It should not be forgotten that single shell nanotube
sam-ples contain tubes of not only different diameters but also
different electronic properties. In any case, both
semicon-ducting and metallic nanotubes will have high surface
ener-gies and, therefore, even if different their wetting
proper-ties will probably fall within the intervals reported here for
g
Cand g
max. When considering the capillarity of single shell
tubes with very small inner diameter (< 1 nm), the fact that
the inner and outer surfaces may have different energies
owing to the asymmetry of the electronic densities
[22]should be taken into account. To gain proper insight into
such issues, it will be necessary to have samples with just
one type of well-defined single shell nanotube, which are
currently not available.
Experimental
Raw nanotubes were sonicated for 1 h in carbon disulfide (25 mL per 10 mg of nanotubes), filtered, and dried before wetting experiments. Pure samples were prepared by following the purification procedure described elsewhere [14]. The annealed nanotubes were first purified and then heated under vacuum (2 ´ 10±6torr) at 100 C for 10 min, followed by 15 min at
500 C, and eventually the temperature was raised to 900 C before natural cooling. The powders were ground in a mortar and divided in small quartz tubes containing 2±3 mg of nanotubes each. The samples were degassed un-der vacuum (5 ´ 10±6torr) and heated for an hour (at 500 C for the raw and
annealed, 120 C for the purified nanotubes) before being transferred under argon to a glove box. The chemical, (the purest quality available from Al-drich Chemical Company Inc., 99.95±99.99 %) to be melted was added on top of the packed nanotubes in an approximately 1:1 volume ratio. The quartz tube was then degassed for another hour at the same temperature as described earlier (or below the melting point of the chemical, depending on its nature) and sealed under vacuum (2±5 ´ 10±6torr).
The sealed quartz tubes were heated from room temperature to 50 C above the melting point of the tested compounds at about 1 C/s and left at the final temperature for 1±4 h.
Received: April 24, 1998 Final version: June 29, 1998 ±
[1] P. M. Ajayan, T. W. Ebbesen, Rep. Prog. Phys., 1997, 60, 1025. [2] P. M. Ajayan, S. Iijima, Nature,1993 361, 333.
[3] E. Dujardin, T. W. Ebbesen, H. Hiura, K. Tanigaki, Science, 1994, 265, 1850.
[4] S. C. Tsang, Y. K. Chen, P. J. F. Harris, M. L. H. Green, Nature, 1994, 372, 159.
[5] D. Ugarte, A. Châtelain, W. A. de Heer, Science, 1996, 274, 1897. [6] M. H. L. Green et al., Chem. Commun., 1998, 347.
[7] P. M. Ajayan, O. Stephan, P. Redlich, C. Colliex, Nature, 1995, 375, 564.
[8] T. W. Ebbesen, H. Hiura, M. E. Bisher, M. M. J. Treacy, J. L. Shreeve-Keyer, R. C. Haushalter, Adv. Mater., 1996, 8, 155.
[9] S. C. Tsang, Y. K. Chen, P. J. F. Harris, M. L. H. Green, Nature, 1994, 372, 159.
[10] J. Israelachvili, Intermolecular and Surface Forces, 2nd edition, Aca-demic Press, London, 1992, 317.
[11] Handbook of Physics and Chemistry (Ed: R. C. Weast), CRC Press, West Palm Beach, FL, 58th edition, 1977±78.
[12] Y. Miyamoto, A. Rubio, X. Blase, M. L. Cohen, S. Louie, Phys. Rev. Lett., 1995, 74, 2993 (the value of surface tension of potassium (g = 395 mN/m) used in this reference corresponds to a very old study, more recent measurements indicate a value of about 100±120 mN/m (see reference 11); A. Rubio, Y. Miyamoto, X. Blase, M. L. Cohen, S. Louie, Phys. Rev. B, 1996, 53, 4023.
[13] T. Guo, P. Nikolaev, A. Thess, D. T. Colbert, R. E. Smalley, Chem. Phys. Lett., 1995, 243, 49.
[14] E. Dujardin, T. W. Ebbesen, A. Krishnan, M. M. J. Treacy, Adv. Ma-ter., 1998, 10, 611.
[15] A. M. Schwartz, C. A. Rader, E. Huey, in Contact Angle, Wettability and Adhesion, Advances in Chemistry Series 43, ACS, Washington, 1963, p. 250.
[16] W. A. Zisman, in Contact Angle, Wettability and Adhesion, Advances in Chemistry Series 43, ACS, Washington, 1963, p. 1.
[17] Notice that by convention the units of gCare in energy per unit area
since it is a characteristic of the solid surface, while the surface tension of a liquid is given in units of force per unit length.
[18] J. C. Charlier, X. Gonze, J. P. Michenaud, Europhys. Lett., 1995, 29, 43.
[19] V. N. Bogomolov, Sov. Phys. Tech. Phys., 1992, 37, 79. [20] S. K. Rhee, J. Am. Ceram. Soc., 1971, 54, 376. [21] V. A. Ogarev, Kolloidnyi Zhurnal, 1978, 40, 153.
[22] X. Blase, L. X. Benedict, E. L. Shirley, S. G. Louie, Phys. Rev. Lett., 1994, 72, 1878.
[23] H. M. Princen, Surface and Colloid Science (Ed: E. Matijevic), vol. II, Interscience, New York, 1969.
[24] B. J. Carroll, J. Colloid Interface Sci., 1976, 57, 488.
Low-Temperature Solution Route to
Molybdenum Nitride**
By Hsin-Tien Chiu,* Shiow-Huey Chuang,
Gene-Hsiang Lee, and Shie-Ming Peng
Transition metal nitrides are technologically important
materials with many interesting properties.
[1±3]Frequently,
these materials are prepared at high temperatures by direct
nitridation or chemical vapor deposition (CVD)
[4]employ-ing N
2or NH
3as the source of nitrogen atoms. Rapid
solid-state synthesis, a highly exothermic self-propagating
reac-tion, represents an alternative route.
[5±9]Here, we wish to
report the synthesis of molybdenum nitride powder,
[10±12]an effective catalyst for desulfurization and
hydro-denitrogenation of hydrocarbons,
[13]via a sol-gel type of
solution process, employing a mixture of Na
2MoO
4, (Me
3-Si)
2NH, Me
3SiCl, and NEt
3in refluxing DME
(1,2-di-methoxyethane, boiling point 358 K). Although the growth
of metal nitride thin films by CVD at 473 K has been
re-ported,
[14,15]this is the first time that a transition metal
ni-tride has been prepared from solution at low temperature.
When Na
2MoO
4was reacted with Me
3SiCl, (Me
3Si)
2NH
and NEt
3in refluxing DME under N
2or Ar atmosphere, a
relatively air-stable black powder 1 precipitated, while a
white solid, identified as Et
3NHCl, deposited on the inner
surface of the condenser. (Me
3Si)
2O was detected as the
major by-product in the solution; no volatile Mo
by-prod-ucts were detected in the reaction mixture. The average
particle size of 1 was determined to be 15±30 nm using
±
[*] Prof. H. T. Chiu, Dr. S. H. Chuang Department of Applied Chemistry National Chiao Tung University Hsinchu, Taiwan 30050 (R.O.C.) G. H. Lee, S. M. Peng Department of Chemistry National Taiwan University Taipei, Taiwan 10764 (R.O.C.)
[**] We thank the National Science Council of Taiwan, the Republic of China (NSC-85-2113-M-009-012) for support.
STEM (scanning transmission electron microscopy,
Fig. 1a). ED (electron diffraction) (Fig. 1b) identifies
over-lapping diffraction signals, which are assigned to randomly
dispersed microcrystals of a cubic-phase material with a =
4.2 and residual NaCl. The lattice parameter of a = 4.2
is close to that of g-Mo
2N, 4.16 . Due to the small particle
size of 1, X-ray diffraction (XRD) only showed reflections
of residual NaCl, the reason being that in general X-rays,
with their wavelengths much longer than that of the
elec-tron beam, can only be used to show diffraction patterns of
crystalline particles with sizes greater than 200 nm.
[16]After 1 was thermally treated at 873 K for 1 h, broad XRD
peaks corresponding to the g-Mo
2N phase were observed.
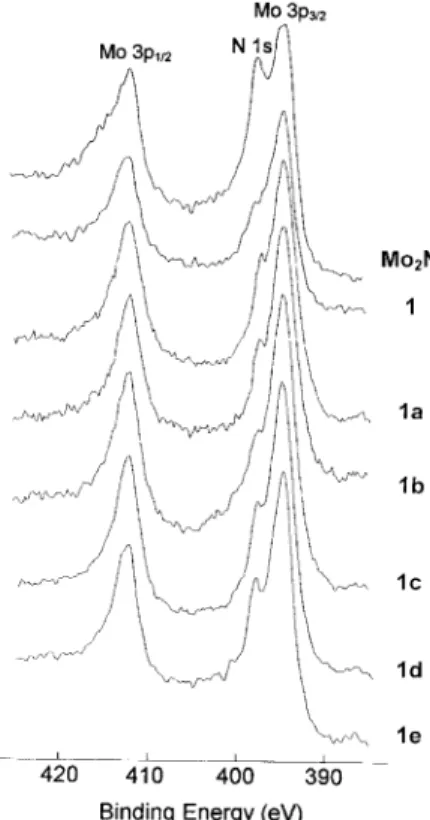
A high-resolution XPS (X-ray photoelectron spectroscopy)
study of 1 showed signals corresponding to Mo(3d
5/2),
Mo(3d
3/2), Mo(3p
3/2), Mo(3p
1/2), and N(1s) electrons at
228.2, 231.4, 394.1, 411.8, and 397.3 eV, respectively. The
signals of C(1s) and O(1s) electrons were observed at 284.1
and 531.0 eV, respectively. They are assigned to
by-prod-ucts, which are yet to be fully separated. The N(1s) signal is
characteristic for a metal nitride material. These data were
compared with a commercial sample of Mo
2N, which
showed corresponding elemental signals at 228.2, 231.3,
394.2, 411.7, and 397.2 eV. In Figure 2, high-resolution
spectra of Mo(3p
3/2), Mo(3p
1/2), and N(1s) electrons are
shown for 1 and the reference sample, thus confirming that
1 contains molybdenum nitride. After NaCl was separated
from 1 by dissolving the salt in water, the insoluble residue,
1a, was collected and dried in vacuo at room temperature.
XPS of 1a indicates that the molybdenum and nitrogen
sig-nals (Fig. 2) differ little from those of 1. Heating 1 in
vacu-um at 673 K, to remove traces of any volatile by-products,
produced a black powder 1b. As shown in Figure 2, the
high-resolution XPS signals of 1b show little difference
either. In addition, XPS signals of NaCl were observed for
1b. Neither 1a nor 1b showed C±H stretching signals in the
IR spectra.
Analyzing these data, where the relative concentrations
of Mo and N were determined from the XPS data by
inte-grating the corresponding signals after curve fitting (with a
commercial sample as a standard), we conclude that the
black powder 1 contains nanosized molybdenum nitride
Fig. 1. a) STEM bright-field image and b) ED (L = 80 cm, l = 0.0336 ) of 1 (ultrasonically irradiated in ethanol).
Fig. 2. High-resolution XPS signals of Mo(3p3/2), Mo(3p1/2), and N(1s)
particles, MoN
x(x = 0.4 0.1). A balanced equation is
pro-posed in Equation 1 to describe the overall reaction
stoi-chiometry. The evolution of dinitrogen molecules has been
proposed but has not yet been confirmed.
Na
2MoO
4+ 2(Me
3Si)
2NH + 4 Me
3SiCl + 2 NEt
3ÿÿÿÿÿÿÿÿ! NoN
x+ 2 NaCl + 4 (Me
3Si)
2O +
DME (reflux)2 NEt
3HCl + (1 ± 0.5x) N
2(1)
In order gain further insight into this reaction, we
investi-gated the system by adding reagents sequentially, thus
al-lowing reaction intermediates to be isolated. The
observa-tions are summarized in Scheme 1. Only Me
3SiCl showed
an initial significant rate of reaction towards Na
2MoO
4in
DME. MoO
2Cl
2(DME), 2, was isolated in high yield.
[17]Reacting 2 with Me
3SiCl, (Me
3Si)
2NH, and NEt
3in
reflux-ing DME generated a black powder 1c, characterized to be
MoN
xby XPS (Fig. 2). Treatment of 2 with (Me
3Si)
2NH
yielded a pale yellow liquid, 3, by distillation, which is yet
to be fully characterized. Compound 3 gradually darkened,
indicating further reaction at room temperature. A known
dimeric nitrido complex [N º Mo(OSiMe
3)
3×NH
3]
2, 4,
crys-tallized from the mixture as a minor product.
[18]Formation
of (Me
3Si)
2O, (Me
3Si)
2NH and a brown precipitate yet to
be characterized were also observed. Addition of NEt
3to 3
enhanced the apparent rate of formation of 4. Addition of
pyridine to 3 resulted in the isolation of another nitrido
complex N º Mo(OSiMe
3)
3×py, 5.
[17]Reacting 3 with Me
3-SiCl, (Me
3Si)
2NH, and NEt
3in refluxing DME generated a
black powder, 1d, shown to be MoN
xby XPS (Fig. 2).
In an aprotic environment, (Me
3Si)
2NLi was allowed to
react with 2 in hexane to form a pale yellow liquid,
formu-lated as a nitrido complex N º Mo(OSiMe
3)
2(N(SiMe
3)
2),
6, in high yield. Contrary to the instability of 3 at room
temperature, 6 showed little sign of decomposition. A
pyri-dine adduct of 6, N º Mo(OSiMe
3)
2(N(SiMe
3)
2)×py, 7, was
crystallized in good yield from hexane. Compound 7 is a
mononuclear five-coordinate complex with a distorted
square pyramidal geometry about the metal center (Fig. 3).
Scheme 1.
Fig. 3. ORTEP drawing of 7, showing the numbering scheme for the non-hy-drogen atoms. Selected bond distances () and angles ( ): Mo±N(1) = 1.640(3), Mo±N(2) = 1.973(3), Mo±N(3) = 2.334(3), Mo±O(1) = 1.921(3), Mo±O(2) = 1.924(2), N(1)±Mo±N(2) = 102.5(1), N(1)±Mo±N(3) = 92.0(1), N(2)±Mo±N(3) = 165.4(1), N(1)±Mo±O(1) = 106.6(1), N(1)±Mo±O(2) = 107.6(1), O(1)±Mo±O(2) = 139.1(1), O(1)±Mo±N(2) = 96.3(1), O(1)±Mo± N(3) = 77.7(1), O(2)±Mo±N(2) = 97.7(1), O(2)±Mo±N(3) = 79.2(1).
The Mo atom lies slightly above the basal plane while
the nitrido ligand occupies the apical position. The
N(1)±Mo distance is short, 1.640(3) . The overall
geom-etry of 7 is closely related to those of 4, 5, and
[N º MoCl
3(N(SiMe
3)
2)]
±.
[19]When 6 was reacted with
Me
3SiCl, (Me
3Si)
2NH, and NEt
3in refluxing DME a black
powder 1e resulted that was identified as MoN
xby XPS
(Fig. 2).
Comparing this observation with the experiment
employ-ing 3 to form molybdenum nitride, little difference exists
except that 6 was first prepared in an aprotic medium and
then exposed to a protic medium. Therefore, we propose
protonation of 6 to be an essential step. This step probably
converts the nitrido ligand to an imido ligand,
[20]then,
through condensation reaction steps, into MoN
x. For 3, in
the presence of a base such as pyridine or NEt
3,
deprotona-tion occurs and the nitrido complexes 4 and 5 are
gener-ated. In Scheme 2, a generalized Mo±N±Mo
polymeriza-tion route is proposed to account for the formapolymeriza-tion of the
molybdenum nitride. Intermolecular condensation
reac-tion, removal of a molecule HY from Mo=NH and Y±Mo
results in a Mo±N±Mo linkage. This step is comparable to
the M±O±M formation pathway proposed for sol-gel routes
to metal oxide materials.
[21]The proposed Mo º N and
Mo=NH species resemble the M±O
±and M±OH species in
a regular sol-gel process. Analogous reactions are known
for Mo±O±Mo and W±N±W connectivity formations.
[20,22]Repeating the condensation step polymerizes the
mono-meric MoN-containing units, such as imido and nitrido
in-termediates, into clusters of oligomers. Ladder structures
found for [(
tBuCH
2
)
2TaN]
5and W
4N
4(NPh
2)
6(OBu)
2may
be viewed as models to represent the initial stages of the
oligomerization.
[23,24]Further polymerization causes these
oligomers to coagulate into nanosized molybdenum nitride
powders.
In order to extend the chemistry to include other metals,
we attempted to prepare tungsten nitride powder by
react-ing Na
2WO
4with Me
3SiCl, (Me
3Si)
2NH, and NEt
3in
re-fluxing DME. The reaction did not proceed probably due
to the high W±O bond strength. However, reacting a
mix-ture of WCl
6, (Me
3Si)
2NH, and NEt
3in refluxing DME
produced a black precipitate. Preliminary characterization
of this black powder by XPS indicated the presence of
tungsten nitride with some residual chloride.
[25]In summary, we have demonstrated a low-temperature
solution route to molybdenum nitride by reacting a mixture
of Na
2MoO
4, Me
3SiCl, (Me
3Si)
2NH, and NEt
3in refluxing
DME. From the molecular complexes observed in this
study, the process shows parallels to the sol-gel processing
of metal oxide materials in many ways. This study extends
the already versatile chemistry of interconversions among
various metal±nitrogen containing complexes by showing
an excellent correlation between molybdenum nitrido
com-plexes and molybdenum nitride. Further investigations to
extend our understanding of the reaction are in progress.
Experimental
All chemicals and solvents were manipulated under dry and oxygen-free N2atmosphere. Reactions carried out under Ar atmosphere showed same
result.
Compound 1: To Na2MoO4 (2.0 g, 9.7 mmol) suspended in DME
(100 mL), NEt3(5.5 mL, 40 mmol), (Me3Si)2NH (4.1 mL, 19 mmol), and
Me3SiCl (11.1 mL, 87.5 mmol) were added sequentially. The mixture
gradu-ally darkened within 1 h, after which it was refluxed for 12 h. During this time, a white solid deposited on the inner surface the condenser. An air-stable black precipitate was collected from the reaction mixture.
Compound 1a: After 1 was washed with H2O, the insoluble black
precipi-tate was collected and dried under vacuum.
Compound 1b: 1 was heated at 673 K under vacuum for 1 h. The black solid was collected.
Binding energies [eV]. 1a: Mo(3d5/2), 228.4; Mo(3d3/2), 231.5; Mo(3p3/2),
394.4; Mo(3p1/2), 411.9; N(1s), 397.0. 1b: Mo(3d5/2), 228.0; Mo(3d3/2), 231.3;
Mo(3p3/2), 394.4; Mo(3p1/2), 411.9; N(1s), 397.0. Based on the intensities of
C(1s) and O(1s) signals, both elements are judged to be insignificant compo-nents. 1c: Mo(3d5/2), 228.3; Mo(3d3/2), 231.5; Mo(3p3/2), 394.4; Mo(3p1/2),
411.6; N(1s), 397.1. 1d: Mo(3d5/2), 228.2; Mo(3d3/2), 231.4; Mo(3p3/2), 394.3;
Mo(3p1/2), 411.6; N(1s), 397.2. 1e: Mo(3d5/2), 228.4; Mo(3d3/2), 231.6;
Mo(3p3/2), 394.4; Mo(3p1/2), 411.8; N(1s), 397.3.
Compound 3: To 2 (5.0 g, 17 mmol) in hexane (100 mL), (Me3Si)2NH
(14.6 mL, 69.2 mmol) was added. After work up, a yellow liquid was iso-lated (3.1 g, 40 % yield based on Mo). Initially, the liquid was formuiso-lated to be (Me3SiN=)2Mo(OSiMe3)2based on NMR spectroscopy evidence [26]. 1H NMR (300 MHz, CDCl
3, 25 C): d 0.11 (s, OSiMe3), 0.20 (s, NSiMe3).
Al-though the two signals appeared to correspond to an equal number of hy-drogen atoms, accurate integrations were difficult to obtain due to the broadening of the signal at 0.11 ppm. Contrary to the literature description of (Me3SiN=)2Mo(OSiMe3)2, 3 showed sign of decomposition and gradually
darkened at room temperature. We speculate that a minor proton-contain-ing molecule coexisted because regardless how carefully we prepared 3, stretchings of O±H (3779, 3730, 3640, 3570 cm±1) and N±H (3404, 3357,
3241, 3161 cm±1) were observed in the IR spectra. The mass spectrometry
(MS) data suggest that ions of three different complexes coexisted when 3 was evaporated into the spectrometer. MS (EI = 12 eV, direct inlet,98Mo):
m/z = 379 (MoNO3Si3C9H27+), 450 (MoN2O2Si4C12H36+), 526 (MoN2O
3-Si5C15H46+± Me + 1). The assignments are m/z = 379, N º Mo(OSiMe3)3; m/
z = 450, (Me3SiN=)2Mo(OSiMe3)2; m/z = 526, a fragment of (Me3SiN=)
Mo(NHSiMe3)(OSiMe3)3. Thus, we speculate that when freshly prepared, 3
was a mixture of (Me3SiN=)2Mo(OSiMe3)2, a major product, and Me3SiOH,
a minor by-product. Upon standing at room temperature, Me3SiOH reacted
with (Me3SiN=)2Mo(OSiMe3)2through several addition±elimination and
si-lyl group migration steps. 3 thus became a mixture of molybdenum com-plexes, including a nitrido complex later isolated as 4 and 5.
Compound 6: To 2 (1.51 g, 5.20 mmol) stirred vigorously in hexane (50 mL), LiN(SiMe3)2 (1.74 g, 10.4 mmol) was added. The mixture was
stirred for 2 h. After work-up, 6 was isolated as a yellow liquid (2.0 g, 85 % yield based on Mo).1H NMR (300 MHz, toluene-d
8, ±20 C): d 0.25 (s, 9H,
NSiaMe3), 0.33 (s, 18H, OSiMe3), 0.54 (s, 9H, NSibMe3);13C NMR (75 MHz,
toluene-d8, ±20 C): d 1.8 (OSiMe3), 2.7 (NSiaMe3), 5.0 (NSibMe3). MS (EI, 98Mo): m/z = 450 (M+).
Compound 7: To 6 (2.0 g, 4.4 mmol) in hexane (50 mL), pyridine (py) (1.5 mL, 19 mmol) was added. After stirring for 18 h, the solvent was re-moved, producing a yellow solid. Recrystallization from hexane yielded yel-low crystals (1.5 g, 64 % based on Mo).1H NMR (300 MHz, toluene-d
8, ±10 C): d 0.14 (s, 18H, OSiMe3), 0.43 (s, 9H, NSiaMe3), 0.67 (s, 9H, NSibMe3), 6.42 (t, 2H, ±NCHCHCH), 6.79 (t, 1H, ±NCHCHCH), 8.44 (d, 2H, ±NCHCHCH);13C NMR (75 MHz, toluene-d 8, ±10 C): d 2.0 (OSiMe3), 2.9 (NSiaMe3), 5.4 (NSibMe3), 124.8 (±NCHCHCH), 137.7 (±NCHCHCH),
150.7 (±NCHCHCH). MS (EI,98Mo): m/z = 379 (M+± py). Crystal
parame-ters of 7 at 298 K: space group P1, a = 10.499(1), b = 10.974(3), c = 13.001(3) , a = 83.03(2) , b = 81.49(2) , g = 80.31(2) , V = 1453.1(5) 3, Z =
2, Dc= 1.206 g/cm3, Rf= 0.036, Rw= 0.034.
Received: April 20, 1998 Final version: June 30, 1998 ±
[1] L. E. Toth, Transition Metal Carbides and Nitrides, Academic Press, New York 1971.
[2] S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, Blackie Academic Professional, Glasgow 1996.
[3] H. O. Pierson, Handbook of Refractory Carbides and Nitrides, Noyes Publications, Westwood 1996.
[4] D. M. Hoffman, Polyhedron 1994, 13, 1169. [5] J. B. Wiley, R. B. Kaner, Science 1992, 255, 1093. [6] E. G. Gillan, R. B. Kaner, Inorg. Chem. 1994, 33, 5693.
[7] A. Hector, I. P. Parkin, J. Chem. Soc., Chem. Commun. 1993, 1095. [8] A. L. Hector, I. P. Parkin, Polyhedron 1995, 14, 913.
[9] A. L. Hector, I. P. Parkin, Chem. Mater. 1995, 7, 1728.
[10] Generally, molybdenum nitride powder is prepared by passing NH3
over MoO3at T > 873 K.
[11] L. Volpe, M. Boudart, J. Solid State Chem. 1985, 59, 332. [12] M. A. Sriram, P. N. Kumata, E. I. Ko, Chem. Mater. 1995, 7, 859. [13] S. Ramanathan, S. T. Oyama, J. Phys. Chem. 1995, 99, 16 365. [14] R. Fix, R. G. Gordon, D. M. Hoffman, Chem. Mater. 1991, 3, 1138. [15] R. Fix, R. G. Gordon, D. M. Hoffman, Chem. Mater. 1993, 5, 614. [16] A. R. West, Solid State Chemistry and Its Applications, Wiley, New
York 1984, p. 51.
[17] H.-T. Chiu, Y.-P. Chen, S.-H. Chuang, J.-S. Jen, G.-H. Lee, S.-M. Peng, Chem. Commun. 1996, 139.
[18] G. S. Kim, C. W. Dekock, J. Chem. Soc., Chem. Commun. 1989, 1166. [19] D. Fenske, A. Frankenau, K. Dehnicke, Z. Anorg. Allg. Chem. 1989,
574, 14.
[20] W. A. Herrmann, S. Bogdanovic, R. Poli, T. Priermeier, J. Am. Chem. Soc. 1994, 116, 4989.
[21] M. Prassas, L. L. Hench, in Ultrastructure Processing of Ceramics, Glasses, and Composites (Eds: L. L. Hench, D. R. Ulrich), Wiley, New York 1984, Ch. 9.
[22] T. E. Glassman, M. G. Vale, R. R. Schrock, Organometallics 1991, 10, 4046.
[23] M. M. B. Holl, P. T. Wolczanski, G. D. Van Duyne, J. Am. Chem. Soc. 1990, 112, 7989.
[24] Z. Gebeyehu, F. Weller, B. Neumüller, K. Dehnicke, Z. Anorg. Allg. Chem. 1991, 593, 99.
[25] Selected binding energies of tungsten nitride powder [eV]: W(4 f7/2),
33.2; W(4 f5/2), 34.5; Cl(2p), 199.2; N(1s), 398.3 (major), 400.6 (minor).
After the powder was heated at 573 K in vacuum for 2 h, the Cl(2p) and the minor N(1s) signals decreased to negligible. The N/W ratio is 0.5.
[26] H. W. Lam, G. Wilkinson, B. Hussain-Bates, M. B. Hursthouse, J. Chem. Soc., Dalton Trans. 1993, 1477.
A Novel Pathway to PbSe Nanowires at Room
Temperature**
By Wenzhong Wang,* Yan Geng, Yitai Qian, Mingrong Ji,
and Xianming Liu
Recently, one-dimensional (1D) structures with
meter diameter, such as nanowires (nanorods) and
nano-tubes, have attracted considerable attention due to their
special properties.
[1±13]Compared with
micrometer-diam-eter whiskers, they are expected to have remarkable
me-chanical properties, including electrical, optical, and
mag-netic properties that are in principle tunable by varying the
diameter and chirality.
[14,15]These new nanoscale materials
have potential applications in both mesoscopic research
and development of nanodevices. Previous work in this
field focused on carbon nanowires and nanotubes, which
were the by-product of fullerene research.
[16]Convention-ally, carbon nanowires or nanotubes can be grown in an arc
discharge at a temperature of 3000 K,
[17,18]by thermal
de-position of hydrocarbons,
[19]or vapor±liquid±solid (VLS)
growth.
[3,4,12]Comparatively little research has been carried
out on other 1D materials and nearly all the previous
meth-ods of preparing nanowires or nanotubes require extreme
conditions. Therefore, one of the important goals of
materi-als scientists is to prepare nanoscale materimateri-als under milder
conditions. Here we report a novel route to PbSe
nano-wires: PbCl
2, Se, and KBH
4were kept in a sealed flask at
room temperature for 4 h using ethylenediamine as the
sol-vent. The study on PbSe is meaningful because it could be
widely used for IR sensors,
[20]solar cells, infrared
detec-tors,
[21]chemical sensors,
[22]and so on. To our knowledge,
the method here is the mildest route so far to produce
nanowires and it is reasonable to assume that other
nano-scale materials can be obtained by a similar process except
that so far only the reactant PbCl
2has been substituted by
other MCl
ncompounds.
In a standard experimental procedure, an appropriate
amount of Se powder, PbCl
2, and KBH
4were placed in a
flask, which was filled with ethylenediamine up to 90 % of
its volume. The flask was then sealed and maintained at
about 10 C for 4 h. The precipitate was filtered and
washed with distilled water, the black product was
col-lected, and, finally, dried in vacuum at about 10 C for 12 h.
X-ray powder diffraction (XRD) was used to
character-ize the product. It was collected on a Rigaku D/max gA
ro-_______________________
±
[*] Dr. W. Wang, Dr. Y. Qian, Dr. M. Ji, Dr. X. Liu Structure Research Laboratory
University of Science and Technology of China Hefei, Anhui 230026 (People's Republic of China) Dr. W. Wang, Dr. Y. Geng, Dr. Y. Qian Department of Chemistry
University of Science and Technology of China Hefei, Anhui 230026 (People's Republic of China)
[**] This work is supported by the Chinese National Natural Science Fund and National Nanometer Materials Climbing Program.