國立台灣大學醫學院分子醫學研究所 在職專班遺傳諮詢組
碩士論文
Graduate Institute of Molecular Medicine College of Medicine
National Taiwan University Master Thesis
卡曼氏症候群患者(Kallmann Syndrome)在 KAL-1 基 因上突變的搜尋與探討—以台大醫院的病患為例 Mutations in KAL-1 gene of Kallmann Syndrome cases in
NTU Hospital
研究生:吳哲宗
英文姓名:Che-Tsung Wu
指導教授:楊偉勛教授 Advisor:Dr. Wei-Shiung Yang
中華民國九十八年六月 June, 2009
國立臺灣大學(碩)博士學位論文 口試委員會審定書
卡曼氏症候群患者(Kallmann Syndrome)在 KAL-1 基 因上突變的搜尋與探討—以台大醫院的病患為例
Mutations in KAL-1 gene of Kallmann Syndrome cases in NTU Hospital
本論文係吳哲宗君(學號 P96448001)在國立臺灣大學 分子醫學研究所完成之碩士學位論文,於民國九十八年七月 一日承下列考試委員審查通過及口試及格,特此證明
口試委員:
楊偉勛教授
(簽名)
(指導教授)
黃天祥教授
蔡文友副教授
系主任、所長 李芳仁教授
(簽名)
誌 謝
我知道在有限的時間下,這份論文還留有許多成長的空間,但這份論文的得 以完成,駑鈍的我單靠個人微薄的力量絕對無法達成,需要感謝的人實在太多….
首先要感謝台大公衛學院副院長陳為堅教授的寬容與鼓勵,讓我在工作之餘 同時還能以在職生就讀遺傳諮詢碩士在職專班。感謝實驗室同仁菁英在實驗室工 作上的分擔與幫忙。另外更要感謝指導教授楊偉勛醫師實驗室的助理語喬。
此外要感謝台大醫學院黃天祥教授、台大小兒科蔡文友醫師在個案收集、以 及研究進度上的指導與幫助。再者,更要感謝指導教授楊偉勛老師的鼓勵與指導,
若沒有老師體恤我工作上與在職生的角色,以及實驗室語喬的鼎力相助,分擔了 我半數的檢體眾多 exons 之 PCR 與定序的工作負擔,我實在無法如期獨立完成所 有檢體之定序。非常感謝語喬實驗室專業技術上的意見與提醒,定序工作上寶貴 經驗的傳承。
謝謝啟盛的耐心與包容,對於我這分生半調子常常提出的ㄅ、ㄆ、ㄇ與 A、
B、C 等級的問題適時的解惑,與不厭其煩的解說,特別是在半夜時分…;美華 在撰寫論文上的諮詢與建議,以及在排版上的大力幫忙。
士儀、Scott 彥彰的支持,讓這兩年的課業與工作在繁瑣中,還能讓生活洋溢 著歡笑與充實感。
謝謝基因飛躍公司彭春琇小姐的專業與耐心。
謝謝台大醫院細胞遺傳檢驗室張麗束學姊的指導,也謝謝淑靜姊的熱心幫忙。
另外,亦要感謝同班同學這兩年的患難與共,班代益智體貼的時時提醒、朝 義的熱忱與撰寫論文的認真態度,適時地令人鼓舞;麗滿撰寫論文與專業上的建 議,使我獲益良多,在在都是不可多得的良師與益友。
最後要感謝我的家人,媽媽的關心與大哥、二哥的鼓勵,你們的支持一直是 我的原動力。
中文摘要
卡曼氏症候群(Kallmann syndrome)患者是一群屬原發性促性腺激素分泌不 足的性腺發育低下症(idiopathic hypogonadotropic hypogonadism)且伴隨嗅覺低 下(hyposmia)或嗅覺喪失(anosmia)的患者。這樣的患者由於下視丘不分泌「促 性腺激素釋放激素」(GnRH),導致腦下垂體分泌黃體激素(LH)、濾泡刺激素
(FSH)不足、或是 LH 與 FSH 雖分泌正常,但性腺對於這兩種激素卻毫無反應 或是反應遲滯,以致性腺激素分泌不足,男性睪丸分泌的睪固酮素(testosterone), 女性卵巢分泌女性激素雌二醇(estradiol)不足,而導致性發育遲滯。一般此症候 群目前已知可分為四類型,KAL1 是 X 染色體上 KAL-1 基因突變的類型,屬於 X 染色體性聯遺傳隱性模式;KAL2 為 FGFR1 基因突變造成的,屬於體染色體顯性 遺傳;另外還有 KAL3(PROKR2 基因)與 KAL4(PROK2 基因)共四類型。
本研究收集台大醫院內科與小兒內分泌專科門診歷年診斷為卡曼氏症候群的 確診病患共八位,其中一位 25 歲女性(個案 3)、其餘七位是年齡 19 到 45 歲不 等的男性,包括一位 5 歲的男童。進行 KAL-1 基因 14 個 exons 的序列分析檢測,
希望找出這些病患於 KAL-1 基因上功能性的突變。
基因序列分析檢測結果發現突變點的位置,多發生在 exon 11(c.1600G>A)
與 exon 12(c.1833C>T),所造成的胺基酸改變為 Val534Ile 與 synonymous mutation
(Ile611),個案 1、2、3、5、7、8 均在此兩點突變,個案 5 除此兩突變點,還 有 exon 14 上的四個突變點,c.1997A>T(Lys666Met)、c.2003G>A(Arg668His)、
以及在 3’端超過中止密碼的*19G>T 與*21G>A。
個案 6 之 5 歲的男童,其突變則比較複雜,從 exon 11(Intron 10 交界處)一 直到 exon 14 共有 37 個突變點位;顯然我們 PCR 反應所擴增的基因片段很可能 並非其 X 染色體的 exon11~14,而是 Y 染色體上 Yq11.2 上不再製造任何蛋白的 pseudogene,KALP。
個案 4 則未找到任何突變的序列或點位。
iv
Abstract
Kallmann syndrome is a developmental disorder characterized by idiopathic hypothalamic hypogonadotropic hypogonadism with olfactory loss (anosmia or hyposmia:with low or poor sense of smell). These patients have delayed or absent puberty due to deficiency of hypothalamic gonadotropin-releasing hormone (GnRH), resulting in low secretion or absence of pituitary luteinizing hormone (LH), follicle-stimulating hormone (FSH). Even though the normal level of LH and FSH secretion may exist, there is no response or delayed response in sex hormone secretion from the gonads of these patients. These result in insufficient secretion of testosterone in male or ovarian secretion of estradiol in female. Currently this syndrome can be divided into four types, KAL1, caused by mutations in the KAL-1 gene located on X chromosome Xp22.3 and inherited in an X-linked recessive mode;
KAL2, caused by mutations in FGFR; KAL3, caused by mutations in PROKR2 gene, and KAL4, caused by mutations in PROK2 genes. The KAL2 and KAL4 are inherited in an autosomal dominant mode except KAL3.
In this study, we have collected a total number of eight patients with Kallmann syndrome in the clinics of Internal Medicine and the Pediatric Endocrine special clinic, in National Taiwan University Hospital. One was a 25-year-old female, and the other seven were males with their age range 19-45, and one 5-year-old boy. In this thesis, searching for mutations among these Kallmann syndrome patients were focused on the KAL-1 gene first. In order to delineate the functional mutations of KAL-1 genes among these patients, sequence analysis of all 14 exons in KAL-1 gene was performed.
We found that two point mutations within the coding sequence (CDS) region in patients (subjects) 1, 2, 3, 5, 7, 8. These two mutations were in the exon 11 (c.1600G>A) and exon 12 (c.1833C>T), leading to the Val534Ile missense mutation
and a synonymous mutation (Ile611). There were more mutations found in exon 14 of the Subject 5 in addition to the previous two point mutations. They were c.1997A>T (Lys666Met), c.2003G>A (Arg668His), *19G>T and *21G>A, and the latter two have been beyond the termination code in the 3 'end.
Subject 6 was a 5-year-old boy with a total of 37 mutations from exon 11 (at the junction between Intron 10 and exon 11) to the exon 14. It might arise from the possible incorrect amplification of the fragments from the pseudogene KALP which is homologue of KAL-1 on Y located in Yq11.2 by PCR reaction, instead of truly amplifying the real exon11-14 of KAL-1 on X chromosome.
We did not find any mutations in the sequence of Subject 4 in KAL-1.
vi
目 錄
口試委員會審定書……… i
誌謝………. ii
中文摘要……… iii
英文摘要………. … iv
第一章 研究背景與動機 ... 1
一、卡曼氏症候群(Kallmann Syndrome)簡介 ...1
二、卡曼氏症候群(Kallmann Syndrome)的臨床表徵與診斷 ...2
三、卡曼氏症候群(Kallmann Syndrome)的分類 ...3
四、研究問題 ...5
五、研究假說 ...5
六、研究動機 ...6
第二章 研究方法 ... 8
一、研究對象 Subjects(Patients) ...8
二、人體 DNA 之萃取(Genomic DNA Extraction)...16
三、聚合酶連鎖反應(PCR amplification) ...17
四、DNA 洋菜膠電泳(Agarose gel electrophoresis)-- PCR 產物的 DNA 品質之確定...24
五、 DNA 序列的定序(DNA sequencing)與比對...25
第三章 結果 ... 26
第四章 討論 ... 39
參考文獻 ... 46
附 錄 ... 49
表目錄
表 1 個案的臨床表徵與診斷 ... 10
表 2 Hardelin et al., 1993 所載 KAL-1 gene 14 個 exons PCR 時所用之 primers pairs ... 20
表 3 個案 1~8 在 KAL-1 基因上 14 exons 的突變情形 ... 26
表 4 個案 5 的突變點位置與造成胺基酸改變的情形 ... 28
表 5 個案 6 KAL-1 基因的所有 exon 之突變情形... 35
表 6 KAL-1 cDNA 區段的 SNP 與胺基酸改變資訊(cSNP)(摘錄自 NCBI, KAL-1 cSNP in SNP database)... 39
表 7 KAL-1 的 14 exons primer pairs match KALP sequence 的情形 ... 44
表 8 KAL-1 基因 14 個 exons 之 CDS 長度與 mRNA 長度參照表... 49
viii
圖目錄
圖 1 個案 1 與個案 2 的家族圖譜 ... 9
圖 2 個案 3 之家族圖譜 ... 12
圖 3 個案 4 之家族圖譜 ... 12
圖 4 個案 5 之家族圖譜 ... 13
圖 5 個案 6 之家族圖譜 ... 14
圖 6 個案 7 的家族圖譜 ... 15
圖 7 個案 8 的家族圖譜 ... 16
圖 8 按照(Hardelin, et al., 1993)所載 PCR thermal cycle 條件之 KAL-1 基因 14 個 exon PCR 產物電泳圖... 19
圖 9 增多循環 cycles 數、縮短反應時間以減少雜 bands 產生與 primer 的殘留 ... 21
圖 10 提高 annealing 溫度,以減少雜 bands 及比較 DMSO 加與否的差異... 22
圖 11 女性自願者之 DNA 檢體的 PCR 產物電泳圖 ... 23
圖 12 最後 KAL-1 基因 14 個 exons 的 PCR 反應條件所呈現的電泳圖... 24
圖 13 個案 3 在 exon 11 上突變點(c.1600)之正股序列... 27
圖 14 個案 3 在 exon 11 上突變點(c.1600)之反股序列... 27
圖 15 個案 5 其 KAL-1 基因的 exon 14 之定序圖譜... 29
圖 16 個案 6 其 KAL-1 基因的 exon 11 之正股定序圖譜... 30
圖 17 個案 6 其 KAL-1 基因的 exon 12 之正股定序圖譜... 31
圖 18 個案 6 其 KAL-1 基因的 exon 13 之正股定序圖譜... 32
圖 19 個案 6 其 KAL-1 基因的 exon 13 之反股定序圖譜... 33
圖 20 個案 6 其 KAL-1 基因的 exon 14 之正股定序圖譜... 34 圖 21 個案 6 在 KAL-1 基因上 14 個 exon 的 PCR 反應產物於 2% agarose 電泳圖
... 37 圖 22 個案 5 與 6 在 KAL-1 基因 exon 10 的 PCR 產物電泳圖 ... 38 圖 23 個案 5 與 6 在 KAL-1 基因 exon 10 的 PCR 產物電泳圖(repeat 重作第二次)
... 38
第一章 研究背景與動機
一、 卡曼氏症候群(Kallmann Syndrome)簡介
卡曼氏症候群患者(Kallmann Syndrome)是一群屬原發性促性腺激素分泌不 足的性腺發育低下症(idiopathic hypogonadotropic hypogonadism;簡稱 IHH)且伴 隨嗅覺低下(hyposmia)或嗅覺喪失(anosmia)的患者。這樣的患者由於下視丘 不分泌「促性腺激素釋放激素」(gonadotropin releasing hormone;GnRH),導致腦 下垂體分泌黃體激素(luteinizing hormone;LH)、濾泡刺激素(follicle stimulating hormone;FSH)不足、或是 LH 與 FSH 雖分泌正常,但性腺對於這兩種激素卻毫 無 反 應 或 是 反 應 遲 滯 , 以 致 性 腺 激 素 分 泌 不 足 , 男 性 睪 丸 分 泌 的 睪 固 酮 素
(testosterone)或女性卵巢分泌女性激素雌二醇(estradiol)不足,而導致性發育 遲滯(Pallais, Caudill, Pitteloud, Seminara, & Crowley, 2007)。
卡曼氏症雖是西元 1944 年德裔醫生 Franz Josef Kallmann 所報告發生於家族性 類無睪症患者的一種嗅覺喪失之促性腺激素分泌不足的性腺發育低下症。但其實 追溯歷史在西元 1856 年,更早於 Kallmann 醫師 90 年的西班牙病理學家 Maestre de San Juan 就曾報告其在解剖一位性腺機能不足的男性病患遺體時,發現未發育的性 器與嗅覺球的喪失之情況。其後,西元 1954 年 De Mosier、西元 1963 年 De Mosier 與 Gauthier 的發現,建議此症可能病因源頭為下視丘區域。之後的研究者才逐漸 揭開卡曼氏症候群在病理上的病因基礎(Fechner, Fong, & McGovern, 2008)。
目前已知位於下視丘的「促性腺素釋放激素」(GnHR)的神經原細胞(neurons)
在人類正常性發育與生育上扮演著關鍵的控制者角色。但有趣的是正常的「促性 腺素釋放激素」(GnHR)的神經原細胞在胚胎發育早期,並不在腦內,而是在嗅 板,其需要倚靠嗅覺神經系統的軸突(axon)的引導,而逐漸從鼻腔移行(migration)
至腦的下視丘部位。若因為基因突變導致這類神經原的細胞移行失敗,便會造成 此類神經原組織發育駐足下視丘失敗,無法正常分泌「促性腺素釋放激素」(GnHR)
於腦下垂體門脈循環系統(pituitary portal system)中。因而無足夠的 GnHR 去刺 激腦下垂體前葉分泌黃體激素(luteinizing hormone;LH)與濾泡刺激素(follicle stimulating hormone;FSH)。而也由於嗅覺神經原細胞亦移行失敗,胚胎在之後的 發育無法讓嗅覺神經纖維正常發育,因此此類患者的嗅覺呈現遲鈍或是喪失的現 象(Herbison, 2007)。
二、 卡曼氏症候群(Kallmann Syndrome)的臨床表徵與診斷
臨床上病患通常是因為性器官短小與隱睪症狀、生長緩慢與體型瘦小或是青 春期第二性徵發育不全,體毛、腋毛與陰毛短少,或是女性原發性無月經而來就 醫。通常伴隨無嗅覺或嗅覺遲鈍。於人口中之發生率,男性約 1/10,000,女性約 1/50,000;男女比約為 5 比 1(Fechner, 2008)。
部分病患甚至伴有聽覺障礙,與腎臟發育不全症狀等其他臨床症狀,包括併 指、短掌骨等骨骼異常,唇、顎裂,先天性心臟病,腸迴轉異常,synkinesia 等不 隨意運動的神經學症狀。X-linked 性聯遺傳的患者可能合併性聯遺傳疾病如魚鱗 癬、智能障礙等(Pallais, et al., 2007, Diagnosis section)。
其臨床表徵包括:
1. 生長緩慢與體型瘦小或是青春期第二性徵發育不全,體毛、腋毛與陰毛短 少。
2. 男性性器官短小,隱睪症狀或是青春期之前的睪丸小於 4 毫升;女性原發 性無經、胸部發育不良,無腋毛陰毛發育。
3. 無嗅覺或嗅覺遲鈍。
在診斷上除了前述青春期男女第二性徵發育不全之臨床徵候以外,還需要血 液內分泌檢查與核磁共振造影檢查。
核磁共振造影檢查:
1. 下視丘與腦下垂體無外傷與異狀。
2. 嗅覺球萎縮或喪失。
3. 無其它器質性下視丘與腦下垂體異常(Pallais, et al.)。
血液內分泌檢查:
1. 黃體激素(LH)與濾泡刺激素(FSH)低下或在正常值,但血液循環中之 性荷爾蒙低下(男性 testosterone 小於 100ng/dl;女性 estradiol(雌二醇 E2)
小於 50pg/ml)。
2. 須測試甲狀腺功能包括 TSH 和 free T4 和泌乳激素(PRL)及 ACTH 誘發 測試。
3. 為與血色素沈著病(hemochromatosis)做鑑別診斷,須檢驗血清鐵、鐵蛋 白及運鐵蛋白(Pallais, et al., 2007)。
三、 卡曼氏症候群(Kallmann Syndrome)的分類
Kallmann Syndrome 1(KAL1),為 X 染色體性聯遺傳(X-linked),主要為 KAL-1 gene 上突變(包含 deletions、missense、nonsense、splice variant mutations),KAL-1 gene 位於 X 染色體 Xp22.3 的位置,包含 14 個 exons,主要負責製造具有 680 個胺 基酸組成的 Anosmin-1 蛋白,其功能主要於神經原細胞與纖維的附著(adhesion)、
軸突樹突的伸長、生長以及與 GnHR 神經原細胞的移行與上皮細胞的成形叢生有 關。目前已知有點突變與整段序列丟失(deletion),14 個 exons 上均有不同類型點 突變與缺失、丟失,沒有突變常發生的熱點(hot spot)傾向(Tsai & Gill, 2006)。
約佔卡曼氏症患者 5%-10%。
Kallmann Syndrome 2(KAL2),為體染色體顯性遺傳,主要為FGFR1 gene上 發生屬於功能喪失(loss of function)之突變(包含deletions、missense、nonsense、
splice variant mutations),而且似乎呈現出半型不足(haplo-insufficiency)的現象
(Hebert, Lin, Partanen, Rossant, & McConnell, 2003)。FGFR1 gene位於染色體 8p11.2-12,包含18個exons,主要負責製造纖維母細胞生長因子接受體-1(Fibroblast Growth Factor Receptor-1;FGFR1),為纖維母細胞生長因子四種接受體之ㄧ,對 於神經系統細胞之間訊息傳遞扮演著重要功能。嗅覺神經系統的嗅覺受體神經原 正常發育需要此受體蛋白與Anosmin-1蛋白的交互作用。已知與嗅球發育發展有 關,具有此基因之突變而導致嗅球發育受阻似乎間接引起「促性腺激素釋放激素 神經原細胞」(GnRH neurons)的移行異常,而造成卡曼氏症候群的症狀。目前已 知FGFR1 gene上發生突變而導致卡曼氏症約佔10%-17%,雖是顯性遺傳模式,但 臨床症狀的表現型卻差異頗大,呈現非完全穿透型式(incomplete penetrance),特 別是在有家族性病史之個案。彼此間表現型的變異性從性腺發育不良且嗅覺喪失 之卡曼氏症到「具有正常嗅覺之促性腺激素分泌不足的性腺發育低下症」
(normosomic IHH)、甚至到青春期發育遲滯(delay puberty)之較輕症狀者都有。
其它臨床表徵包括缺牙、隱睪、唇顎裂、單側腎發育不全等。目前研究者懷疑FGFR1 應該與KAL-1 gene在共通的路徑上具有某種交互作用,它們在「促性腺激素釋放激 素神經原細胞」(GnRH neurons)的發育與移行(migration)都扮演著重要角色
(Pitteloud, et al., 2006)。
Kallmann Syndrome 3(KAL3),為體染色體隱性遺傳,主要為 PROKR2
(prokineticin receptor-2)gene 上發生突變(missense 與 nonsense mutations),約佔 卡曼氏症患者 5%。PROKR2 基因有 2 個 exons,目前此基因突變在人類功能性上 的效應仍不清楚,只知在基因剔除鼠(knock out mice)實驗上,發現會導致嗅球 喪 失 , 與 下 視 丘 缺 乏 合 成 有 關 「 促 性 腺 素 釋 放 激 素 」( gonadotropin-releasing
hormone)的神經原細胞相關之嚴重的生殖系統萎縮。
Kallmann Syndrome 4 ( KAL4 ), 為 體 染 色 體 顯 性 遺 傳 , 主 要 為 PROK2
(prokineticin-2)基因突變(missense 與 nonsense mutations,以及 translation start sites 的改變 alterations),約佔卡曼氏患者不到 5%。 PROK2 基因具有 4 個 coding exons,
還包含一個 alternative exon 3。正常的 PROK2 基因的產物就是 PROKR2 的 ligand
(
Pallais, et al., 2007)
。四、 研究問題
目前台大醫院內科與小兒科內分泌門診所診斷為卡曼氏症的患者,雖然臨床 表徵、核磁共振造影 MRI 檢查、內分泌檢測都是確診個案,但這些個案在以往內 科黃天祥教授與小兒科蔡文友醫師的研究(黃教授之病人 4 位,蔡醫師之病人 3 位),嘗試從其基因序列中找出是否有目前文獻已知 KAL-1 基因的突變,但都未能 找到。
五、 研究假說
1. 可能剛好國內的患者其先天基因上的突變,便非文獻上所常描述之 KAL-1~KAL-4 的基因突變(僅佔 20%-25%),而是另外的 75%-80%未知 罕見的其他基因之突變所造成,如:GnRH、GnRH-R、GPR54、KISS1 等,
或是其他(Trarbach, Silveira, & Latronico, 2007)。
2. 僅由 DNA 序列分析(Sequence analysis)的方式檢測,對於女性個案因 不同 X 染色體上是否有某單一 X 染色體(父方或是母方)缺失或丟失一
段,因基因劑量(gene dosage)不同而致病(haplo-insufficiency),此法 無法檢測出。
這是 DNA 序列分析的限制。
無法如:螢光原位雜交(FISH)、或是微陣列晶片 array-CGH 去比對發現 出是否有某一 X 染色體具有微缺失,以及確切缺失的位置(Pallais, et al., 2007)。
六、 研究動機
若非實驗條件未能充分配合,以致無法再現檢測成果。也或許是台大門診所 診治之個案並非目前已知之 KAL-1~KAL-4 的基因突變(僅佔所有卡曼氏症患者的 20%-25%)。
另外,PCR 的反應條件未達再現性之需求。
PCR 所用的 Taq polymerase 為低、缺乏 proof reading 的特性
(Hardelin, et al., 1993)所記載僅 primers 序列與 PCR cycle 的溫度、時間 條件,並未述及其用何種 Taq polymerase 或是其他種 PCR 聚合酶,以及 其他鹽類、離子(如:MgCl2)濃度的特殊條件。
PCR 產物定序 sequencing 時,反應條件未達再現性之需求。
PCR 產物純度、量,不足夠達再現性之需求。
所用的 Taq polymerase 未達條件或是其實應該用他種 PCR 聚合酶。
此外,也有可能以前蔡醫師所檢測參考的 PCR 之 primers 以及檢測基因片段 方式,並非全部 KAL-1 基因的 14 個 exons 均搜尋(Parenti, et al., 1995)。因此,本 研究嘗試從其之前 KAL-1 基因檢測方法的 Protocol 與 PCR 的條件、Primer 的序列 重新審視,企圖重新按照(Hardelin, et al., 1993)檢測的條件再確認檢測一遍,以
作為是否朝卡曼氏症其他基因突變(另外 75%-80%)的方向去尋找的基礎。
若是因為改換正確聚合酶、PCR 條件,PCR 產物純度提高,而能找到 KAL-1 基因上的突變最好。若一再確認上述假說中所提及之因素已排除,仍找不到 KAL-1 的基因突變,或許可作為將來學弟、學妹們或台大其他研究者往 KAL-2~KAL-4 或 其他基因突變的方向去努力思考與搜尋的基礎。
第二章 研究方法
原則上希望尋找卡曼氏症候群(Kallmann Syndrome)的患者,然後在這些符 合診斷個案的DNA序列中尋找是否有國外文獻所探討已知KAL-1 gene的突變。所 以,需要採集個案的DNA,本研究以周邊靜脈血採血3ml,經萃取成Genomic DNA 之後,再設法以DNA為模板作聚合酶連鎖反應PCR。由於目前已知KAL-1 gene上的 突變Mutation所造成的KAL1型的卡曼氏患者,在KAL-1 gene的14個exons上並未發 現突變的好發頻率較高的區段(hot spots)(Tsai & Gill, 2006),因此所有14個exons 均要搜尋看是否有PCR產物,再經sequencing定序,去比對是否有突變序列或點位。
一、 研究對象 Subjects(Patients)
個案主要是由台大醫院內科黃天祥教授與小兒科蔡文友醫師的內分泌門診收 案,符合青春期性腺發育不全(血中性腺激素低下)、下視丘腦下垂體腺無解剖 學上之異常且嗅覺喪失(嗅球的MRI,發現嗅球萎縮或喪失),血液中濾泡刺激素 FSH與黃體素LH在正常範圍或是低下的Kallmann Syndrome卡曼氏症候群患者診 斷。本研究主要鎖定在KAL-1基因的突變搜尋,屬於性聯遺傳的卡曼氏症患者
(X-linked Kallmann Syndrome),一般而言會是以男性患者居多。經門診回診時,
告知詳情徵求同意參加研究者,在簽署(未成年者由監護人簽署)臨床研究同意 書之後,於門診追蹤回診時收案進入本研究。但由於卡曼氏症候群患者原本發生 率就不高(萬分之一)目前總共收案進入本研究者,黃天祥教授門診有四位(個 案1~4)與蔡文友醫師門診有三位(個案5~7)病患個案,蔡克嵩醫師門診一位,
共八位病患同意進入本研究。詳細臨床表徵請參照表1。
個案1與個案2是一對32歲的雙胞胎兄弟,於19歲兵役體檢時發現性發育不
足,第二性徵不明顯,外生殖器短小,於民國84年10月21日轉至台大住院,經血 液內分泌檢查、核磁造影檢查、心理測驗檢查等,確診為卡曼氏症候群患者。個 案1(雙胞胎中之哥哥),不僅嗅覺喪失,從五歲因其智力溝通發展緩慢,才發現 其聽覺損傷,需戴助聽器,19歲時於台大施作心理測驗,WAIS智力測驗語言智商 68,操作智商84,總智商數73,介於智能不足邊緣,但不排除是因為從小聽力缺 損導致影響學習。個案2則智力正常,高中成績中等,大學聯考數學還達40分,除 了聽力正常外,其餘臨床表現與個案1同,兩位個案均長有「多發性骨性軟骨瘤」
(multiple exostosis)。個案1與個案2家族圖譜請參見圖1。
圖 1 個案 1 與個案 2 的家族圖譜
表 1 個案的臨床表徵與診斷
表1 個案的臨床表徵與診斷
個案3是一位25歲女性,青春期時無初經而來台大診斷為卡曼氏症。嗅覺喪失,左 耳自小聽覺不好。家族圖譜請參見圖2。目前已知個案3的哥哥也是符合卡曼氏症 診斷,但其個人並未同意進入本研究。
圖 2 個案 3 之家族圖譜
個案4是一位45歲男性,於35歲時因第二性徵不明顯,外生殖器短小,亦具有 男性女乳化特徵。來台大確診為卡曼氏患者。家族圖譜請參見圖3。
圖 3 個案 4 之家族圖譜
個案5是一位19歲男性,於民國78年剛出生時,於署立新竹醫院因外生殖器短 小與隱睪症狀轉診至台大,隱睪症狀後經手術使其睪丸正常沉降至陰囊中,但之 後睪丸發育仍有「男性假性雌雄同體陰陽人」(male pseudohermaphroditism)的表 徵,但腎臟超音波檢查兩側腎臟並無異常,經一連串檢查之後確診為卡曼氏症候 群患者。家族圖譜請參見圖4。
圖 4 個案 5 之家族圖譜
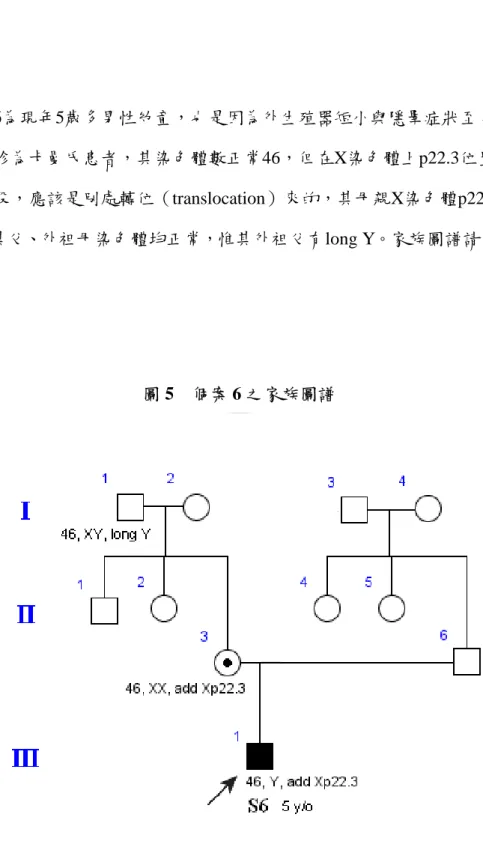
個案6為現年5歲多男性幼童,也是因為外生殖器短小與隱睪症狀至台大診 治,經確診為卡曼氏患者,其染色體數正常46,但在X染色體上p22.3位置多一段 染色體片段,應該是別處轉位(translocation)來的,其母親X染色體p22.3也是多 一小段,其父、外祖母染色體均正常,惟其外祖父有long Y。家族圖譜請參見圖5。
圖 5 個案 6 之家族圖譜
個案7為現年20歲男性,其因13歲青春期發育遲滯,14歲時仍然身材矮小而至 台大醫院診斷,經確診為卡曼氏症患者。其染色體為46,XY,9q12+,16qh+,long Y,第9號染色體上q12的位置,顯微鏡下染色質有稍微多一些,另外第16號染色體 q臂近中心點位置,也是顯微鏡下異質染色質(heterochromatin)有稍微多一點點;
Y染色體亦然。家族圖譜請參見圖6。
圖 6 個案 7 的家族圖譜
個案8為內科蔡克嵩醫師之病人,現年35歲已婚男性。於民國93年7月因咽膜 炎鼻漏不止求助台大,經MRI診斷才發現其蝶竇底部有先天缺陷,而造成腦脊髓液 鼻漏,經開刀以「蝶竇蝶鞍修補術」將其蝶竇底部組織修補好。住院期間因其MRI 檢查發現其蝶竇(sphenoid sinus)組織有疑似腦下垂體腺瘤(pituitary adenoma)
的跡象,為進一步確診,才發現其性發育不全,經蔡克嵩醫師診斷為卡曼氏症。
其性發育不全在蔡克嵩醫師門診持續補充雄性激素(testosterone),民國95年結婚,
希望能生育子女,於民國95年9月開始作「人類停經激素/絨毛膜促性腺激素」
(HMG/HCG)治療,期望能促進精子發育,但一直未能成功。家族圖譜請參見圖 7。
圖 7 個案 8 的家族圖譜
二、 人體 DNA 之萃取(Genomic DNA Extraction)
以EDTA為抗凝劑之5ml的真空採血管,採週邊靜脈血液檢體3ml全血,送實驗 室萃取DNA。檢體運送至實驗室之後,採用Qiagen Puregene DNA Blood Kit D-50K 試劑組萃取之。
將全血加入尖底塑膠離心管中,加入三倍體積之紅血球破壞溶液(RBC Lysis
Solution)上下搖晃混合10次,靜置5分鐘於室溫(15-25度C),讓RBC破裂溶解完 全。然後以2,000g(3,000rpm)離心10分鐘,讓白血球沉澱下來,小心倒掉上層液,
然後用殘留的溶液約200ul,加入Cell Lysis Solution 3ml混合,將白血球沉澱震動混 合均勻,以利白血球溶解破裂,另外再加入15ul的RNase A solution,上下搖晃混合 25次,置於37度C,15分鐘,再用冰浴降溫三分鐘。然後加入三分之ㄧ體積(1ml)
的蛋白質沉澱溶液Protein Precipitation Solution,高速強烈震動20秒,離心5分鐘於 2,000g(3,000rpm)。然後將上層液小心加入於盛有10ml体積的100%異丙醇 isopropanol新的乾淨離心管中。然後輕微上下顛倒搖晃50次直到有絲狀(DNA)產 生。然後再次離心5分鐘於2,000g(3,000rpm)。倒掉上層液,再加入70%的乙醇
(ethanol),上下搖晃數次去清洗(wash)DNA的沉澱物(pellet),離心5分鐘於 2,000g(3,000rpm),然後小心地倒掉上層液,將殘留液體吸乾,讓DNA pellet晾 乾至少5分鐘。回溶DNA於DNA Hydration Solution中,然後儲存於-20℃冰箱中保 存。
三、 聚合酶連鎖反應(PCR amplification)
按照(Georgopoulos, et al., 1997;Hardelin, et al., 1993)所記載聚合酶連鎖反 應PCR的條件,個案的Genomic DNA各取100ng當作聚合酶連鎖反應PCR的模板
(Template),用來擴增放大(amplification)KAL-1基因的14個exons的聚合酶連 鎖反應(PCR)合成產物DNA片段(amplicon)。而KAL-1基因的14個exons之每個 PCR反應所用的Primers序列則參考(Hardelin, et al., Table 2)所記載,參見表2。
PCR 反 應 的 溫 度 條 件 則 如 前 ( Georgopoulos, et al. ) 文 獻 所 描 述 的 : 94°C denaturation,30秒;60°C annealing(除了exon 1在此步驟為63°C;exon 14在此步 驟溫度為55°C外,其餘exon 2-13均在60°C annealing,30秒,以避免反應產生大分 子量的雜band產物);72°C extension,30秒;總共31個循環cycles。Primers 使用
100 nM 的濃度each。針對exons 1 與10,PCR反應條件須要加入10% DMSO,以避 免在擴增exon 1產物時會有次級結構產生,而在擴增exon 10時會與Y染色體上 homologous的pseudogene產生非專一性結合。此 protocol 的 modification 理由簡 略說明如下:
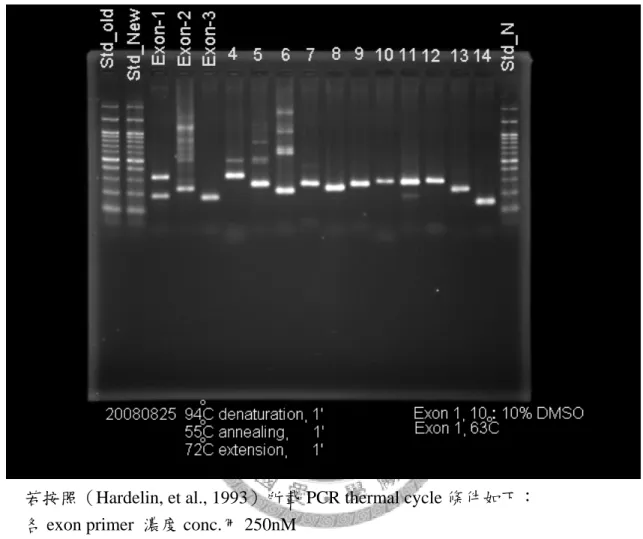
1. 原條件會有雜 bands 產生。Primer dimers 殘留。見圖 8。
結果 14 個 exons PCR 產物的 agarose 電泳膠圖(見圖 8),顯示除了 exon1 會在 175bp、300bp 以上各有一產物外,exon 2、4、5、6、7 則在產物的 後面有雜 bands,exon 11 則在產物前端有一依稀可見的較短之雜 band,顯 然需要提高 annealing 溫度,或是縮短一點反應溫時間;所有各 lane 產物 最前端有霧狀 primer dimers 殘留,可能亦需要增多循環 cycles 數,或是調 低 primer 加入的濃度量。
2. 增加 cycle 數,縮短一點反應時間,以減少雜 bands 產生與 primer 的殘留。
見圖 9,利用兩位男性自願者之 DNA 做 PCR 反應模板,增多循環 cycles 數、縮短反應循環 cycles 時間各為:
94°C denaturation, 40”;
55°C annealing, 30”;(exon 1 在此步驟為 63°C)
72°C extension, 40”;
總共 34+1 個循環 cycles Exon 1 & 10 with 10% DMSO added
結果雜 bands 明顯變少,但仍不盡理想。不過透過檢體來自不同兩位男性 自願者之 DNA,exon 1 之產物均會出現兩條產物,各在 175bp 與大於 300bp 左右的長度。顯示該兩條產物,應該是在 exon1 此 primer pairs sequence 與 溫度條件下之現象。
圖 8 按照(Hardelin, et al., 1993)所載 PCR thermal cycle 條件之 KAL-1 基因 14 個 exon PCR 產物電泳圖
若按照(Hardelin, et al., 1993)所載 PCR thermal cycle 條件如下:
各 exon primer 濃度 conc.用 250nM 94°C denaturation, l min;
55°C annealing, l min;(exon 1 在此步驟為 63°C)
72°C extension, l min;
總共 30+1 個循環 cycles
Exon 1 & 10 with 10% DMSO added
表 2 Hardelin et al., 1993 所載 KAL-1 gene 14 個 exons PCR 時所用之 primers pairs
圖 9 增多循環 cycles 數、縮短反應時間以減少雜 bands 產生與 primer 的殘留。
3. 提高 annealing 溫度,以減少雜 bands,及比較 DMSO 加與否的差異。見圖 10。
為避免 primer dimers 殘留,改以較低之 primer 濃度 100nM 94°C denaturation, 30”;
60°C annealing, 30”;(exon 1 在此步驟為 63°C)
72°C extension, 30”;
總共 30+1 個循環 cycles
上:DNA of M1(male volunteer #1)Exon 1 & 10 with 10% DMSO added 下:DNA of M2(male volunteer #2)without any DMSO(exon 1 在此步驟為
63°C)
顯示 primer dimers 及雜 bands 已消失,若 exon 1 未加 DMSO,則雖無 175bp 之雜 band,但該出現 329bp 的產物則無法順利產生;反而產生大於 1Kbp 的產物。
圖 10 提高 annealing 溫度,以減少雜 bands 及比較 DMSO 加與否的差異
圖11為女性自願者之DNA檢體的PCR產物電泳圖,exon 14的產物很微弱,顯 然60°C並不適合exon 14之反應,應該繼續維持在55°C。
圖 11 女性自願者之 DNA 檢體的 PCR 產物電泳圖
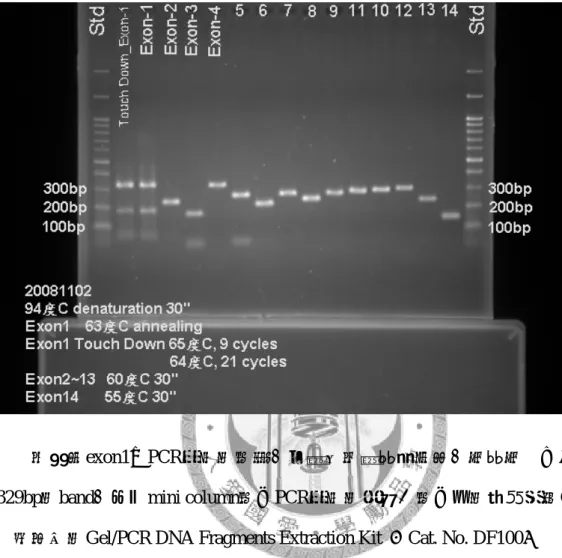
另外如圖12,由於annealing溫度由55°C提高至60°C,為免exon 2-13 PCR的反 應產物產率不佳,MgCl2的濃度需要3mM,比一般1.5mM多一倍。因為exon 1用分 斷放大(先以高一點溫度65°C,9 cycles;再以低一點溫度64°C,反應完剩餘之21 個循環數)擴增的方式,依然無法去除175bp左右長度的雜band。遂決定用切膠的 方式。
圖 12 最後 KAL-1 基因 14 個 exons 的 PCR 反應條件所呈現的電泳圖
為確保exon1之PCR產物的純度,在跑完洋菜膠電泳後,切膠切下分子量長度 329bp的band,再過mini column純化PCR產物的處理。純化用的試劑組採Geneaid 旭基科技的Gel/PCR DNA Fragments Extraction Kit (Cat. No. DF100)
四、 DNA 洋菜膠電泳(Agarose gel electrophoresis)-- PCR 產物的 DNA
品質之確定
由前述PCR amplification所產生的PCR產物DNA,於水平電泳槽,以TBE buffer
(Tris-Borate-EDTA buffer)溶液為電泳電解液,各取少許DNA的PCR產物(約 5~8ul)與5X之loading dye混合均勻,loading在2% agarose gel的well裡。於50~150V,
40~400mA 下 跑 2%agarose gel 電 泳 25~30 分 鐘 , 然 候 將 整 片 agarose gel 浸 置 於 Ethidium Bromide(簡寫EtBr)中5~15分鐘染色,再用水浸泡或沖洗5分鐘。EtBr
會結合DNA分子,於UV燈照射下於agarose gel產生螢光的帶(band)。若有band 表示DNA分子還完整(intact),若不見band出現,則表示該檢體PCR產物未產生、
DNA量太少、濃度太稀或者已經裂解,品質堪慮。
DNA Ladders Standard
使用Bioman廠牌DM100之100bp的DNA Ladders standard marker,能提供100、
200、300、400、500、600、700、800、900、1k、1.5k、3kbps各個長度的片斷。
含6X loading dye 六倍濃度的染劑。在agarose gel 電泳圖上以Std標記。
五、 DNA 序列的定序(DNA sequencing)與比對
PCR產物經純化,送定序服務,委託業界較具信譽之基基因因飛飛躍躍科科技技有有限限公公司司與與 明明欣欣生生物物科科技技有有限限公公司司,由ABI-sequencer 3130xl或3730毛細管電泳方式將DNA鹼 基序列定出來,以作為與KAL-1基因14個exons正常基因序列的比對之用。
將定序結果之序列文字檔,利用NCBI的BLAST( Basic Local Alignment Search Tool)比對工具,比對各研究對象(個案)的DNA個別exon序列與KAL-1基因genomic DNA database的最新版本序列(Ensembl 50: Jul 2008 - currently www.ensembl.org)
在DNA nucleotide序列的異同,試圖找出突變位置;再佐以比對定序波型圖譜,若 有不明確波型(peak),則再以反股PCR產物,送再次定序以確認該DNA鹼基點位 之正確性。
每位研究對象(個案)之KAL-1基因的14個exon之PCR產物,均以BLAST之 bl2seq工具、人工比對(參照波型圖)以及BioEdit程式作序列文字檔的再次比對等,
共三道手續以玆確認。
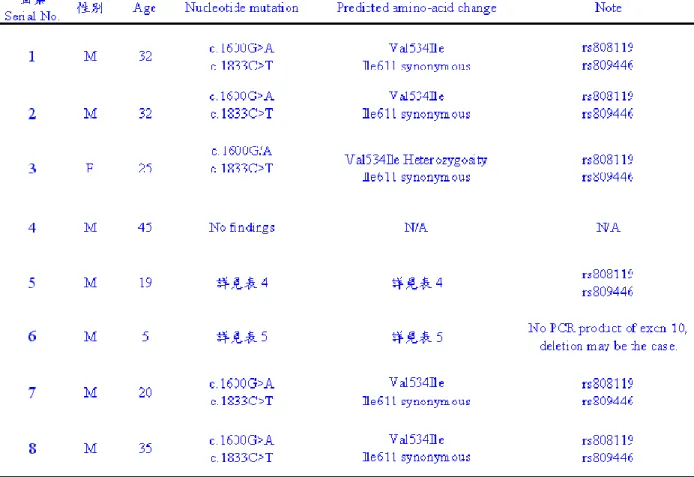
第三章 結果
我們在個案1、2、3、5、7、8的KAL-1基因之exon 11與exon 12,均發現具有 兩個共同的點突變,exon 11(c.1600G>A)與exon 12(c.1833C>T)。其中exon 11 的c.1600突變點位置造成胺基酸的改變為Val534Ile。另一點位於exon 12的c.1833則 並未改變原本該位置上之胺基酸,Isoleucine(Ile,異白胺酸),乃是屬於silent mutation的synonymous mutation。參見表3。
表 3 個案 1~8 在 KAL-1 基因上 14 exons 的突變情形
突變點位置於cDNA位置的計數,參見附錄表8。
比較特殊的是個案3因為是女性,在exon 11(c.1600)的突變屬兩不同X染色 體對偶基因上各為鹼基G或A的Heterozygosity現象,參見圖13、14。c.1600與c.1833 此兩點即SNP rs808119、rs809446(參見表6)。
圖 13 個案 3 在 exon 11 上突變點(c.1600)之正股序列
圖 14 個案 3 在 exon 11 上突變點(c.1600)之反股序列
個案4則14個exon均未找到任何一突變的序列。
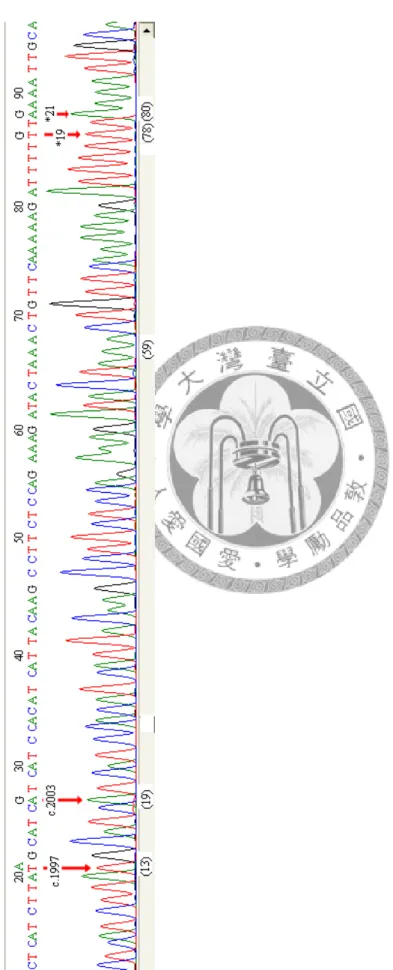
個案5除具有前述exon 11(c.1600G>A)與exon 12(c.1833C>T)此兩突變點外,
還有exon 14上的四個突變點,c.1997A>T(Lys666Met)、c.2003G>A(Arg668His),
以及在3’端超過exon 14的第59個鹼基位置的中止密碼(termination code),往下游
(downstream)方向19個鹼基位置,即第78個鹼基位置上亦有突變點*19G>T,與 第80個鹼基位置上(往下游(downstream)方向21個鹼基位置)亦有突變點*21G>A,
個案5之exon 14上的定序圖譜,詳見圖15。預測因突變而改變胺基酸之對照情形,
參見表4。
表 4 個案 5 的突變點位置與造成胺基酸改變的情形
Exon Nucleotide mutation Predicted amino-acid change
11 c.1600G>A Val534Ile
12 c.1833C>T Ile611 synonymous
14 c.1997A>T Lys666Met
c.2003G>A Arg668His
*19G>T at 78th nucleotide position of exon14, already beyond the 59th nucleotide(termination code).
*21G>A at 80th nucleotide position of exon14, already beyond the 59th nucleotide(termination code).
圖 15 個案 5 其 KAL-1 基因的 exon 14 之定序圖譜
圖15 個案5其KAL-1基因的exon 14之定序圖譜
個案6的KAL-1基因突變位置主要發生在從intron 10與exon 11交界處開始,至exon 14共37處突變,其intron 10與exon 11之突變點位置共有10處,參見其定序圖譜,圖 16。
圖 16 個案 6 其 KAL-1 基因的 exon 11 之正股定序圖譜
圖16 個案6其KAL-1基因的exon 11之正股定序圖譜
個案6的exon 12則共有突變點5處,參見其exon 12定序圖譜,圖17。
圖 17 個案 6 其 KAL-1 基因的 exon 12 之正股定序圖譜
圖17 個案6其KAL-1基因的exon 12之正股定序圖譜
個案6的exon 13(包含intron 12與exon 13交界處)則共有突變點18處,參見其exon 13 定序圖18、19。(g.IVS12-23G>C不在圖18此正股定序圖範圍內;包含於反股定序圖中)
圖 18 個案 6 其 KAL-1 基因的 exon 13 之正股定序圖譜
圖18 個案6其KAL-1基因的exon 13之正股定序圖譜
圖 19 個案 6 其 KAL-1 基因的 exon 13 之反股定序圖譜
圖19個案6其KAL-1基因的exon 13之反股定序圖譜
個案6的exon 14(含3’端UTR區域)則共有突變點4處,參見其exon 14定序圖譜,圖20。
圖 20 個案 6 其 KAL-1 基因的 exon 14 之正股定序圖譜
圖20 個案6其KAL-1基因的exon 14之正股定序圖譜
而個案6其突變點預測可能造成胺基酸之改變,參見表5。
表 5 個案 6 KAL-1 基因的所有 exon 之突變情形
Exon Nucleotide mutation Predicted amino-acid change Intron10 g.IVS10-30G>C -
g.IVS10-1G>C -
11 c.1454A>G Asn485Ser
c.1458C>T Tyr486 synonymous
c.1464T>G Ile488Met
c.1471G>C Asp491His
c.1474C>G Leu492Val
c.1516C>T Arg506Trp
c.1525delA Frame shift
c.1587G>C Lys529Asn
12 c.1678G>T Val560Phe
c.1694T>C Ile565Thr
c.1699G>A Gly567Ser
c.1708T>A Ser570Thr
c.1825T>C Phe609Pro
Intron12 g.IVS12-23G>C -
g.IVS12-16T>C -
g.IVS12-13G>A -
13 c.1848T>C His616 synonymous
c.1856T>C Leu619Pro
c.1871T>G Leu624Arg
c.1890C>T Tyr630 synonymous
c.1891C>T Arg631X
c.1908G>A Val636 synonymous
c.1932G>T Pro644 synonymous
c.1939A>C Ile647Leu
Exon Nucleotide mutation Predicted amino-acid change
c.1946C>T Thr649Ile
c.1947G>A Thr649Ile
c.1952G>A Arg651Gln
c.1955C>T Thr652Met
c.1957delC Pro653Trp
c.1958C>T Glu654Lys
c.1960_1961insA Leu655 synonymous
14 c.1997A>T Lys666Met
c.2003G>A Arg668His
*19G>T
at 78th nucleotide position of exon14, already beyond the 59th nucleotide(termination code).
*21G>A
at 80th nucleotide position of exon14, already beyond the 59th nucleotide(termination code).
此外,個案6的PCR反應產物,一直未見exon 10的band(見圖21、22與23),
由兩位有經驗的不同實驗操作者,總共PCR反應4次,有三次絲毫不產生exon 10的 band,僅有一次有微弱的band,而且僅是一點點少許螢光,很可能是PCR反應擴增 到Y染色體上的pseudogene KALP的同源homologous exon。很可能個案6在exon 10 就整個丟失(deletion)了。
圖 21 個案 6 在 KAL-1 基因上 14 個 exon 的 PCR 反應產物於 2% agarose 電泳圖
圖 22 個案 5 與 6 在 KAL-1 基因 exon 10 的 PCR 產物電泳圖
圖 23 個案 5 與 6 在 KAL-1 基因 exon 10 的 PCR 產物電泳圖(repeat 重作第二次)
第四章 討論
個案1、2、3、5、7、8均在KAL-1基因的exon 11(c.1600G>A)與exon 12
(c.1833C>T)發生點突變,其中exon 11的c.1600突變點位置造成胺基酸的改變為 Val534Ile,就是在KAL-1 gene的蛋白質Anosmin-1的肽胜鏈(popypeptide)上的第 534個胺基酸位置(residue)由Valine(Val,纈胺酸)變成Isoleucine(Ile,異白胺 酸)。Valine與Isoleucine均屬非極性疏水性胺基酸,且沒有很大的官能基或官能團 在其支鏈上,導致的蛋白質3D結構上構形的大改變之機率可能不大;另一點位 c.1833則位於exon 12相對應是在Anosmin-1的肽胜鏈(popypeptide)上的第611個胺 基酸位置,但是並未改變原本該位置上之胺基酸,Isoleucine(Ile,異白胺酸);
乃是屬於silent mutation的synonymous mutation。NCBI、Ensembl等public database 顯示這兩突變點均是屬於蠻常見的多型性(common polymorphism),參見表6。
表 6 KAL-1 cDNA 區段的 SNP 與胺基酸改變資訊(cSNP)(摘錄自 NCBI, KAL-1 cSNP inSNP database)
這兩點經查美國國家衛生研究院(NIH)生物科技資訊中心(NCBI)的公共 資料庫,發現此兩突變變異點,實際上是屬單核甘酸多型性的變異點SNP(Single nucleotide polymorphism)。此兩點即分別為c.1600(rs808119)與c.1833(rs809446), 見表6。且後者rs809446的C minor allele的allele frequency既使在不同研究團隊與資 料庫中,華人漢族裔的allele frequency高達31.2-34.4 %。然而前者c.1600G>A
(rs808119)目前尚無其allele frequency資料在NCBI或相關的資料庫中。
個案1與個案2是一對雙胞胎兄弟,目前單由病歷資料無法判斷他們是屬於同 卵雙胞亦或是異卵雙胞胎,由於他們均有長「多發性骨性軟骨瘤」(multiple exostosis),會令人往同卵雙胞方面推測,但在聽覺上,有一位是正常、另一位則 自小聽障需輔以助聽器。目前已知「遺傳性多發性骨性軟骨瘤」(hereditary multiple exostosis)的致病基因EXT1、EXT2各位於8q24.11-q24.13與11p12-p11(Schmale, G.
A. et al., 2008),似乎與X-linked的KAL-1較無直接關聯。兄弟兩者KAL-1基因序列 分析結果亦是exon 11(c.1600G>A)與exon 12(c.1833C>T)發生點突變。但已知 這兩突變點屬多型性,因此這二點的突變,可能並不是引致這兩兄弟卡曼氏症的 原因。
個案3其實也是發現在前述兩鹼基位置突變,只是因為個案3是女性,具有兩 個X染色體,序列分析的結果發現其exon 11的變異點rs808119(c.1600G/A)呈現 Heterozygosity,即一個X上的點位是G,另一X染色體上的點位是A。個案3亦是嗅 覺喪失(Anosmia),青春期無月經來潮,性腺發育不全,亦有聽覺障礙,不過是 單側。
就個案1、2與3而言,或許是前述假說段落所提及之理由,才使得內科黃天祥 教授與小兒科蔡文友醫師之前在對於他們的基因檢測,一直未能找到KAL-1的基因 上的突變。另外因為個案3是女性,若其症狀是為KAL-1基因突變造成的,由於是X 染色體的性聯遺傳,女性是患者機率上較男性低。必須是其哥哥也是確診屬於 KAL1才成立。不然其極可能是其他(如FGFR1,KAL-2)基因的突變所造成(Franco,
B. et al., 1991)。目前已知個案3的哥哥也是符合卡曼氏症診斷,但其個人並未同意 加入本研究。
個案5、個案7與個案8也是具有前述exon 11與exon 12兩點突變(c.1600G>A)
(c.1833C>T),即c.1600(rs808119)與c.1833(rs809446)兩SNP。個案5另外在 exon 14還有四個突變點,c.1997A>T(Lys666Met)、c.2003G>A(Arg668His)、
以及在3’端的*19G>T與*21G>A。c.2003G>A(Arg668His)有報告說(Georgopoulos, et al., 1997)其實是屬於在健康的人也會有的多型性(polymorphism);c.1997A>T
(Lys666Met)目前則尚未找到有報告其與任何臨床表徵直接相關。而這兩點 c.1997A>T(Lys666Met)、c.2003G>A(Arg668His)在NCBI與Ensembl等公共資 料庫裡尚未有任何資料顯示其已被確認為SNP。*19G>T與*21G>A雖然不直接負責 胺基酸密碼轉譯,不過也許對於KAL-1基因的調控有關,這有待將來進一步研究與 探討。此前述個案5、7、8這三位患者,除個案5突變點較多外,個案7以及個案8 與稍前述及的個案1、2與個案3,他們的臨床症狀與表徵,似乎在KAL-1基因的CDS
(coding sequence)上找不到突變點與其直接相關。
個案7的染色體Karyotype報告為46,XY,9q12+,16qh+,long Y,經諮詢與 委請台大醫院細胞遺傳檢驗室張麗束學姊再次對Karyotype的照片圖影本作確認判 讀,雖然其9q與16q靠近centromere位置有點染色質較深較黑,但都還是屬於正常 族群的polymorphism的範圍內,Y染色體其實也並未長過18號染色體,也只是染色 質稍深一點點,仍屬正常Y。就個案7其染色體圖判讀,外觀仍是屬於正常46,XY 的Karyotype。
比較奇怪的是個案4,與個案1、個案2一樣有男性女乳化(Gynecomastia)症 狀、生殖器發育遲滯(micropenis, little axilary & pubic hair)、嗅覺喪失(Anosmia)
以及聽覺障礙(Auditory (Hearing) impairment)等卡曼氏症的臨床表徵,但整個 KAL-1基因的14個exon之序列卻檢查不出任何異常或突變的地方。顯然可能不是 KAL-1基因上的CDS突變造成他的症狀,亦或者是丟失的exon片段,剛好PCR反應
到Y染色體上的KALP pseudogene上的homologous region,所以,我們會以為找不到 突變點,雖然這樣機率不高,但還是無法完全排除其可能性,除非我們進一步用 FISH或是array-CGH的技術再做進一步確認。
此外,序列分析結果呈現最複雜的,莫過於個案6這位5歲的小朋友,從exon 11
(Intron 10交界處)一直到exon 14共有37個突變點位,很顯然要在一次受精卵的發 育就有這麼多的突變似乎不太能,既使是來自於父母,那父或母早就有症狀,怎 麼能夠讓性腺發育到能孕育下一代(個案),病歷上未特別記載父母是否有因性腺 發育遲滯而接受賀爾蒙治療,所以,推測PCR反應的產物擴增到了Y染色體上的同 源pseudogene KALP上的機率極大。KALP是KAL-1在Y染色體上同源演化的對偶基 因,經過 靈長類演化的過程,變成Y染色體上Yq11.2上不再製造任何蛋白的 pseudogene。除了KALP僅總共有11個exons,無KAL-1的exon 3、exon 8與exon 9,
KALP與KAL-1在序列上具有高度相似性89.7~98.9%(Del Castillo, I., et al. & Incerti, B. et al., 1992)。
另 外 , 個 案 6 的 病 歷 記 載 個 案 的 染 色 體 檢 查 報 告 Karyotype 為 46, Y, add (X)(p22.3),即Y染色體正常,而X染色體p22.3位置多嵌上了一段不知來自於何處 的染色體小片段。其母親則是46, XX, add (X)(p22.3)也是有一段染色體小片段嵌入 Xp22.3的位置上,其父為46, XY正常,而其外祖母是46, XX,外祖父是46, XY, long Y。
很可能個案多嵌上了這一段染色體小片段於Xp22.3,是來自於其母親給的X染 色體。而其母親在Xp22.3位置多的這一小片段,有可能是來自於外祖母的減數分 裂,也有可能是來自外祖父的減數分裂;若是來自於外祖父,可能是外祖父的Y,
於減數分裂重組(recombination)時,轉位(translocate)黏上了X染色體,給了個 案母親。而個案母親add (X)(p22.3)的X又傳給了個案。所以個案的X染色體上的 p22.3位置,可能因這嵌入的片斷剛好就是其外祖父Yq11.2上的同源KALP區段,因 為減數分裂重組時,轉位(translocation)嵌上了X染色體,透過個案母親遺傳而來。
才會定序結果是如此紊亂,突變位置點位眾多。也有可能嵌入了來自別處基因的 片斷,破壞了原Xp22.3位置KAL-1的完整性,造成我們PCR反應反而擴增到Yq11.2 上的同源pseudogene KALP上了。
但個案KAL-1的exon 10很顯然是整個丟失(deletion),而我們在PCR反應 所得產物,從Intron 10與exon 11交界一直到exon 14所PCR反應擴增到的產物片 段,便已經是突變紊亂的現象。因此,很可能個案6的KAL-1基因從exon 9以後,即 exon10一直到exon 13(或exon 14)就整個大片段丟失,才會造成我們PCR反應擴增不 到Xp22.3上的KAL-1,反而擴增(amplified)到Y染色體上的KALP的同源區段去了。
參見表7,14 exons primer pairs match KALP sequence的情形,顯示利用原本KAL-1 的14 exons的14對primers,真的去比對是否吻合(match)Y染色體的KALP基因區 段時,發現exon 2、exon 5、exon 11、exon 13的reverse strand的primer sequence序列,
還完全吻合到KALP基因區段相對應同源的exon區域;就算是其他未完全吻合的 exon primers,如: exon 6、exon 7、exon 10、exon 11的forward strand,以及exon 12 的reverse strand也僅差1個鹼基;exon 5的forward strand、exon 10的reverse strand以 及exon 12的forward strand,也僅差別2個鹼基。因此,初步判斷,在某些恰好的溫 度、或其他條件符合下,PCR反應可能也無法排除不放大擴增到Yq11.2的KALP基 因之區段。亦或者是這Yq11.2恰好是經重組轉位,嵌入了Xp22.3位置的片段,所以 我們PCR一直擴增到此處的產物。
表 7 KAL-1 的 14 exons primer pairs match KALP sequence 的情 形
Exon Forward (F) or Reverse (R) strand
Similarity of primer in nucleotide within KALP
2 R Perfect match
5 F ---Cg
---TT
R Perfect match
6 F a---
g---
7 F ---ACTggg
---TCTggg
10 F ---AgC
---ggC
R ggAAgT---
AgAAgC----
11 F AATATg---
AATATT---
R Perfect match
12 F ---CCAgTC--- ---TCAgTT---
R ---TgT---
---CgT---
13 R Perfect match
14 F ATGTT---
ATGTg---
至於個案6的exon 14定序結果是屬於KAL-1的產物還是Y上的KALP的產物,則 需要進一步釐清,因為單從個案6的突變點眾多,從Intron 10與exon 11交界一直到 exon 14,我們也許乍看會推測應該是個案6從exon 10就丟失一直到exon 14,甚至 因為前述個案5其exon 14也有四個突變點,「c.1997A>T(Lys666Met)、c.2003G>A
(Arg668His)、在3’端的*19G>T與*21G>A」與個案6的exon 14突變情形一樣,進 而 也 推 斷 個 案 5 可 能 也 與 個 案 6 一 樣 擴 增 到 KALP 。 但 前 已 提 到 exon 14 上 的 c.2003G>A(Arg668His)可能是一般的多型性(Georgopoulos, et al., 1997),所以,
也許個案6的exon 14還在,才會擴增到此4點。而且,有趣的是當我們將KAL-1與 KALP在公共資料庫的Genomic sequence最後的exon序列放在一起比對BLAST,發 現兩同源exon高達96%的相似序列,其CDS不同的點位居然就是前述個案5與個案6 的exon 14之四個突變點。不過,縱然如此,我們還是無法就此確認我們PCR擴增 到的個案5與6的exon 14產物,就是來自其KALP的產物。假若是,那為何另外六位 個案在同樣PCR條件,exon 14產物並沒有發現這四個突變點;也許我們可以找exon 14前後(Intron 13與exon 14之3’-UTR)比較特殊unique,能區別KAL-1與KALP基 因的序列,重新設計primer,來重新PCR擴增最後exon產物定序之,說不定可以釐 清。因此,個案5與個案6的exon 14之四個突變點,值得我們進一步去探討與確認。
要證明前段敘述所執「PCR擴增到Yq11.2的KALP基因之區段」的假說,則還 需要許多工作,例如:利用FISH或是array-CGH的技術再去定位個案6的add (X)(p22.3)片段,以及其KAL-1基因的Intron 10與exon 11交界一直到exon 14,是否 還存在,然後再與個案母親以及外祖父一一作比對、定位,才能進一步一一排除,
確定是前述的哪種可能性產生的現象。
此外,也可利用Long range PCR,乾脆將個案6的KAL-1基因整個複製下來,或 是將個案6的KAL-1基因整個cloning下來,再來重新設計primer,re-sequencing,確 認與驗證其KAL-1基因上的區段序列,是否如我們先前的定序結果般地如此紊亂,
突變眾多。但這些都是需要投入更多時間的工程。
參考文獻
Albuisson, J., Pêcheux, C., Carel, J. C., Lacombe, D., Leheup, B. Lapuzina, P., et al.
(2005) Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2).
Human Mutation, 25(1), 98-99.
Ballabio, A., Bardonit, B., Carrozzo, R., Andria, G., Bick, D., Campbell, L., et al. (1989) Contiguous gene syndromes due to deletions in the distal short arm of the human X chromosome. Proceedings of the National Academy of Sciences of the United States of America, 86, 10001-10005.
Del Castillo, I., Cohen-Salmon, M., Blanchard, S., Lutfalla, G. & Petit, C. (1992) Structure of the X-linked Kallmann syndrome gene and its homologous pseudogene on the Y chromosome. Nature Genetics, 2, 305-310.
den Dunnen, J. T. & Antonarakis, S. E. (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Human Mutation. 15, 7-12.
den Dunnen, J. T. & Antonarakis, S. E. (2001) Nomenclature for the description of human sequence variations. Hum Genet 109,121–124.
Fechner, A., Fong, S. & McGovern, P. (2008) A review of Kallmann Syndrome:
Genetics, pathophysiology, and clinical management. Obstetrical and Gynecological Survey, 63(3), 189-194.
Franco, B., Guioli, S., Pragliola, A., Incerti, B., Bardoni, B., Tonlorenzi, R., et al. (1991) A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature, 353, 529-536.
Georgopoulos, N. A., Pralong, F. P., Seidman, C. E., Seidman, J. G., Crowley, W. F. Jr.
& Vallejo, M. (1997) Genetic heterogeneity evidenced by low incidence of KAL-1 gene mutations in sporadic cases of gonadotropin-releasing hormone deficiency.
Journal of Clinical Endocrinology and Metabolism, 82(1), 213-217.
Guioli, S., Incerti, B., Zanaria, E., Bardoni, B., Franco, B., Taylor K., et al. (1992)
Kallmann syndrome due to a translocation resulting in an X/Y fusion gene. Nature Genetics, 1, 337-340.
Hardelin, J. P., Levilliers, J., Del Castillo, I., Cohen-Salmon, M., Legouis, R., Blanchard, S., et al. (1992) X chromosome-linked Kallmann syndrome: Stop mutations validate the candidate gene. Proceedings of the National Academy of Sciences of the United States of America, 89, 8190-8194.
Hardelin, J. P., Levilliers, J., Blanchard, S., Carel, J. C., Leutenegger, M.,
Pinard-Bertelletto, J. P., et al. (1993) Heterogeneity in the mutations responsible for X chromosome-linked Kallmann syndrome. Human Molecular Genetics, 2(4), 373-377.
Hardelin, J. P., Levilliers, J., Young, J., Pholsena, M., Legouis, R., Kirk, J., et al. (1993) Xp22.3 Deletions in isolated familial Kallmann's Syndrome. Journal of Clinical Endocrinology and Metabolism, 76(4), 827-831.
Hebert, J. M., Lin, M., Partanen, J., Rossant, J. & McConnell, S. K. (2003) FGF signaling through FGFR1 is required for olfactory bulb morphogenesis.
Development (Cambridge, U.K.) 130, 1101–1111.
Herbison, A. E. (2007) Genetics of puberty. Hormone Research, 68(suppl 5), 75–79.
Hershkovitz, E., Loewenthal, N., Peretz, A. & Parvari, R. (2008) Testicular expressed genes are missing in familial X-linked Kallmann syndrome due to two large different deletions in daughter's X chromosomes. Hormone Research, 69(5), 276–283.
Incerti, B., Guioli, S., Pragliola, A., Zanaria, E., Borsani, G., Tonlorenzi, R., et al. (1992) Kallmann syndrome gene on the X and Y chromosomes: implications for
evolutionary divergence of human sex chromosomes. Nature Genetics, 2, 311-314.
Pallais, J. C., Caudill, M., Pitteloud, N., Seminara, S. & Crowley, W. F., Jr. (May 23, 2007) Kallmann Syndrome on GeneReviews. Retrieved March 25, 2008, from http://www.ncbi.nlm.nih.gov/books/bv.fcgi?highlight=Kallmann%20syndrome&rid
=gene.chapter.kms
Pallais, J. C., Caudill, M., Pitteloud, N., Seminara, S. & Crowley, W. F., Jr. (May 23, 2007) Hypogonadotropic Hypogonadism Overview on GeneReviews. Retrieved
March 25, 2008, from
http://www.ncbi.nlm.nih.gov/books/bv.fcgi?highlight=Kallmann%20syndrome&rid
=gene.chapter.ihh-ov
Parenti, G., Rizzolo, M. G., Ghezzi, M., Di Maio, S., Sperandeo, M. P., Incerti, B., et al.
(1995) Variable penetrance of hypogonadism in a sibship with Kallmann Syndrome due to a deletion of the KAL gene. American Journal of Medical Genetics, 57, 476-478.
Pitteloud, N., Acierno, J. S. Jr., Meysing, A., Eliseenkova, A. V., Ma, J., Ibrahimi, O. A., et al. (2006) Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proceedings of the National Academy of Sciences of the United States of America, 103(16), 6281-6286
Pitteloud, N., Zhang, C., Pignatelli, D., Li, J., Raivio, T., Cole, L. W., et al. (2007) Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proceedings of the National Academy of Sciences of the United States of America, 104(44),
17447–17452.
Sato, N., Katsumata, N., Kagami, M., Hasegawa, T., Hori, N., Kawakita, S., et al. (2004) Clinical assessment and mutation analysis of Kallmann Syndrome 1 (KAL1) and Fibroblast Growth Factor Receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. The Journal of Clinical Endocrinology & Metabolism, 89(3), 1079–1088.
Schmale, G. A., Wuyts, W., Chansky, H. A. & Raskind, W. H. (2008) Hereditary Multiple Exostoses on GeneReviews. Retrieved July 1, 2009, from
http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=ext
Trarbach, E. B., Silveira, L. G. & Latronico, A. C. (2007) Genetic insights into human isolated gonadotropin deficiency. Pituitary, 10(4), 381-391.
Tsai, P.S. & Gill, J. C. (2006) Mechanisms of Disease: insights into X-linked and autosomal-dominant Kallmann syndrome. Nature Clinical Practice Endocrinology and metabolism, 2(3), 160-171.
附 錄
表 8 KAL-1 基因 14 個 exons 之 CDS 長度與 mRNA 長度參照表
Gene Exon length (bp ) CDS Nucleotide
position Note length of mRNA
KAL1 5'-UTR 150 150
1 207 357 357
2 48 255 405
3 63 318 468
4 223 541 691
5 185 726 876
6 130 856 1006
7 206 1062 1212
8 145 1207 1357
9 147 1354 1504
10 95 1449 1599
11 172 1621 1771
12 221 1842 1992
13 142 1984 2134
14 59 2043 4180 2193
Total 2043
各exon鹼基長度參考KAL-1 gene genomic DNA database的最新版本序列
(Ensembl 50: Jul 2008 - currently www.ensembl.org)