參數有:(1)各階段的再沸器熱 量;(2)夾帶劑連續進料時的流量;(3)出產丙酮產 物流(P1)時的回流比。搜尋到的最佳化操作参數由 表一及圖十四可知為:再沸器熱量為 0.36GJ/hr,
夾帶劑連續進料流量為 24kmol/hr,及 P1 出料回流 比為 1.8。
決定第一不纯物(S1)時的回流比的目標函數 Φ1 為:
Φ1=S1 結束的時間*S1 中甲醇量
此目標函數的物理意義:為尋找一個可以用最短時 間將塔頂的甲醇濃度提升到預定值,及所回收第一 不純物中最少甲醇含量的回流比,意即在不同再沸 器熱量、不同回流比下,尋找一個最小的 Φ1。對 於 S1 中所模擬結果,由圖十五可知出產第一不純 物時的 Q 為 0.22GJ/hr,回流比為 7。
決定甲醇產物流(P2)時回流比的目標函數為:
時間 出料結束後的的總蒸餾 甲醇產物流(P2)
0.5
CAP PMethanol
= +
由於本步驟不需要加入夾帶劑,所以是一個單純的 多成分分離的批次蒸餾,所以目標函數可以使用 Luyben[6]的生產力因子(capacity factor, CAP)來作 為目標函數。圖十六為不同 Q 與回流比和 CAP 的 關係。由結果可知出產甲醇產物流(P2)時的 Q 為 0.15GJ/hr 及回流比選為 20。
表一、搜尋最佳參數的不同操作條件 操作條件 再沸器熱量
(GJ/hr)
夾帶劑流量 (kmol/hr) A 0.35 22 B 0.35 23 C 0.35 24 D 0.36 23 E 0.36 24 F 0.36 25 G 0.37 24 H 0.37 25 I 0.37 26
R
0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 2.6
profit
500 600 700 800 900 1000 1100 1200
A B C D E F G H I
圖十四、最佳化目標函數在不同操作條件下的比較
R
6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5
13.5 14.0 14.5 15.0 15.5 16.0
Q =0.2(G J/hr) Q =0.21 Q =0.22 Q =0.23 Q =0.24
圖十五、回收第一不純物中不同 Q 與 R 的關係
R
14 16 18 20 22 24
CAP
0.0785 0.0790 0.0795 0.0800 0.0805 0.0810 0.0815
Q=0.13(GJ/hr) Q=0.14 Q=0.15 Q=0.16 Q=0.17
圖十六、甲醇產物流在不同 Q 及回流比的結果
以 上 結 果 可 與 固 定 各 階 段 再 沸 器 熱 量 在 0.2GJ/hr 的結果相比較,從表二得知在改變各階段 再沸器熱量所探討的最佳化結果當中,對於 P1 過 程中將再沸器熱量由 0.2GJ/hr 改為 0.36GJ/hr 加上 其他變數最佳化探討之後得知第一輕成分的回收 比雖然不如原固定再沸器熱量的結果,但是在回收 第一輕成分所花的時間卻明顯的減少,而對於收集 Φ1
甲醇產物流過程由 0.2GJ/hr 改為 0.15GJ/hr,第二 成份的回收在回收比及時間上都比原結果差,而最 後總時間也只是減小些許時間。對於這個結果不如 當初我們所預期藉由尋找各操作階段最佳的再沸 器熱量來達到更好的模擬結果,推測可能的原因在 於 P1 過程中將再沸器熱量提升了近一倍,雖然獲 得不錯的結果卻降低了 P2 回收的效率。
表二、兩種最佳化結果比較
Q=0.2GJ/hr 改變再沸器熱量 全回流時 3.17 1.57 P1 結束時 5.34 2.71 P1 回流比 2.5 1.8 P1 濃度 0.95 0.95 P1 回收比 0.684 0.617 P2 結束時 12.26 11.83 P2 回流比 17.5 20 P2 濃度 0.92 0.92 P2 回收比 0.271 0.24 P3 濃度 0.99 0.99 P3 回收比 0.924 0.951 總時間 12.26 11.83
固定各操作階段再沸器熱量的最佳化操作 圖 十 七 表示 不 同 再 沸 器熱 量 ( 不 一 定使 用 0.2GJ/hr) 與各操作階段時間的關係,隨著再沸器 熱量越大,各階段的時間都有明顯減少的趨勢。在 產量部份,從圖十八可知隨著再沸器熱量愈大,丙 酮與水的產量變動並不明顯,但是甲醇卻明顯減 少。圖十九為不同再沸器熱量與回流比的關係,隨 著再沸器熱量愈大,丙酮產物流最佳操作回流比並 不明顯,而甲醇產物流必須操作在高回流比但卻沒 有獲得較好的結果。因此對於改變再沸器熱量雖然 會使系統操作總時間減少,卻不利甲醇成份的分 離。
再沸器熱量 (GJ/hr)
0.10 0.12 0.14 0.16 0.18 0.20 0.22 0.24 0.26 0.28 0.30 0.32
時間(hr)
0 2 4 6 8 10 12 14 16 18
全回流時間 P1結束時間 總時 間
圖十七、不同再沸器熱量與各產物回收比的關係
再沸器熱量 (GJ/hr)
0.10 0.12 0.14 0.16 0.18 0.20 0.22 0.24 0.26 0.28 0.30 0.32
回收比(kmol/kmol)
0.0 0.2 0.4 0.6 0.8 1.0
P1回收比 P2回收比 P3回收比
圖十八、不同再沸器熱量與各產物回收比的關係
再沸器熱量 (GJ/hr)
0.10 0.12 0.14 0.16 0.18 0.20 0.22 0.24 0.26 0.28 0.30 0.32
回流比(kmol/kmol)
0 10 20 30 40 50
P1回流比 P2回流比
圖十九、不同再沸器熱量與回流比的關係
4.2 乙酸甲酯-環己烷-四氯化碳在一般批次萃取蒸 餾之最適化操作
由圖二十的 RCM 圖可以看出,利用一個簡單 的蒸餾塔分離任何組成三成份的混合物,在塔頂所 能的得到的都是最輕的乙酸甲酯與環己烷的共沸 物,而無法得到高純度的乙酸甲酯;但是利用在塔 的中間板連續加入夾帶劑的方式,就可以在塔頂得
到高純度的乙酸甲酯。原理為在連續加入夾帶劑 (四氯化碳)後,四氯化碳在萃取段將環己烷吸收後 帶至塔底,進入精餾段的物流為乙酸甲酯、四氯化 碳與極少量的環己烷,而乙酸甲酯與四氯化碳就在 精餾段中進行分離,在塔頂得到高純度的乙酸甲 酯;由於四氯化碳的沸點介於乙酸甲酯與環己烷之 間,所以在分離的順序與前節不同,在本系統中得 到產物的順序為:(1)高純度的乙酸甲酯;(2)不纯 物 1;(3)高純度的四氯化碳;(4)不纯物 2;(5)環己 烷。圖二十一為此批次萃取蒸餾系統固定回流比之 控制圖。
圖二十、乙酸甲酯,環己烷和四氯化碳在一大氣壓 的 RCM 圖
圖二十一、本批次萃取蒸餾系統固定回流比控制圖
動態模擬結果
系統第一階段所使用的最佳化目標函數為:
*
=乙酸甲酯產量 乙酸甲酯價格-夾帶劑花費-再沸器熱量花費P1
獲利 結束時間
此目標函數的物理意義為:每單位時間的獲利,本 系統用此目標函數搜尋的參數有(1)各階段的再沸 器熱量;(2)夾帶劑連續進料時的流量;(3)出產乙 酸甲酯產物流(P1)時的回流比。表三為不同操作條
件的簡介;圖二十二為以這些操作條件所得到的最 佳化目標函數的比較,圖中的 X 軸為回流比(R)、
Y 軸為目標函數。搜尋到的最佳化操作参數為:Q 為 0.22GJ/hr 及夾帶劑連續進料流量 2 kmol/hr。
表三、不同操作條件
操作條件 Q(GJ/hr) 夾帶劑流量 (kmol/hr)
A 0.21 2
B 0.21 3
C 0.22 2
D 0.22 3
E 0.23 2
F 0.23 3
R
5.2 5.4 5.6 5 .8 6.0 6 .2 6.4 6.6 6.8 7.0 7.2
profit
-4 0 -2 0 0 2 0 4 0 6 0 8 0 10 0 12 0 14 0 16 0
A B C D E F
圖二十二、最佳化目標函數在不同操作條件下的比 較
決定第一不纯物(S1)時的回流比的目標函數 Φ1 為:
Φ1=S1 結束的時間*S1 中甲醇量
此目標函數意義為尋找一個可以用最短時間將塔 頂的四氯化碳濃度提升到預定值及所回收第一不 純物中最少四氯化碳含量的回流比,意即在不同再 沸器熱量、不同回流比下,尋找一個Φ1 的最小值。
但在改變 P1 過程中的再沸器熱量之後,卻無法如 固定再沸器熱量為 0.12GJ/hr 時可獲得四氯化碳的 預定濃度 0.92,此處只能收集到組成 0.87 的四氯 化碳。由上式的目標函數經由圖二十三最佳化過程 可得出產第一不純物時的 Q 為 0.13GJ/hr 及回流比