國立台灣大學生命科學院生態學與演化生物學研究所 碩士論文
Institute of Ecology and Evolutionary Biology College of Life Science
National Taiwan University Master Thesis
微小核酸對大岩桐花朵對稱性的調控
Global analysis of small RNAs for controlling floral symmetry in Sinningia speciosa
粘雅淇
Ya-Chi Nien
指導教授 : 王俊能 博士 指導教授 : 陳荷明 博士
Advisor: Chun-Neng Wang, Ph.D.
Advisor: Ho-Ming Chen, Ph.D.
中華民國 107 年 8 月
August, 2018
中文摘要
花朵性狀的多樣發育變化形成開花植物在地球上繁盛出近三十萬種的多樣性,
其中,演化出花朵兩側對稱性的物種,具有相異的背腹側花瓣形態,不但使花形變 異更特化,花朵傳粉性狀特化也促進植物與昆蟲共演化。然而,對於建立背腹側花 瓣極性的分子機制,目前學界尚未完全知曉。MicroRNAs (miRNAs)為一種內生的
單股、非編碼的小片段RNA,長度約 20-24 個鹼基對。許多研究發現,miRNAs 參
與生物的基因表現調控,但目前尚未有研究在背腹側花瓣中探討miRNAs 的調控。
本實驗以野生型的大岩桐原種 (Sinningia speciosa) 兩側對稱花為材料,結合次世
代定序與生物資訊分析,發現 157 個 miRNAs 表現於背腹側花瓣中。其中,不僅
包含尚未在苦苣苔科發表過的conserved miRNAs,同時也有 95 個未曾在其他物種
發表的novel miRNAs。有趣的是,我們發現 miR157 與 miR390 在背腹側花瓣有差
異性表現,兩者在腹側花瓣皆偵測到較高的表現量。透過降解體定序,我們分別確 認miR157 與 miR390 的目標基因為 SPLs 與 TAS3。SPLs 曾被報導透過調控 auxin- responsive genes 來決定花器的大小;TAS3 則是藉由產生 ta-siRNAs,降解 ARF3, ARF4,同樣調控 auxin-responsive genes,以建立葉片上下表面的極性。轉錄體定序
資料顯示,大岩桐的SPLs 與 ARF3 同樣也具有背腹側表現差異。接著,我們進行
stem-loop qPCR,再次驗證了定序資料的結果。綜合以上,本研究顯示 miR157/SPLs
與miR390/ARF3 的調控路徑參與大岩桐花瓣發育。同時,此二路徑藉由調控下游
的auxin-responsive genes,可能促使背腹側花瓣相異的形態發生。進一步,我們的
發現也顯示,原先參與葉片上下表面極性的分子調控,可能擴展適應到建立花原基 背腹側的極性。本實驗由轉譯後層次探討植物背腹側花瓣的差異分化,及其對於後 續兩側對稱性的貢獻,有助於我們對於植物體花朵對稱性有更完整且全面的了解。
關鍵字:大岩桐、兩側對稱性、微小核糖核酸、次世代定序
Abstract
Establishment of dorsi-ventral polarity is essential for zygomorphic flower development, which contributes to the evolution of specialized pollination syndrome thus enhances species richness. But the mechanisms underlying the polarity establishment remain unclear. MicroRNAs (miRNAs) are embedded in the regulatory networks of plant development, but direct evidence for miRNAs regulating dorsi-ventral petal morphogenesis has been lacking.
Here, we investigated the dorsi-ventrally expressed miRNAs in the zygomorphic flower of Sinningia speciosa, an economically important ornamental plant. Using a computational pipeline, we identified 157 miRNAs in developing petals, including both conserved miRNAs that are reported for the first time in S. speciosa, as well as 95 novel miRNAs that are not found in other plants. Interestingly, we discovered that miR157 and miR390 appeared more abundant in ventral petal than dorsal part. A combination of PARE analysis allowed us to validate their targets, respectively. SPLs, which are reported to be involved in regulating the floral organ size by controlling auxin-responsive genes, and TAS3, which have been invoked in the control of the specification of abaxial/adaxial
polarity of leafs by generating ta-siRNAs, which in turn cause degradation of the AUXIN RESPONSE FACTOR 3 (ARF3) and ARF4 mRNAs. Intriguingly, our transcriptome data indicated that both SPLs and ARF3 show dorsi-ventral expression pattern. A similar result
was also observed when we experimentally validated the expression patterns by qPCR.
Our findings indicated that the regulation of miR157/SPLs may regulate the auxin- responsive genes, thus contribute to the different morphogenesis of dorsal and ventral petals. As for miR390/ARF3, the establishment of the ta-siRNA polarity is similar to the findings in leaves, which is a crucial determinant of abaial/adaxial polarity development.
As a result, our finding raises an intriguing possibility of co-option in the development of leaf abaxial-adaxial polarity into floral dorsi-ventral zygomorphy.
Keywords: Sinningia, zygomorphy, next-generation sequencing, microRNAs
目錄
中文摘要 ... I Abstract ... II 目錄 ... IV 圖目錄 ... V 表目錄 ... VI
Introduction ... 1
Results ... 6
Discussion ... 32
Conclusion ... 43
Materials and Methods ... 44
Reference ... 49
Supplementary data ... 53
Index of Figures
Figure 1 | The pipeline of sRNA-Seq analysis and DE-miRNA identification ... 7
Figure 2 | Size distribution of clean reads in sRNA-Seq ... 8
Figure 3 | Normalized expression levels of known and novel miRNAs in gloxinia ... 10
Figure 4 | A list of dorsoventrally expressed miRNAs (DE-miRNAs) ... 12
Figure 5 | The precursor of 7 DE-miRNAs can form stable stem-loop structure. ... 12
Figure 6 | T-plot of the targets cleaved by Ssp-miR157a/b, Ssp-miR390 and their pairwise target regions ... 21
Figure 7 | Expression pattern of Ssp-miR157a/b, SsSPL12 and auxin-responsive genes ... 22
Figure 8 | Experimental validation of miR157 expression level between dorsal and ventral petals .. 23
Figure 9 | Measurement of the cell length of dorsal and ventral petal ... 24
Figure 10 | Alignment of TAS3 sequences corresponding to ta-siRNAs from orthologs of 8 species . 26 Figure 11 |The expression level of Ssp-miR390 from transcriptome-Seq data ... 29
Figure 12 | The relative expression level of Ssp-miR390 and SsARF3 ... 29
Figure 13 | A model for the potential miR157/SPLs in zygomorphic flower development ... 29
Figure 14 | A comparison of the miR390/TAS3/ta-siARF pathway in leaf of Arabidopsis and rice with in the flower of Gloxinia ... 29
Index of Tables
Table 1 | Summary of the sRNA-Seq result ... 7
Table 2 | The 5’ terminal nucleotides and length of the 95 Gloxinia-specific miRNAs. ... 11
Table 3 | Target genes of DE-miRNAs in the gloxinia PARE library ... 14
Table 4 | 5’D7+ and 5’D8+ ta-siRNAs and their pairwise target region of ARF genes. ... 28
Introduction
Small RNAs (sRNAs) are key regulators of gene expression in many eukaryotic organisms (Pais et al. 2011). These molecules, mostly ranging from 21 to 24 nucleotides in length, affect all levels of developmental information in plants (D'Ario et al. 2017). A special class of sRNAs, known as microRNAs (miRNAs), can regulate the stability of the encoded transcripts for translation into functional proteins (Rubio-Somoza and Weigel 2011). Comparing to animal miRNAs, which have often hundreds of targets, plant miRNAs tend to have fewer targets, often with regulatory function, such as transcription factors and F-box protein (Rhoades et al. 2002, Jones-Rhoades and Bartel 2004). This places miRNAs in a central position within gene expression programs underlying plant development.
Flower symmetry is considered a morphological novelty that contributed significantly to the rapid radiation of the angiosperms, which already puzzled Charles Darwin and prompted him to name this phenomenon an ‘abominable mystery’ (Busch and Zachgo 2009). An important step in the diversification of angiosperms was the development of monosymmetric (zygomorphic) flowers from ancestrally polysymmetric (actinomorphic) flowers (Hileman et al., 2014). To generate a zygomorphic flower, a dorsoventral axis is established early in the floral meristem so that a specific position can
be assigned to each organ. (Blazquez et al. 2006)
Detailed flower developmental genetic studies in a few model species (e.g.
Antirrhinum majus, snapdragon) have provided a foundation for deep insights into floral
symmetry evolution (Hileman, 2014). However, it appears that previous research in this field might not have regarded the possibility of miRNA, a key regulator within gene expression programs, participating the establishment of zygomorphic flower. In addition, the current model for establishing zygomorphy may not have been completely detailed.
In certain zygomorphic flowers, various morphological differences exist in the dorsal and ventral petals, including cell size, cell number, pigmentation and epidermal microstructure etc. However, the current model for zygomorphy may not completely explain how floral differentiation along dorsi-ventral was established. Regarding that miRNAs have great significance for plant development, the main issues addressed in this research are to determine whether miRNAs have a role in establishing zygomorphic flowers.
Genuine miRNAs can be identified through detecting both the miRNAs itself and the corresponding cleaved targets using Parallel Analysis of RNA Ends (PARE) (Lin et al., 2016). Also, identification of miRNA targets is essential to better understanding the roles of miRNAs. By using PARE, target RNA cleavage products can be cloned and deeply sequenced (Jeong et al., 2013). In brief, the miRNA-mediated target cleavage is
an endonucleolytic site-specific cleavage that cuts within the target RNA strand between nucleotides 10 and 11 relative to the 5’ end of the miRNA sequence. The single – nucleotide resolution of PARE data can therefore provide detailed information about site- specific cleavages guided by the individual miRNAs in the samples examined. PARE has been a powerful approach for global targets identification and validation in diverse plant species (Lin et al. 2016, Li et al. 2017).
Sinningia speciosa (gloxinia), a member of the Gesneriaceaea, is an economically
important ornamental plant. The cultivation of gloxinia species began in the early 1800s when specimens from Brazil were brought into Great Britain (Dong et al., 2018). Since that time, gloxinia has become one of the most well-known species in the genus, and after 200 years of cultivation and breeding, the phenotypes of modern cultivars have diverged radically from those of their ancestors. The ancient gloxinia characterized by zygomorphic flowers, a derived condition within angiosperms, has reverted to actinomorphic symmetrical flowers several times during the evolutionary path, independently (Dong et al., 2018). Although some genetic studies have been carried out, miRNAs in the gloxinia have not been investigated. In addition, of Gesneriaceaea family, not a single species has been studied with a focus on global target identification using PARE data. With access to the genomic data, combined with the flexible range of material option offered by various symmetrical types, gloxinia is a powerful model for studying
the establishment and transition of floral symmetry.
Here, to better understand gloxinia miRNAs and their targets, we performed sRNA and PARE sequencing of dorsal and ventral petal. We collected developmentally staged young petals and generated two biological replicates for dorsal and ventral petal for sRNA sequencing, as well as 2 PARE libraries. The primary goal of the screen was to determine whether miRNA is a key player in building floral symmetry, and to further identify the targets of these miRNAs. Combined with the draft genomic data, we expected to capture miRNAs express in the developing petals on a global scale, including not only conserved miRNAs that have not been reported in gloxinia, but also novel miRNAs that are unique to Sinningia. Next, the dorsoventrally expressed miRNAs (DE-miRNAs) can stand out by comparing the sRNA-Seq of dorsal and ventral petals. Further, a combination of PARE analysis allowed us to validate the targets of DE-sRNAs. In the following step, since miRNAs regulate target expression negatively, we expect to observe antagonistic expression pattern of the targets in our transcriptome data. Finally, to verify our findings in the RNA sequencing data, the expression profile of DE-miRNAs and their targets were detected using qRT-PCR. Based on the findings, we propose a potential functional module that is mediated through miRNAs, the differential expression pattern of their targets are built across dorsal and ventral petals, thus leading to the distinct morphogenesis of petals, and eventually contribute to the formation of the zygomorphic symmetry. This is the first
study that comprehensively characterizes miRNAs and their associated target genes involved in the distinct morphogenesis of petals. This study provides novel insights that post-transcriptional regulation is also an important aspect of sculpting the zygomorphic floral symmetry.
Results
Overview of the Deep Sequencing Datasets
High-throughput small RNA sequencing (sRNA-Seq) is a powerful technology with high resolution for identifying and characterizing small RNAs expressed in an organism.
Genome sequence information is also indispensable to confirm that read sequences originated from the endogenous sequence, and were not artifacts or derived from contaminating organisms. In order to identify the miRNAs in gloxinia dorsal and ventral petals at a global level, we performed sRNA-Seq using the Illumina system and used computational analysis to obtain a reliable profile of Gloxinia ‘Espirito santo’ small RNAs with reference to the draft genome of Gloxinia ‘Avenida Niemeyer’.
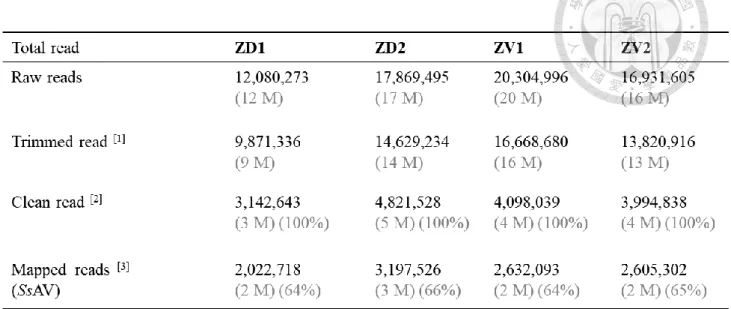
We obtained sequence data from dorsal (seq#ZD1, ZD2) and ventral petal (seq#ZV1, ZV2) of stage 5 floral bud (Supplementary Figure S1). A total of 12 to 20 million raw reads were generated from each library. (Table 1). The total numbers of trimmed reads, ranging from 18 to 36 nucleotides in length, were accessed after pre-processing. In the pre-processing, we precluded low quality reads, adaptor tags, RNAs smaller than 18 nucleotides and the structural RNAs (ie. rRNAs, tRNAs, snRNAs, snoRNAs), using the Bowtie aligner searched against the Rfam database and the genomic data of Gloxinia.
These high-quality clean reads were aligned to the genomic sequence to exclude the non- endogenous reads. The mapping rate is around 60% for 4 libraries. The unmapped sequences that did not match any position on the genome were not analyzed further and were presumed to be sequences from sequencing errors, or unclassified contaminant sequences.
Table 1 | Summary of the sRNA-Seq result
Figure 1 | The pipeline of sRNA-Seq analysis and DE-miRNA identification
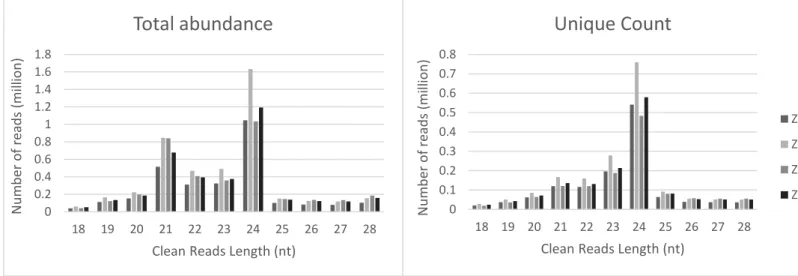
Regarding the size distribution of our mapped reads, most mapped reads (34%) were 24 nt in length. The second largest group (20%) contains reads of 21 nt in length. When the reads were collapsed into unique reads by assigning each unique read count, the amount of 21-nt reads is decrease. The finding suggested that for 21-nt reads, it is the richness, but not the diversity that contributes to the high abundance. As for the 24-nt reads, the collapse did not affect their dominance. The result indicated that, the 24 nt peak was comprised by large quantity of low-copy unique reads. This is consistent with previous reports indicating that the majority of small RNAs are 21 and 24 nt. While the 21 nt peak had the high richness of reads, the 24 nt peak was comprised by the highest number of low-copy unique reads (Hackenberg et al. 2016). It has been reported that most sRNAs functioning as small interfering RNAs (siRNAs) are 24 nt long (Rajagopalan et al. 2006); the appearance of such large numbers of 24 nts RNAs in the four small RNA libraries indicated that siRNAs accounted for a large proportion in the sRNA datasets.
These results are in agreement with previous report on cotton (Wang et al. 2015), rice (Li et al. 2011), maize (Kang et al. 2012), and Medicago truncatula (Szittya et al. 2008).
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8
18 19 20 21 22 23 24 25 26 27 28
Number of reads (million)
Clean Reads Length (nt)
Unique Count
ZD1 ZD2 ZV1 ZV2 0
0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8
18 19 20 21 22 23 24 25 26 27 28
Number of reads (million)
Clean Reads Length (nt)
Total abundance
Figure 2 | Size distribution of clean reads in Sinningia speciosa sRNA-Seq
(A) The size distribution of the clean reads calculated by the total abundance reads.(B) The size distribution of the clean reads calculated by the unique count. (non-redundant reads)
The mapped reads were used to perform the miRNA prediction by ‘miRDeep2’.
MiRDeep2 is a tool for identifying novel and known miRNAs in deep sequencing data, which performs a series of miRNA characterizations, such as mapping to reference genome sequence, discovery of miRNA clusters and prediction of MIRNA loci based on the criteria. The analytical pipeline is demonstrating in Figure 1.
By allowing three base mismatches and less than 20 genomic hits, we identified potential MIRNA loci in the genome. In a second step, potential miRNA precursors (pre- miRNAs) are excised from the genome using the read mappings as guidelines. Next, the set of known Viridiplantae mature miRNAs, retrieved from miRBase, release 21 (June 2014), are given to the module as a known miRNAs reference. In the following step, the reference is mapped against the potential pre-miRNAs. The mapped pre-miRNAs are designated as conserved pre-miRNAs, the remaining as gloxinia-specific pre-miRNAs.
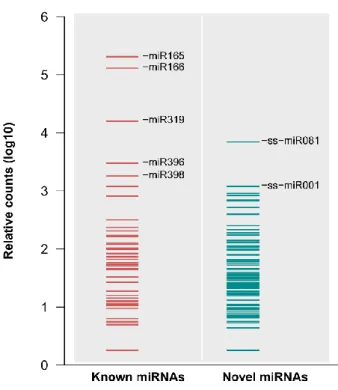
In total, 62 known miRNAs, belonging to 45 conserved miRNA families, were found to be expressed in gloxinia developing petals. The expression level of miRNAs varied among identified families. The most abundant sequence in our sequencing sets was the mature product of miR165 and miR166, which are the ancient miRNAs that repress the expression of the cognate C3HDZ transcription factor family (HD-ZIPIII) in land plants.
(Zhong et al., 2007)
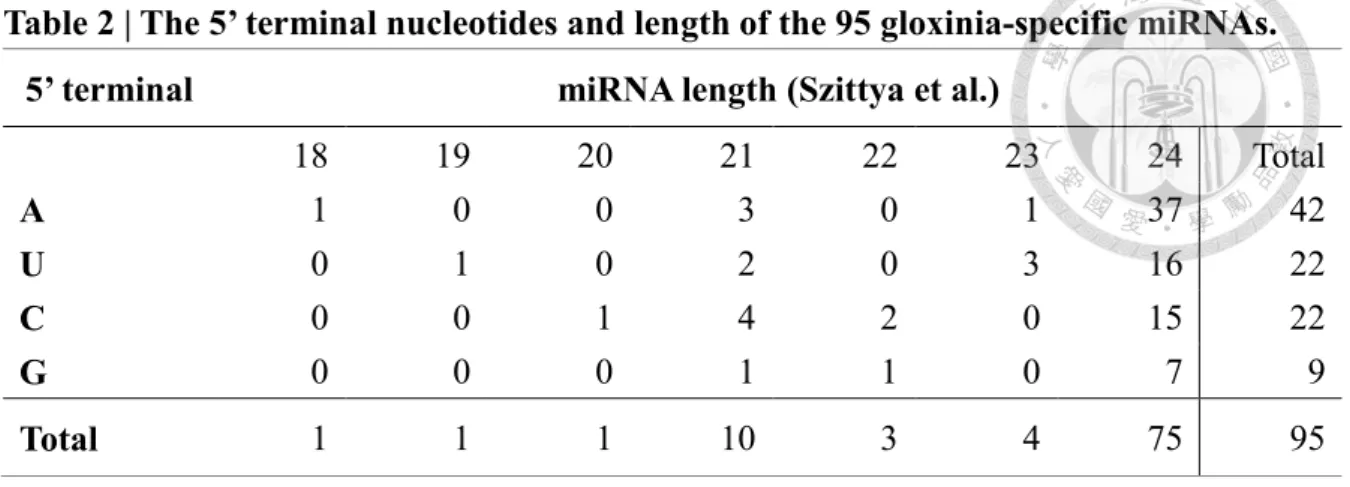
One of the most crucial advantages of high-throughput sequencing technology is that it can facilitate the detection of species-specific miRNAs with extremely low expression level. In addition to the 62 known miRNAs described above, we also identified 95 Gloxinia-specific, non-conserved miRNAs. These Gloxinia-specific miRNA candidates were named temporarily using the SspmiR-number format, before being submitted for an official designation.The lengths of the newly identified miRNAs ranged from 18 to 24 bp;
24 nt miRNAs comprised the largest category followed by 21 nt miRNAs, and other
categories were much less frequent (Table 2), which is consistent with the observation that miRNAs are typically 21 or 24 nt in plants (Wang et al. 2016). Additionally, first nucleotide bias analysis of novel miRNAs showed that adenosine (A, 44.2%) was the most prominent nucleotides at the 5’ terminus and uracil, cytosine (U, C, 23.2%) are tie for the second place, whereas guanine lag far behind the other (G, 9.5%). (Table 2).
Figure 3 | Normalized expression levels of known and novel miRNAs in gloxinia
A logarithmic graph showing the normalized expression levels of miRNAs detected in gloxinia. Red bars represent known miRNAs, and gray bars represent novel miRNAs. Abundant non-conserved miRNAs were expressed in addition to conserved miRNAs
Table 2 | The 5’ terminal nucleotides and length of the 95 gloxinia-specific miRNAs.
5’ terminal miRNA length (Szittya et al.)
18 19 20 21 22 23 24 Total
A
1 0 0 3 0 1 37 42U
0 1 0 2 0 3 16 22C
0 0 1 4 2 0 15 22G
0 0 0 1 1 0 7 9Total
1 1 1 10 3 4 75 95Characterization of dorsoventrally expressed miRNAs (DE-miRNAs)
All identified miRNAs were listed in (Supplementary Table S1). To explore the dorsoventrally expressed miRNAs (DE-miRNAs), using the quantifier module in the miRDeep2, we obtain the normalized reads count of ZD1, ZD2, ZV1, ZV2, respectively.
Next, using DESeq2 package, we perform statistical analysis between ZD and ZV sRNA- Seq data. We discovered 7 DE-miRNAs when setting the p-value cutoff at 0.05, all of the DE-miRNA precursors can form the stem and loop structures (Fig. 5). Among them, 2 Gloxinia-specific miRNAs (Ss-novel-miR039-5p and Ss-novel-miR053-5p) express higher level in the dorsal petal, the other 5 known miRNAs (Ss-miR157-5p-a, Ss- miR157-5p-b, Ss-miR166-3p, Ss-miR390-5p-b, Ss-miR5654-3p) show more abundant expression in the ventral petals. The different expression pattern of DE-miRNAs indicate that these miRNAs may play influential regulatory roles in the differential morphogenesis of dorsal and ventral petals.
Figure 5 | The precursor of 7 DE-miRNAs can form stable stem-loop structure.
We have established the structures of 7 DE-miRNA precursors using bioinformatics tool.
The precursors can be partitioned into mature, star and loop part based on the reads mapping to it. Each parts are specified with red, blue and yellow color, respectively.
Figure 4 | A list of dorsoventrally expressed miRNAs (DE-miRNAs)
The list shows the ID, sequence and length of the 7 DE-miRNAs. The length is range from 21-24.
Two of the Gloxinia-specific DE-miRNAs express higher level in the dorsal petal and the other 5 known DE-miRNAs show more abundant expression in the ventral petals.
High throughput deep PARE sequencing reveals the targets of DE-miRNAs
Confirmation of miRNA targets was a prerequisite to better understand the functional roles of miRNAs. In previous studies, miRNA targets were investigated mainly by bioinformatics prediction and few miRNAs target were experimentally validated. To elucidate the functions of miRNAs in dorsoventrally establishment, we predicted their potential regulatory targets using Parallel Analysis of RNA Ends (PARE). As shown in
Table 3, Ssp-miR157a-5p-a/b targets the Squamosa promoter-binding-like proteins (SPLs)
family, including SPL1, SPL9, SPL12, SPL16 (Fig. 5A-5D), suggesting that miRNAs in one family can target multiple member in a gene family.Ta-siRNA-producing locus 3 (TAS3) genes, which produce noncoding transcripts that are transformed into short 21-nt ta-siRNAs by RNA silencing pathways, had specific cleavage sites that were located in the complementary sequences of Ssp-miR390a-5p-b (Fig. 5E, 5F). For Ssp-miR166h-3p and Ssp_novel_miR053, there is only a single significant target unannotated function in both NCBI and TAIR database. No target genes were found for the remaining one known miRNA (Ssp-miR5654-3p) and two species- specific miRNAs (Ssp_novel_miR001-a.1 and Ssp_novel_miR039), suggesting that the miRNAs with no predicted targets may suppress gene expression by different mechanisms including the inhibition of translation.
Table 3 | Target genes of DE-miRNAs in the gloxinia PARE library
NAa, not available
miRNA Target Function Score Category P-Value
Ssp-miR157a-5p-b Contig6205 Squamosa promoter-binding-like protein 12 (SPL12)
1 0 0.002
Contig22546 SPL9 1 0 0.003
ZD-2_p_00036035 SPL16-like isoform X1 2 0 0.007
Contig5998 SPL1-like 4 0 0.01
Ssp -miR157a-5p-a ZV-12_p_00000148 SPL1 1 0 0.0004
Contig6205 SPL12 2 0 0.002
Contig22546 SPL9 2 0 0.004
ZD-2_p_00036035 SPL 16-like isoform X1 3 0 0.007
Ssp -miR166h-3p Contig5497 NAa 14 1 0.049
Ssp _novel_miR039 NA NA NA NA NA
Ssp -miR390a-5p-b Contig326 TAS3 4 0 0.002
Contig326 TAS3 3 4 0.038
Ssp -miR5654-3p NA NA NA NA NA
Ssp _novel_miR053 ZV-12_p_00000029 NA 8 2 0.570
Contig6432 NA 14 2 0.961
Contig20887 NA 10 2 0.995
Ssp _novel_miR001- a.1
NA NA NA NA NA
A characteristic feature of miRNA-guided cleaving was that the cleavage takes place precisely between the 10th and 11th nt from the 5’ end of miRNA in the complementary region of the target transcript. Therefore, cleaved RNA targets should have distinct peaks of degradome tags at the predicted cleavage site relative to other regions of the transcript.
Systematic identificaion of miRNA targets was accomplished by analyzing the 20 and 21 nt tags with the CleaveLand pipeline for miRNA target identification. The abundance of tags for each of the transcript were presented in the form of target plots (T-plot). The T- plot shows SPLs were precisely mapped from the 10th and 11th position of complementary sequences at the 5’ end of the miRNA, showing evidence of cleavage by Ssp-miR157a- 5p-a and Ssp-miR157a-5p-b. On the other hand, Ssp-miR390 show two target sites on one single transcript. However, only the site approach to the 3’ can be cleaved. We found a mismatch located at the 10th and 11th nucleotide between 5’ target site and Ssp-miR390.
(Red shaded). Compared with A. thaliana and rice, miR157 and miR390 targets were found to be conserved in gloxinia, indicating that the miRNA-target relationship was evolutionary conserved.
(A) ZD PARE data – Ssp-miR157a
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::::::::::::::
SsSPL1 862 AUGCUCUCUAUCUUCUGUCAA 882
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::: :::::::::::
SsSPL9 3474 GUGCUCUCUCUCUUCUGUCAA 3494
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::: :::::::::::
SsSPL12 2194 GUGCUCUCUCUCUUCUGUCAA 2214
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::: ::::::::::
SsSPL16 1282 GUGCUCUCUCUCUUCUGUCAU 1302
Ssp-miR157a / SsSPL1 Ssp-miR157a / SsSPL9
Ssp-miR157a / SsSPL12 Ssp-miR157a / SsSPL16
(B) ZD PARE data – Ssp-miR157b
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::: :::::::::::::
SsSPL1 793 CAGCUCCCUCUCUUCUGUCAU 813
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::::: ::::::::::::::
SsSPL9 3474 GUGCUCUCUCUCUUCUGUCAA 3494
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::::: ::::::::::::::
SsSPL12 2194 GUGCUCUCUCUCUUCUGUCAA 2214
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::::: :::::::::::::
SsSPL16 1282 GUGCUCUCUCUCUUCUGUCAU 1302
Ssp-miR157b / SsSPL1
Ssp-miR157b /
Ssp-miR157b / SsSPL12 Ssp-miR157b / SsSPL16
(C) ZV PARE data – Ssp-miR157a
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::::::::::::::
SsSPL1 862 AUGCUCUCUAUCUUCUGUCAA 882
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::: :::::::::::
SsSPL9 3474 GUGCUCUCUCUCUUCUGUCAA 3494
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::: :::::::::::
SsSPL12 2194 GUGCUCUCUCUCUUCUGUCAA 2214
miR157a 21 CACGAGAGAUAGAAGACAGUU 1 ::::::::: ::::::::::
SsSPL16 1282 GUGCUCUCUCUCUUCUGUCAU 1302
Ssp-miR157a / SsSPL1 Ssp-miR157a / SsSPL9
Ssp-miR157a / SsSPL12
Ssp-miR157a / SsSPL16
(D) ZV PARE data – Ssp-miR157b
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::: :::::::::::::
SsSPL1 793 CAGCUCCCUCUCUUCUGUCAU 813
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::::: ::::::::::::::
SsSPL9 3474 GUGCUCUCUCUCUUCUGUCAA 3494
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::::: ::::::::::::::
SsSPL12 2194 GUGCUCUCUCUCUUCUGUCAA 2214
miR157b 21 CACGAGUGAGAGAAGACAGUU 1 :::::: :::::::::::::
SsSPL16 1282 GUGCUCUCUCUCUUCUGUCAU 1302
Ssp-miR157b / SsSPL1 Ssp-miR157b / SsSPL9
Ssp-miR157b / SsSPL12 Ssp-miR157b / SsSPL16
(E) ZD PARE data – Ssp-miR390
miR390 21 CCACGAUAGGGAGGACUCGAA 1 ::::::::::. :::::::::
SsTAS3 295 GGUGCUAUCCUACCUGAGCUU 315
miR390 21 CCACGAUAGGGAGGACUCGAA 1 ::::::::::::::::
SsTAS3 566 UUGUCUAUCCCUCCUGAGCUG 586
(F) ZV PARE data – Ssp-miR390
miR390 21 CCACGAUAGGGAGGACUCGAA 1 ::::::::::. :::::::::
SsTAS3 295 GGUGCUAUCCUACCUGAGCUU 315
miR390 21 CCACGAUAGGGAGGACUCGAA 1 ::::::::::::::::
SsTAS3 566 UUGUCUAUCCCUCCUGAGCUG 586
Ssp-miR390 / SsTAS3 Ssp-miR390 / SsTAS3
Ssp-miR390 / SsTAS3 Ssp-miR390 / SsTAS3
Figure 6 | T-plot of the targets cleaved by Ssp-miR157a, Ssp-miR157b, Ssp-miR390 and their pairwise target regions
The T-plots show the distribution of 3’ end of the degradome tags within the full-length of the target mRNA sequence (bottom). The black arrowhead indicates the 10th and 11th positions of the miRNA. The alignment shows the miRNA with a portion of its target sequence. The two dots indicate matched RNA base pairs; one dot shows a GU mismatch whereas no dots represent other types of mismatch.
Expression correlation between Ssp-miR157 and its Targets
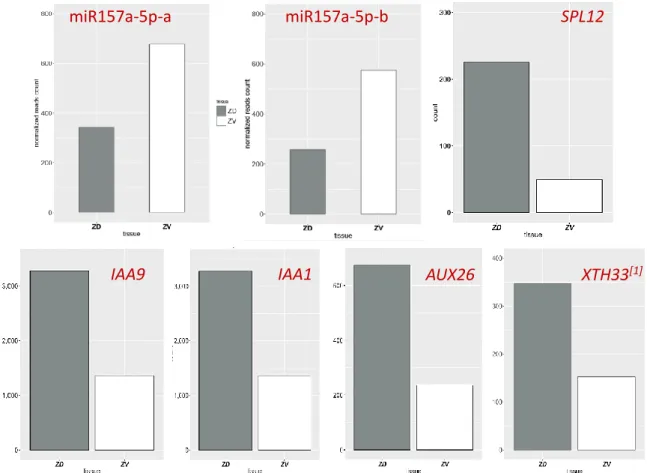
To assess the influence of the miRNAs on their targets, transcriptome-Seq analysis was used to quantify the transcriptional levels of the target genes. As shown in Fig. 6, SPL12, one of the miR157 target genes, the transcript level was negatively correlated with the miR157 levels. The miR157/SPLs was reported to regulate the auxin responsive genes, and thus influence the cell elongation of petals (Liu, Tu et al. 2017). Based on the findings, we checked the expression level of several auxin-responsive genes in our transcription data. Intriguingly, we discovered IAA9, IAA14, AUX26 and XTH33 transcripts levels are positively correlated with the SPL12 level, demonstrating greater expression level in the dorsal petal.
miR157a-5p-a miR157a-5p-b miR157a-5p-b
SPL12
XTH33
[1]AUX26 IAA1
4 IAA9
Figure 7 | Expression pattern of Ssp-miR157a/b, SsSPL12 and auxin-responsive genes
The gray and white bars indicate the expression level from the transcriptome-Seq result of dorsal and ventral petal, respectively.[1] XTH33: xyloglucan endotransglucosylase hydrolase protein 33
Validate the dorsoventrally expression of Ssp-miR157 by stem-loop qRT-PCR
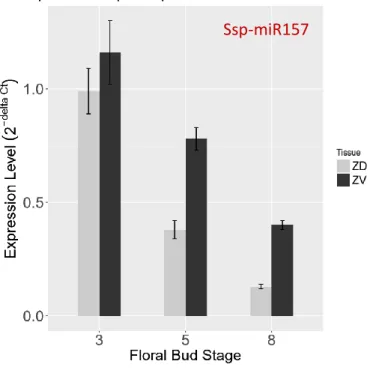
To confirm the expression level result we obtained in the RNA sequencing data, we employed stem-loop qRT-PCR to detect the expression level of Ssp-miR157. To investigate the spatial expression trends, total RNA was prepared from samples of three developmental stages of gloxinia dorsal and ventral petals (Supplementary Figure S1).
As shown in Fig. 7, the result not only reconfirms the high expression level of Ssp- miR157 in the ventral petals at stage 5, but also shows the spatial fluctuation according to the floral bud development. Interestingly, we discovered that the dorsoventrally differential expression could not be detected until the floral buds reached stage 5.
Ssp-miR157
Figure 8 | Experimental validation of miR157 expression level between dorsal and ventral petals
The gray and black bars indicate the expression level of miR157 in the dorsal and ventral petals from stem- loop qRT-PCR results, respectively. The x represents the floral bud in different developmental stages.Consistent with our NGS data, the Ssp-miR157 show higher expression in the ventral petal at stage 5. In addition, the expression level demonstrates temporal fluctuation along with the floral development.
24
Finally, cause the miR157/SPL12/Auxin-responsive genes show dorsoventral polarity, we want to clarify what aspect of the flower might be influenced by the axis . The previous research done in cotton plants indicated that the axis affected the cell size in the petals (Liu et al. 2017). Based on the study, we measured the cell size of the dorsal and ventral petals of Gloxinia. Surprisingly, the cell length show difference between dorsal and ventral petals. The dorsal petal composed of the larger cells than the ventral part (Fig. 8). The result indicated that Ssp-miR157, via its mediation of the cleavage of SPLs mRNA, may be critical for regulating the differential cell expansion level of dorsal and ventral petal. Thus contributes to the different length of the floral tube, create the asymmetry of corolla, which is a crucial factor to decide a zygomorphic flower (Wang et al. 2015).
A B
B B B B B B B B B B
Figure 9 | Measurement of the cell length of dorsal and ventral petal.
Regarding different developmental stages (Fig. 8-A), we measured the tube length and cell size of tube of dorsal and ventral petal, respectively (Fig. 8-C, Fig. 8-D). The measuring distance of floral tube of dorsal and ventral petal are indicated in red and blue curves, respectively (Fig. 8-B). Cells are larger in the dorsal tube, and the trend become more noticeable when floral bud enter its later stages.
Cloning and Analysis of miR390-Targeted TAS3 Gene in Gloxinia
Among plant TAS genes, the most well studied is TAS3; its transcript bears two target sites of miR390, generating ta-siRNAs via the so-called “two-hit” mechanism (Axtell et al., 2006). The resulting conserved ta-siRNA is known as “ta-siARF” as it targets AUXIN RESPONSIVE FACTOR (ARF) genes (Allen et al., 2005; Axtell et al., 2006). In this study, we found Ssp-miR390 showed dorsoventrally expression pattern, however before exploring further, it is prerequisite to confirm the existence of miR390/TAS3/ta-siARF pathway in the Gloxinia petal. Based on the sequences of TAS3 in our transcriptome data from Sinningia speciosa ‘Carangola’, the full length cDNA of TAS3 was obtained through RT-PCR using gloxinia ‘Espirito santo’ cDNA as the template.
The sequence of SsTAS3 between the two binding sites was 274 bp, and the Ssp- miR390-guided cleavage site was predicted to be located near the 3’ end of the SsTAS3 transcript. Ssp-miR390 interacts in a non-cleavage mode with a second site near the 5’
end, which was found to have a conserved mismatch at the center of the binding region (Fig. 5E, 5F, Fig. 9).
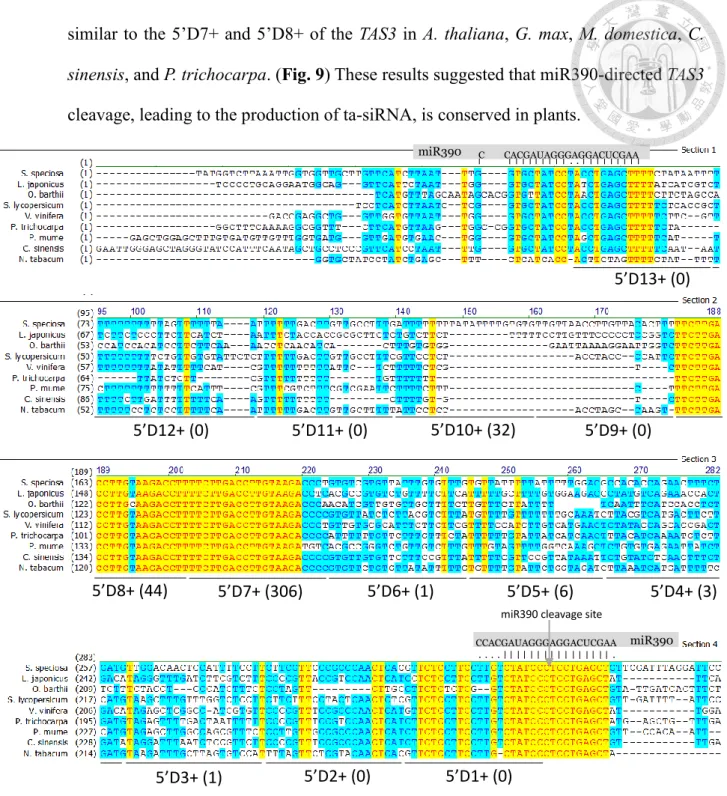
MiR390-guided cleavage was shown to set the 21-nucleotided phase for ta-siRNA precursor processing. Here, according to the Ssp-miR390-guided cleavage site, thirteen potential ta-siRNAs with the 21-nucleotides phase were predicted to be produced from miR390-guided SsTAS3 cleavage (Fig. 9). To confirm the production of ta-siRNAs from SsTAS3, we mapped the reads from sRNA-Seq back to the SsTAS3 using Bowtie aligner.
A large proportion of SsTAS3-derived small RNAs began within one nucleotide of the residues predicted by cleavage at either the 5’ or 3’ site. In phase, the 21-nucleotides positions on the 5’ side of the miR390 cleavage site were named 5’D1+, 5’D2+, and so on. Among them, TAS3_5’D7+ and 5’D8 showed high similarity in sequence, with only one nucleotide difference at the 3’ end of the ta-siRNAs. Further, the sequence is also
26
similar to the 5’D7+ and 5’D8+ of the TAS3 in A. thaliana, G. max, M. domestica, C.
sinensis, and P. trichocarpa. (Fig. 9) These results suggested that miR390-directed TAS3
cleavage, leading to the production of ta-siRNA, is conserved in plants.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
. .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
C CACGAUAGGGAGGACUCGAA
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
.
.
.
.
.
.
.
.
. .
.
.
.
.
.
.
.
.
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
CCACGAUAGGGAGGACUCGAA
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390
miR390 .
.
.
.
.
.
.
. .
.
.
.
.
.
.
. .
.
.
.
.
.
.
. .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
. miR390 cleavage site
miR390 cleavage site
miR390 cleavage site
miR390 cleavage site
miR390 cleavage site
miR390 cleavage site
miR390 cleavage site
miR390 cleavage site
miR390 cleavage site
5’D1+ (0) 5’D1+ (0) 5’D1+ (0) 5’D1+ (0) 5’D1+ (0) 5’D1+ (0) 5’D2+ (0)
5’D2+ (0) 5’D2+ (0) 5’D2+ (0) 5’D2+ (0) 5’D2+ (0) 5’D3+ (1)
5’D3+ (1) 5’D3+ (1) 5’D3+ (1) 5’D3+ (1) 5’D3+ (1)
5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D4+ (3) 5’D5+ (6)
5’D5+ (6) 5’D5+ (6) 5’D5+ (6) 5’D5+ (6) 5’D5+ (6) 5’D5+ (6) 5’D5+ (6) 5’D5+ (6) 5’D5+ (6) 5’D6+ (1)
5’D6+ (1) 5’D6+ (1) 5’D6+ (1) 5’D6+ (1) 5’D6+ (1) 5’D6+ (1) 5’D6+ (1) 5’D6+ (1) 5’D6+ (1) 5’D7+ (306)
5’D7+ (306) 5’D7+ (306) 5’D7+ (306) 5’D7+ (306) 5’D7+ (306) 5’D7+ (306) 5’D7+ (306) 5’D7+ (306) 5’D7+ (306) 5’D8+ (44)
5’D8+ (44) 5’D8+ (44) 5’D8+ (44) 5’D8+ (44) 5’D8+ (44) 5’D8+ (44) 5’D8+ (44) 5’D8+ (44) 5’D8+ (44)
5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D9+ (0) 5’D10+ (32)
5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D10+ (32) 5’D11+ (0)
5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D11+ (0) 5’D12+ (0)
5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0) 5’D12+ (0)
5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0) 5’D13+ (0)
Figure 10 | Alignment of TAS3 sequences corresponding to the TAS3 ta-siRNAs from orthologs of 8 species.
Three reported regions of nucleotide conservation: an ~42 nt region corresponding to the TAS3_ta-siRNAs that target ARF3 and ARF4 (ta-siARFs), two 21-nt region corresponding to the 3’ and 5’ miR390 complementary sites.
GenBank accession numbers are as follows: L. japonicus (AK338955), O. barthii (GQ420228), S. lycopersicum (JX047545), V. vinifera (FQ386573), P. trichocarpa (XM_006378492), Prunus mume(XR_513520), C. sinensis (XR_371831), Nicotiana tabacum (FJ804751)
Gloxinia ta-siRNA Abundance Analysis and their target prediction
Among the thirteen ta-siRNAs, the reads of TAS3 5’D7+ and 5’D8+ siRNAs were most abundant, with 306 and 44 reads (Fig. 9). This result was similar to previous findings in which the reads of TAS3 5’ D7+ and 5’D8+ ta-siRNAs were abundant, the so-called ta- siARFs, while the expressions of other ta-siRNAs were very low. The 5’D1+/D2+/D9+/D11+/D12+ and D13+ siRNA was not detected in gloxinia sRNA-Seq.
These results further proved that Ssp-miR390 was sufficient to trigger secondary ta- siRNAs biogenesis, and miR390-guided TAS3 cleavage with the 21-nt phase was conserved in plants. In addition, the TAS3-derived ta-siARFs were one of the main products from the cleavage event.
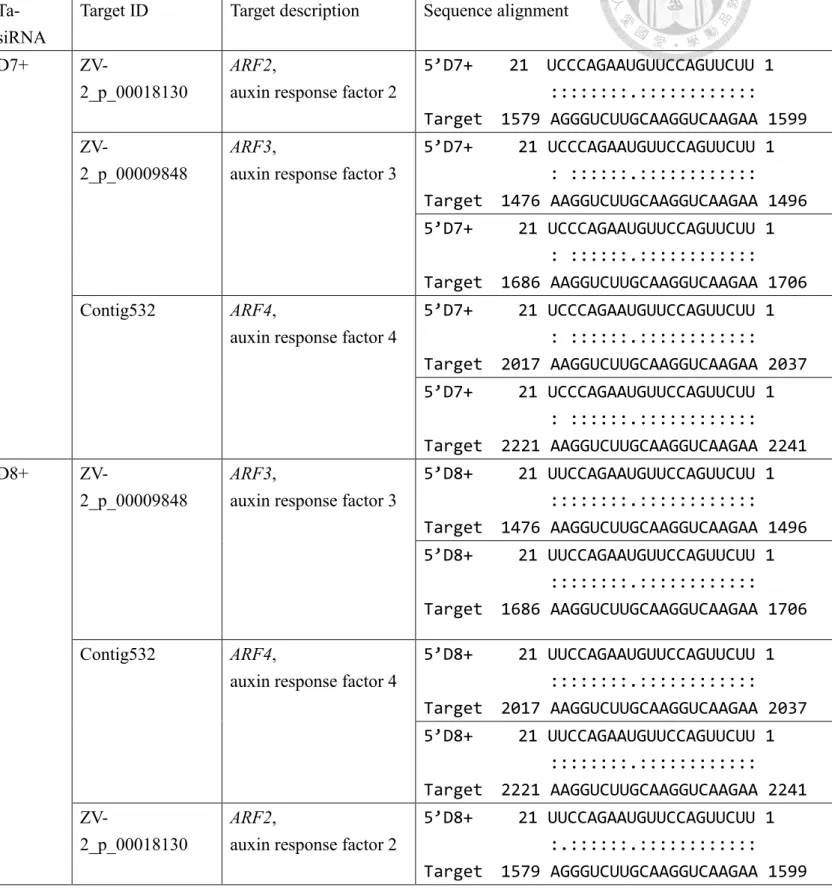
The TAS3-derived ta-siARF specifically targets ARF genes of Clade B (ARF2/3/4).
(Xia et al., 2017). This miR390-TAS3-ARF pathway is of critical function in the regulation of plant growth and development, including leaf morphology, developmental timing and patterning, and lateral root growth (Garcia et al., 2006; Fahlgren et al., 2006; Adenot et al., 2006; Marin et al., 2010; Hunter et al., 2006). Ta-siARFs interact with target homologous mRNAs and guide cleavage by the same mechanism as plant miRNAs. Here, to identify the target of SsTAS3_ta-siARFs, the 5’D7+ and 5’D8+ siRNAs, were matched against nucleic acid sequence of the Gloxinia ARF gene families using psRNATarget analysis. The result indicated that the 5’D7+ and 5’D8+ ta-siRNAs could both target ARF2/-3/-4 from gloxinia (Table 4). In addition, SsARF3 and -4 contained two complementary sites to the TAS3_5’D7+ and 5’D8+, the result was consistent with the previous studies in A. thaliana, G. max (Hu et al. 2013) and longan (Lin et al. 2015).
Table 4 | 5’D7+ and 5’D8+ ta-siRNAs and their pairwise target region of ARF genes.
Ta- siRNA
Target ID Target description Sequence alignment
D7+ ZV-
2_p_00018130
ARF2,
auxin response factor 2
5’D7+ 21 UCCCAGAAUGUUCCAGUUCUU 1 ::::::::.::::::::::::
Target 1579 AGGGUCUUGCAAGGUCAAGAA 1599 ZV-
2_p_00009848
ARF3,
auxin response factor 3
5’D7+ 21 UCCCAGAAUGUUCCAGUUCUU 1 : ::::::.::::::::::::
Target 1476 AAGGUCUUGCAAGGUCAAGAA 1496 5’D7+ 21 UCCCAGAAUGUUCCAGUUCUU 1 : ::::::.::::::::::::
Target 1686 AAGGUCUUGCAAGGUCAAGAA 1706 Contig532 ARF4,
auxin response factor 4
5’D7+ 21 UCCCAGAAUGUUCCAGUUCUU 1 : ::::::.::::::::::::
Target 2017 AAGGUCUUGCAAGGUCAAGAA 2037 5’D7+ 21 UCCCAGAAUGUUCCAGUUCUU 1 : ::::::.::::::::::::
Target 2221 AAGGUCUUGCAAGGUCAAGAA 2241
D8+ ZV-
2_p_00009848
ARF3,
auxin response factor 3
5’D8+ 21 UUCCAGAAUGUUCCAGUUCUU 1 ::::::::.::::::::::::
Target 1476 AAGGUCUUGCAAGGUCAAGAA 1496 5’D8+ 21 UUCCAGAAUGUUCCAGUUCUU 1 ::::::::.::::::::::::
Target 1686 AAGGUCUUGCAAGGUCAAGAA 1706 Contig532 ARF4,
auxin response factor 4
5’D8+ 21 UUCCAGAAUGUUCCAGUUCUU 1 ::::::::.::::::::::::
Target 2017 AAGGUCUUGCAAGGUCAAGAA 2037 5’D8+ 21 UUCCAGAAUGUUCCAGUUCUU 1 ::::::::.::::::::::::
Target 2221 AAGGUCUUGCAAGGUCAAGAA 2241 ZV-
2_p_00018130
ARF2,
auxin response factor 2
5’D8+ 21 UUCCAGAAUGUUCCAGUUCUU 1 :.::::::.::::::::::::
Target 1579 AGGGUCUUGCAAGGUCAAGAA 1599
Validation of ta-siRNA-guided cleavage or target gene SsARF2, -3 and -4 mRNA in gloxinia through PARE analysis
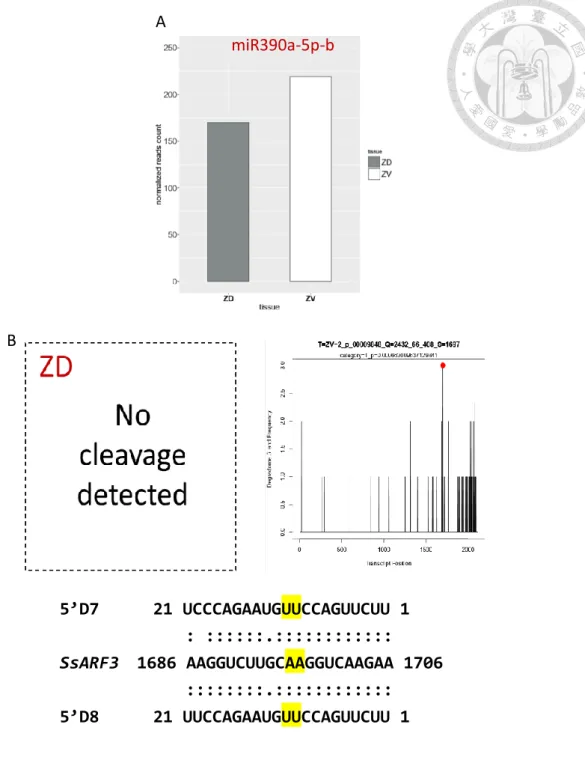
Three ARF genes have been validated as the target of ta-siARFs in Arabidopsis (Xia et al. 2012). In our study, all of them were predicted to be cleaved by ta-siARF through psRNATarget (Table 4). Further, to verify the cleavage event, we take ta-siARFs as input to the Cleaveland4 pipeline analysis, and examine if we can find the cleavage on SsARF2, -3 and -4. Surprisingly, only the cleavage signal of SsARF3 was detected, and the cleavage event specifically happened in the ventral petals (Fig. 10-B). The reason for no detection of cleavage on ARF2 and ARF4 may be correlated with the algorithm in CleaveLand4.
CleaveLand4 pipeline only looks for evidence of slicing at position 10 relative to the aligned small RNA. In short, the cleavage at position 11 or 9 is not relevant to CleaveLand4. Previous study in Longan showed that ta-siARF perform cleavage at position 9 complementary to miR390 on ARF4 (Lin et al. 2015). As a result, to further verify the presence of the cleavage other than 10th position, other experiments should be carried out. (eg. RLM-5’ RACE). On the other hand, the finding of the cleavage event that only happens in the ventral petal is concur with our DE-sRNAs results that miR390 is highly expressed in the ventral petal (Fig. 10-A). The above findings clearly indicated that ta-siARF cleaved the SsARF3 in the Gloxinia petal, and the cleavage event may be more dramatic in the ventral petal. Which lead to the expectation that SsARF3 expression level should also be dorsoventrally different.
30
5’D7 21 UCCCAGAAUGUUCCAGUUCUU 1 : ::::::.::::::::::::
SsARF3 1686 AAGGUCUUGCAAGGUCAAGAA 1706 ::::::::.::::::::::::
5’D8 21 UUCCAGAAUGUUCCAGUUCUU 1
5’D7 21 UCCCAGAAUGUUCCAGUUCUU 1 : ::::::.::::::::::::
SsARF3 1686 AAGGUCUUGCAAGGUCAAGAA 1706 ::::::::.::::::::::::
5’D8 21 UUCCAGAAUGUUCCAGUUCUU 1 miR390a-5p-b
B B B B B B B B B B B B B B
A A A A A A A A A A A A A A A A
Figure 11 | (A) The expression level of Ssp-miR390 from transcriptome-Seq data.
(B) The PARE data of SsTAS3_5’D7+, _5’D8+ cleavage on SsARF3 in dorsal and
ventral petal of Gloxinia.
Expression profiling of miR390/SsARF3 in gloxinia
The target genes for other DE-miRNA, Ssp-miR390, is confirmed to be SsTAS3, which have been invoked in the control of the specification of abaxial/adaxial polarity of leaves by generating ta-siRNAs, which in turn cause degradation of the AUXIN RESPONSE FACTOR 3 (ARF3) and ARF4 mRNAs (Allen et al. 2005). Thus, we cloned and investigated the expression level of SsARF3. Consistent with our expectation, SsARF3 was found in a complementary expression pattern to the Ssp-miR390 (Fig. 11).
In the early stage of floral bud development, we found Ssp-miR390 expressed evenly in the dorsal and ventral petal, causing the uniformly distribution of the SsARF3. As the growth of the floral bud continued, the Ssp-miR390 starts to accumulate in the ventral petal. The stronger degradation in ventral petal lead to the higher level of transcript in the dorsal petal. Moreover, the dorsoventrally pattern continue during the later developmental stage.
Ssp-miR390
SsARF3
Figure 12 | Graphs showing the relative
expression level of Ssp-miR390 (The
positive direction of y) and SsARF3
(The negative direction of y). Detected
by stem-loop qRT-pPCR and qRT-PCR.Discussion
A striking aspect of flowering plant diversity is the variation in flower symmetry.
From an ancestral form of actinomorphy, multiple evolutionary transitions have contributed to instances of non-radial forms, including zygomorphic symmetry. Research has centered on understanding the developmental mechanisms that establish patterns of zygomorphic symmetry. Small RNAs comprises a highly conserved gene regulatory system among land plants. It is reported as a key player in plant developmental timing (Pasquinelli et al., 2002, Rougvie et al., 2005, Moss et al., 2007), senescence (Ellis et al.
2005), leaf morphogenesis (Aida et al. 1997, Palatnik et al. 2003, Todesco et al. 2010), organ polarity (Juarez et al. 2004, Kidner and Martienssen, 2004), vascular development (Todesco et al. 2010), and root architecture (Marin et al. 2010). However, no research had been conducted to investigate the correlation of sRNAs to the floral symmetry. In this study, we have provided a better understanding of Gloxinia miRNAs in several ways. The deep sequencing of small RNA libraries as well as PARE libraries from dorsal and ventral petals allowed for the global analysis of miRNAs expressed in Gloxinia petals. The miRNAs identified include both conserved miRNAs that have not been reported in Gloxinia, as well as Gloxinia-specific miRNAs that have not been found in other plants.
Thus, these miRNAs may function in gene regulatory mechanisms specific to Gloxinia or those shared with related species. Our analysis uncovered a total of 157 miRNAs of conserved and Gloxinia-specific miRNAs. Further, to identify the miRNAs that may participate in the establishment of different morphogenesis of dorsal and ventral petal in Gloxinia zygomorphic flower, we compare the sRNA-Seq data of dorsal and ventral petal.
Interestingly, we discovered 7 dorsoventrally expressed miRNAs (DE-miRNAs). Next, to validate the targets of these DE-miRNAs, PARE sequencing was performed in this
study to detect the exact cleavage site of each miRNA to its target. The result showed that SPLs and TAS3 had major PARE sequences corresponding to cleavage sites by miR157 and miR390, respectively.
An integrated approach for characterizing Gloxinia dorsoventrally expressed miRNAs
Here, we combined transcriptome, small RNA, PARE and genomic sequencing data to develop a pipeline for the identification of Gloxinia miRNAs and their targets.
Transcriptome sequences capture the complete set of transcripts in a specific cell or tissue for a particular developmental stage or condition. Thus, integrating the transcriptomic sequence in miRNAs prediction pipeline can preclude the interruption from false-positive hairpin structures such as those predicted from genomic DNA (Lin et al. 2016). In addition, our pipeline also joined the draft genome of gloxinia, which can serve as a guard to eliminate the misassembled transcripts or short contig sequences, to boost accuracy of the analysis prediction (Lin et al. 2016). As a result, 62 conserved and 95 novel miRNAs were identified using RNA sequencing combined with draft genome. When compared to other plants, the number of miRNAs identified from Gloxinia is less than well genomic- sequenced plants like rice (713), maize (321) and Brachypodium distachyon (525), but more than those plants without genome sequencing data, like Elaeis guineensis (6), Festuca arundinacea (15), Saccharum officinarum (16) (miRbase database release 21).
The first 5’ -end nucleotide of miRNA is a major determinant for sorting the miRNA into a particular AGO complex (Lin et al. 2016). In Arabidopsis, AGO2 and AGO4 preferentially recruit small RNAs with a 5’ terminal adenosine, whereas AGO1 harbors microRNAs (miRNAs) that favor a 5’ terminal uridine. AGO5 predominantly binds small RNAs that initiate with cytosine (Mi et al. 2008). And 5’ –terminal ‘G’ occurs at the lowest
frequency. Regarding our 5’ terminal nucleotides analysis in Gloxinia, the result is in agreement with previous findings.
Illuminating the function of miRNAs requires efficient approaches to identify their target genes. Originally, plant miRNA targets were studied via computational prediction, which is based on the perfect sequence complementary between a miRNA and the target mRNA or sequence conservation among different species. However, targets prediction often has a high level of mismatch in miRNA-mRNA pairs and every single predicted gene must be verified independently. This one-at-a-time isolation of target cleavage remnant is laborious, time-consuming and costly. To overcome the limitation, a new method called Parallel Analysis of RNA Ends (PARE) has been successfully established to identify small RNA targets at a global scale. The method includes deep sequencing, bioinformatic analysis and 5’rapid amplification of cDNA ends (5’ RACE). It has been used for global identification of miRNA-target RNA pairs in numerous plant species, such as A. thaliana (Li et al. 2010), Oryza sativa (Liu et al. 2014), mulberry (Li et al. 2017), purple false brome (Jeong et al. 2013) and liverwort (Lin et al. 2016). An evaluation from PARE allows us to identify miRNA-mediated cleavage (unique and sharp peaks at the 10th and 11th position of the target site) from natural or artificial mRNA decay (an ambiguous pattern dispersed through the transcript.) (Lin et al. 2016). In our study, through the help of PARE data, we identified the targets of DE-miRNAs. Among the 7 DE-miRNAs, miR157 and miR390 conveyed cleavage on SPLs and TAS3, respectively.
The SQUAMOSA promoter-binding protein-like transcription factors 1 (SPL1), SPL9, SPL12 and SPL16 had major PARE sequences corresponding to cleavage sites by miR157.
Indicating that one miRNA may regulate many genes as its targets. The finding is consistent to the previous study that the relationships between miRNAs and their targets may not be one-to-one, should be a multiple-to-multiple relationships. (Hashimoto et al.
2013). On the other hand, the remaining 5 DE-miRNAs, Ssp-miR166h-3p, Ssp_miR5654-3p, Ssp_novel_miR001-a.1, Ssp_novel_miR039, Ssp_novel_miR053 show ambiguous dispersal through the transcript. In short, they seem to not convey a clear and specific cleavage event on any of the target. However, a previous report show that At-miR396 triggers decay of Arabidopsis SHORT VEGETATIVE PHASE mRNA by translation inhibition, resulting in multi-peaks in PARE profile (Yang et al. 2015). Under this circumstance, the multiple peak pattern will fail to pass the criteria for being regarded as a specific miRNA-mRNA pair in our pipeline. So, we cannot exclude the possibility that, instead of downregulating the target by cleavage, the remaining 5 DE-miRNAs might perform miRNA-mediated translation inhibition.
MiR157/SPLs may be Involved in the Regulation of differential cell size in Gloxinia Dorsal and Ventral petal
SPL transcription factors have been reported as miR156/miR157 targets in many species (Wang et al., 2015, Liu et al. 2017). Our PARE sequencing also demonstrated that Ssp-miR157 can target several members in the SsSPL gene family, including SPL1, SPL9, SPL12 and SPL16. In the other hand, we discovered Ssp-miR157 was expressed higher in the ventral petal, and one of its targets, SPL12 demonstrated the negative correlated transcript level. The result suggests the presence of the dorsoventrally miR157/SPL in Gloxinia zygomorphic flowers. Additionally, in all the differentially expressed genes between the dorsal and ventral petal, only the SPL transcription factors were predicted as Ssp-miR157 targets. Therefore, it is reasonable to presume that SsSPL12 as Ssp-miR157 targets play crucial roles in petal development of Gloxinia. In a delicate experiment done in the cotton plant (Gossypium hirsutum), the team demonstrates that over-expression of GhmiR157 in cotton could arrest cell proliferation and cell expansion, which repressed
floral organ development and reduced the final organ size. (Liu et al. 2017). Regarding the dorsoventrally expression of miR157/SPL12 module, we measured the cell size of the dorsal and ventral petal, respectively. Indeed, in Gloxinia ventral petal, where the Ssp- miR157 expressed abundantly, the cells are smaller than the dorsal petal. However, what is the genetic module that mediates between the miR157/SPLs to the cell size difference?
In cotton plant, the team found several auxin-inducible genes were down-regulated in the miR157 over-expression line compared with the control (Liu et al. 2017). However, free IAA content was not reduced in the over-expression line. Therefore, decreased auxin signaling in the over-expression lines was not due to IAA content but defects in signal transduction. The mechanism of how the miR157/SPL functions in auxin signal transduction is still unclear. However, a hypothesis has been proposed , stating that the miR157/SPL may regulate the MADS-box genes, and possibly serve as AUXIN- RESPONSE FACTOR (ARF)-like or ARF partner-like molecules to transduce auxin signaling, thus contributing to cell growth (Liu et al. 2017). To state more clearly, it was reported that the MADS-box transcription factor, including SEPALLATA3 (SEP), APETALA1 (AP1) and AGAMOUS (AG), could bind several ARF recognition motifs. For example, ARF2 was among the proteins that were enriched in AP1 and AG IP samples. In our transcriptome data, we found two MADS-box genes, SEP and AGL6, showed dorsoventrally expression. In the future, the expression profile should be examined in further detail by qRT-PCR. Finally, we propose a possible regulatory network of differential morphogenesis of dorsal and ventral petal in the Gloxinia zygomorphic flower (Fig. 12). At the early stage of floral bud emergence, the expression level of Ssp-miR157 is evenly distributed among each floral primordia, as the result, no significant difference in cell size between the dorsal and ventral petal, thus the entire floral bud seems perfectly symmetrical. As the floral bud enter its mediate stage, the Ssp-miR157 start to accumulate
in the ventral petal, creating a higher level of its target, SsSPL12 mRNA in the opposite dorsal petal. Highly expressed miR157-targeted SsSPL12 may activate transcription of MADS-box genes, such as orthologs of AtSEP and AtAGL6. These MADS-box transcription factors, or some unknown factors, may bind auxin response motifs of downstream gene promoters to transduce auxin signaling. Activated auxin signaling may further regulate downstream auxin-responsive genes to trigger bigger cell size in the dorsal petal. Thus contribute to the different length of the floral tube, create the asymmetry shape in dorsal and ventral petal, which is a crucial factor to decide a zygomorphic flower. The dorsoventrally pattern of Ssp-miR157/SPL12 maintain until the late floral developmental stage. As a result, the difference between dorsal and ventral petal getting greater, the zygomorphic state is assured until the flower bloom.
ZD
ZV
?
MADS-box
Auxin-responsive genes
SPL12
Auxin-responsive genes
Figure 13 | A model for the potential miR157/SPLs in the regulatory network of zygomorphic flower development.
As the floral bud enter its mediate stage, the Ssp-miR157 start to accumulate in the ventral petal, create a higher level of its target, SsSPL12 mRNA in the opposite dorsal petal. Highly expressed miR157-targeted SsSPL12 may activate transcription of MADS-box genes, such as orthologs of AtSEP and AtAGL6. These MADS-box transcription factors, or some unknown factors, may bind auxin response motifs of downstream gene promoters to transduce auxin signaling. Activated auxin signaling may further regulate downstream auxin-responsive genes to trigger bigger cell size in the dorsal petal. Thus contribute to the different length of the floral tube, create the asymmetry shape in dorsal and ventral petal.
MiR390/ TAS3/ ARF3 could affect the establishment of Dorsal-Ventral Polarity In Addition to the Leaves.
An important feature of RNA silencing in plants is that it can be amplified, notably in a selective manner: in some cases, RNAs targeted by sRNAs, in parallel to their degradation, become templates for RNA-DEPENDENT RNA POLYMERASES (RDRs), resulting in de novo synthesis of dsRNA. This dsRNA is in turn processed by DCLs into so-called ‘secondary siRNAs’, which amplify the original sRNA signal and allow silencing of RNA related in sequence to the primarily targeted RNA. Trans-acting siRNAs (ta-siRNAs) from a highly conserved class of plant sRNA that characterizes this type of amplification mechanism. MiR390-mediated cleavage of TAS3 ncRNAs triggers the recruitment of SUPPRESSOR OF GENE SILENCING 3 (SGS3) and RDR6, resulting in the production, by DCL4, of 21-nt ta-siRNAs acting to silence complementary mRNA in trans. The resulting conserved ta-siRNAs are known as ‘ta-siARF’ as it targets AUXIN RESPONSE FACTOR (ARF) gene (Lin et al. 2015).
To date, there are two kinds of TAS3 genes described in plants; they both contain two target sites for miR390, but TAS3-short (TAS3S) can be cleaved at both sites, producing a single, centrally-located ta-siARF. While in TAS3-long (TAS3L), only the 3’ target site is cleavable and sets the phase of ta-siRNA production, generating two ta-siARFs. 5’
miR390 target site of TAS3L is usually non-cleavable because of the presence of a central mismatch (Xia et al. 2015). In our study, when examining the PARE data, we validate the target of Ssp-miR390 is truly SsTAS3. Similar to TAS3 in other species, SsTAS3 have two miR390 target sites. Intriguingly, the 3’ site show a sharp and clear cleaved signal, whereas none of the reads were found to truncated at the 5’ target site. When aligning the sequences of Ssp-miR390 and SsTAS3, we found a central mismatch at the 10th and 11th nt from the 5’ end of miRNA in the complementary region of the target transcript. Thus,
we confirmed the cleavage event of Ssp-miR390 on SsTAS33’ site. In addition, the central mismatch at 5’ target site allow us to classify our SsTAS3 to the TAS3L type. The absent of cleavage signal on TAS3S is identical to the findings in Mimulus corolla, suggesting that the TAS3S gene is not expressed in the flower (Ding et al., 2018). Next, to examine the production of ta-siARFs, the reads from sRNA-Seq are mapped against the SsTAS3.
We found two abudant 21-nt mapped reads that were in phase with the 3’ site; These two ta-siRNAs show high sequence similarity to each other, and also identical to the ta- siARFs of Arabidopsis ta-siARFs. These results validate the production of ta-siARF from SsTAS3, and the 3’ site is the main trigger site of ta-siARF generation. The above evidence proved the existence of conserved miR390/TAS3/ta-siARF in the Gloxinia petal.
Regarding the dorsoventrally expression pattern of Ssp-miR390, we hypothesized that the differential morphogenesis of dorsal and ventral petal may mediated through the tasiARF-ARF pathway. Because Ssp-miR390 expresses higher in the ventral petal, our hypothesis makes two clear predictions: (i) The cleavage event on SsARFs should be more dramatic in the ventral petal. (ii) The abundance of SsARFs transcripts should be lower in the ventral petal compared to that in the dorsal petals. To test the first prediction, we examined the PARE data, intriguing, the cleavage signal of ta-siARF on SsARF3 could only be found in ventral petal. The results confirmed the first prediction. To test the second prediction, qRT-PCR measurement in Gloxinia showed that SsARF3 appeared more depleted in the ventral petal, supporting our second prediction.
After confirmation of miR390/ARF3 being dorsoventrally expressed in Gloxinia, we are curious about the biological role it plays in the establishment of the zygomorphic flower. MiR390/ARF3 has been reported to participate in distinct aspect of plant development. This includes leaf polarity, lateral root growth, somatic embryo development and the formation of corolla tube (Marin et al., 2010, Rubio-Somoza and