Printed in Great Britain 0 1993 Pergamon Press Ltd
OSMIUM(VI) COMPLEXES OF SULPHONYL AMID0
LIGAND. CRYSTAL STRUCTURES OF
WA-HUNG LEUNG*
andERNEST KIN-FAI CHOW
Chemistry Department, Hong Kong University of Science and Technology,
Clear Water Bay, Kowloon, Hong Kong
and
SHIE-MING
PENGt
Department of Chemistry, National Taiwan University, Taipei 10764, Taiwan, R.O.C.
(Received 8 February 1993 ; accepted 5 March 1993)Abstract-Interactions
of [PPh4]2[0s02C14] and [Bu”,Nl[OsNCl~] with LiZL [H2L = 1,2-
bis(p-toluenesulphonylamido)benzene]
gave binuclear osmium(V1) compounds (PPh,),[Osz
04LZ(p-OH)d
(1)and OS~N~L&-OH)~ (2), respectively. The structures of both compounds
have been determined by X-ray crystallography. The crystal structure of the [OsZ04LZ@-
0H),12-
anion contains two trans-0s02(L)
units bridged by two OH groups; the
OS-0(0x0)
bond length is ca 1.74 %i. The crystal structure of 2 contains two OsN(L) units
bridged by two OH groups
; the Os-N(nitrido)bond length is ca 1.56 8.
Multianionic chelating ligands are of current inter-
est because they can stabilize metal ions in high
oxidation states. High-valent metal complexes with
deprotonated
amido ligands, e.g. those of man-
ganese(V),’ iron(
cobalt(IV),3 nickel(III)4 and
copper(III),’ are well documented. However, there
are relatively few studies on complexes of sulphonyl
amido groups. As the sulphonyl group is more elec-
tron-withdrawing
than the carbonyl group one
might expect that sulphonyl amido ligands can be
deprotonated more readily and hence could display
interesting coordination chemistry. To this end we
set out to investigate the chemistry of complexes
containing
chelating sulphonyl
amido ligands,
e.g. bis-(toluenesulphonylamido)benzene
(H,L), as
shown in Fig. 1. The unique features of the ligand
H2L are
: (a) it is bidentate and dianionic ; (b) it canstabilize highly oxidizing metal centres due to the
* Author to whom correspondence should be addressed.
t Author
to whom inquiries concerning X-ray crys-
tallography should be addressed.
strong a-donating deprotonated
sulphonyl amido
groups
; and (c) it is sterically bulky. Recently, thecrystal structures of some copper complexes of H2L
have been described.6 We herein report the synthesis
and crystal structures of two osmium(V1) com-
pounds of the ligand.
EXPERIMENTAL
The ligand H2L,& IpPh4]2[Os02C14]7 and [Bu”,N]
[OsNC14]* were prepared according to the litera-
ture procedures. Organic solvents were purified by
standard methods and distilled before use. ‘H
NMR spectra were recorded on a JEOL EX 400
spectrometer. Chemical shifts (6) were reported ref-
erenced to Si(CH,),.
IR spectra (Nujol) were
Fig. 1.
1635
1636 WA-HUNG LEUNG et al.
obtained on a Perkin-Elmer
1600 FT-IR spectro-
photometer. Elemental analyses were performed by
MEDAC Ltd (Middlesex, U.K.).
Preparation of
[PPh,]2[0s204Lz(p-0H)Z]
(1)To a suspension of H2L (0.4 g, 0.96 mmol) in
THF (20 cm3) at -40°C was added 2 equivalents
of Bu”Li
(ca1.25 cm3 of a 1.6 M solution in
hexanes). The resulting mixture was warmed to
room temperature and stirred for 1 h. To this solu-
tion was added ~Ph&[0sOzC14] (0.51 g, 0.5 mmol)
under nitrogen and the reaction mixture was stirred
at room temperature overnight. After the removal
of volatiles the dark red residue was extracted with
acetonitrile/ether.
Slow evaporation of the extract
at room temperature afforded red crystals, which
were collected and washed with ether (yield 0.25 g).
‘H NMR (CD,CN)
: 2.34 (s, 12H,p-CHJ,5.96 (br,
s, 2H, OH) and aromatic protons. IR (Nujol)/cn-
’ :v(O-H)
3437; v(S=O) 1152, 1108; ~(0~0,) 859.
Found: C, 53.2; H, 4.0; N, 3.3%. Calc. for [OsZ
Cs8H78N40,4P2S4]: C, 53.2; H, 4.0; N, 2.8%.
Preparation of
OS~N~L~(~-OH)~ (2)
The lithium salt of HIL was prepared as for
[OS~O&OH)~]~-. To a THF solution of Li2L (pre-
pared from 0.41 g H2L and 1.25 cm3 Bu”Li) was
added [Bun4NJ[0sNC14] (0.29 g, 0.5 mmol). The
reaction mixture was stirred overnight and evap-
orated to dryness. The dark yellow residue was
extracted with THF. Careful addition of hexane to
the extract afforded the crude product, which was
recrystallized from THF/ether/hexane
to give yellow
crystals (yield 0.2 g). ‘H NMR (CD&N) : 2.29 (s,
6H, p-CH3); 6.28, 6.83, 7.06, 7.81 (all broad
singlets, 12H, aroamtic
protons).
IR (cm-‘):
v(O-H)
3450; v(S=O) 1150, 1126; v(OsN) 986.
Found:
C, 61.5; H, 5.5; N, 11.0. Calc. for
[OS~C&,H~~N~O,&]: C, 62.3; H, 4.9; N, 10.9%.
X-ray crystallographyDiffraction measurements were performed on an
Enraf-Nonius
CAD-4 diffractometer. Lattice par-
ameters of
1were obtained from 25 reflections with
20 angles in the range 17.00-21.00”, whereas cell
dimensions of complex 2 were determined from 25
reflections with 28 angles in the range 11.06-20.40”.
All reflections were collected for Lorentz, polar-
ization and adsorption effects. All data reduction
and refinement were performed using the NRCC-
SPD-VAX packages. The structures were solved
by direct methods and refined by full-matrix least-
squares
; all non-hydrogen atoms were refined withanisotropic thermal parameters. Both complexes
1and 2 crystallize in the triclinic system with the
space group
Pi.
Hydrogen atoms on the organic
ligands were calculated in idealized positions and
were included in the structure factor calculation.
The combined data collection and refinements are
given in Table 1. Selected bond lengths and angles
for complexes
1and 2 are given in Tables 2 and 3,
respectively.
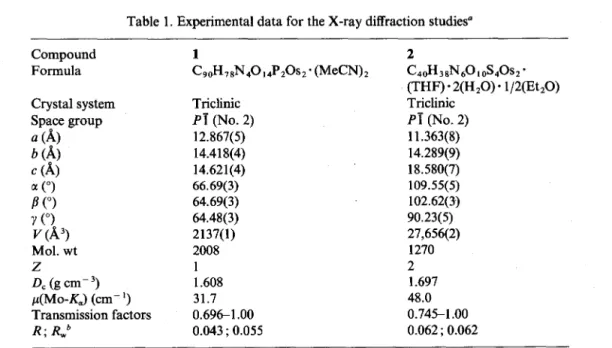
Table 1. Experimental data for the X-ray diffraction studies” Compound Formula Crystal system Space group 0 (A) b (A) c (A) a 0 B (“) Y (“) v (A’) Mol. wt Z & (g cm- ‘) p(Mo-&) (cm- ‘) Transmission factors
R;R,,,b
1C90H78N4014P20s2.tMeCN),
Triclinic Pi (No. 2) 12.867(5) 14.418(4) 14.621(4) 66.69(3) 64.69(3) 64.48(3) 2137(l) 2008 1 1.608 31.7 0.6961.00 0.043 ; 0.055 2 C,oH,~N,G,&Gsz- (THF)*2(H,O)- 1/2(Et,O) Triclinic Pi (No. 2) 11.363(8) 14.289(g) 18.580(7) 109.55(5) 102.62(3) 90.23(5) 27,656(2) 1270 2 1.697 48.0 0.745-l .oo 0.062 ; 0.0620 Features common to all determinations : I(Mo-K,) = 0.7107 A; temperature = 297 K. b
R = Z~~Fo~-~Fc~~/E~Fo'ol,
R,
= ~w(F,-F,)~/ZW~~~]"~.
OS”’ complexes of sulphonyl amido ligand
Table 2. Selected bond lengths (A) and angles (“) for the [Os204Lz@-
OH),]‘- anion
1637
Os-o(1)
1.740(6)
Os-O(2)
1.747(7)
Os-O(3)
2.051(6)
OS-N( 1)
2.022(6)
0(1)--0s--o(2)
O(l)-Os-O(3)
0(3)-Os--o(3’)
0(3)-Os-N(1)
0(4)_-S(ltiN(l)
Os-O(3)---Osa
OS-N(l)-S(1)
N(l)_-C(l)--C(2)
175.7(3)
86.7(3)
71.3(2)
175.7(2)
110.8(4)
108.7(3)
123.9(4)
129.1(7)
S(lW(4)
1.428(7)
8(1)-N(l)
1.646(6)
N(l)_-C(l)
1.40(l)
OS-N(l)-C(l)
O(l>--os-N(1)
N( 1 )-OS-N(~)
0(3)-Os-N(2)
N(l)_-S(l>-C(7)
O(4)-S(lFN(l)
0(4)--8(1)-C(7)
N(l)-C(l)--C(6)
118.4(5)
92.5(3)
78.3(3)
105.9(2)
106.6(3)
110.8(4)
107.7(4)
114.2(7)
RESULTS AND DISCUSSION
Interaction
of [PPh&[0s02Cl,]
with 2 equiv-
alents of the lithium salt of H,L [H2L = 1,2-bis(p-
toluenesulphonylamido)benzene]
in THF gave the
air-stable binuclear hydroxo-bridged
osmium(VI)-
dioxo compound
(l),isolated as its [pPh,]+ salt. A
monomeric compound could not be obtained even
when excess ligand was used, presumably because
the ligand is too bulky for formation of a planar
bischelate complex. The IR spectrum of
1does not
show any sharp N-H
band, indicating that the
sulphonyl amido group is in the deprotonated form.
The broad band near 3470 cn-
’ can be assigned tothe v(O-H) stretch. The Os=O stretch at 845 cm-
’is within the range expected for trans-0~0~ com-
pounds.
The binuclear structure of
1was conlirmed by
an X-ray diffraction
study. A diagram of the
[OszO,L&OH),]*-
anion is shown in Fig. 2;
selected bond lengths and angles are given in Table
2. The geometry around each osmium is best
described as octahedral with the two 0x0 groups as
axial ligands and L and two hydroxo groups as
equatorial ligands. The two truns-OsOzL units are
bridged by the two hydroxo
ligands with the
OS-O(H)--OS’
bond angle of ca 108.7”. A similar
structure has been observed for the binuclear oxo-
bridged 0~~O~py+~ The Os-O(oxo)
bond lengths
(ca 1.74 A) are normal by comparison with trans-
osmium(W)-dioxo
compounds.
The Os-N(sul-
phony1 amido) bond length of cu 2.03 A is similar
to that for Os-N(amido)
found in a related
osmium(VI)-nitrido
compound of a tetradentate
amido ligand. lo Of note, the four toluenesulphonyl
groups of the ligands adopt a “two-uptwo-down”
geometry.
Treatment of [Bu”&J[OsNCL,] with 2 equivalents
of Li2L gave the neutral dimeric osmium(VI)-
nitrido compound 2. Again, the steric bulk of the
ligand L precludes the formation of the monomeric
bischelate complex. The presence of the bridging
hydroxo group is indicated by the broad IR band
at 3450
cn- ‘.
The OS-N stretch at 986 cm-’ is
Table 3. Selected bond lengths (A) and angles (“) for 0s2N2L,(p-OH),
(2)
Os( 1)-O(9)
1.99(2)
Os( l)-N( 1)
1.97(2)
OS(~)-N(5)
1.52(2)
OS(~)-N(6)
1.61(2)
0(9)-0s(1)-0(10)
74.5(7)
0(9)--Os( l)-N( 1)
92.4(7)
0(9)-Os(l)-N(2)
140.2(7)
0(9)-Os(l)--N(5)
110.0(9)
N(l)-Os(l)---N(2)
85.6(7)
N(l)-OS(~)-N(5)
113.1(9)
N(2)-Os(l)--N(5)
109.5(9)
N(l)_-C(l)_-C(6)
112(2)
WI-W)
1.60(2)
S(l>--o(l)
1.46(2)
N(l)-C(1)
1.47(3)
W)-W)_W)
O(l)-8(1)----c(7)
W-W-c(7)
Os(l>--o(9)-0s(2)
OS(~)-N(l)-S(1)
OS(~)-N(lW(1)
N(lW(l)_-C(2)
S(l)--C(7)--c(8)
105(l)
108(l)
106(l)
104.2(7)
124(l)
116(2)
126(2)
120.7(8)
1638 WA-HUNG LEUNG et al.
Fig. 2. A perspective view of the [Os,O,L&-OH)$- anion.
normal for nitrido compounds. The ‘H NMR spec-
trum of 2 is consistent with the solid-state structure.
The structure of 2 has been determined by X-
ray crystallography.
An ORTEP drawing of 2 is
shown in Fig. 3 and selected bond lengths and
angles are given in Table 3. The geometry around
each osmium is square pyramidal with the nitrido
group as an axial ligand. The two Os(N)L units
are bridged by the two :hydroxo ligands with
the two nitrido ligands on the same side. The
Os(l)---O(H)--Os(2)
bond angle of 104.2” and
Os-N(amido)
bond length of 1.973 A are similar
to those for
1.The Os-N(nitrido)
bond length
is normal for osmium(W)-nitrido
compounds.
’ ’It might be noted that this is the tirst structure of
a binu~l~r osmi~-~t~do
~ompo~d.
The reactivity of these two osmium complexes is
being studied.
Acknowledqemertts-We thank the Hong Kong Uni- 5. S. T. Kirsey Jr, J. A. Neubecker and D. W. versity of Science and Technology, Hong Kong Research
Grants Council and the National Science Council of the Republic of China for support. W.-H.L. thanks Prof. Cm-Ming Che for helpful discussions.
6.
7. 8.
REFERENCES
9. 1. T. J. Collins, R. D. Powell, C. S. Slebodnich and
E. S. Uffelman, J. Am. Chem. Sot. 1990,112,899.
2. T. J. Collins, K. L. Kostka, E. Munich and E. S. 10. Uffelman, J. Am. Chem. Sot. 1990,112,5637.
3. F. C. Anson, T. J. Collins, R. J. Coots, S. L. Gibson and T. G. Richmond, J. Am. Chem. Sot. 1984,106,
5037. 11.
4. L. Fabbrizzi, T. A. Kaden, A. Perotti, B. Seghi and L. Siegfried, Znorg. Chem. 1986,25, 321.
Margerum, J. Am. Chem. Sot. 1979,101, 1631. (a) H.-Y. Cheng, P.-H. Cheng, C.-F. Lee and S.-M. Peng, Znorg. Chim. Actu 1991, 181, 145; (b) H.-Y. Cheng, G.-H. Lee and S.-M. Peng, Znorg. Chim. Acta 1992, 191,25.
P. A. Shapley, H. S. Kim and S. R. Wilson, Organometallics 1988, 7, 928.
W. P. Griffith and D. Pawson, J. Chem. Sot., Dalton Trans. 1973, 1315.
A. M. R. Galas, M. B. Hursthouse, E. J. Behrman, W. R. Midden, G. Green and W. P. Griffith, Trans. Met. Chem. 1981,6, 194.
(a) C. J. Barner, T. J. Collins, B. E. Mapes and B. D. Santersiero, Znorg. Chem. 1986, 25, 4322 ; (b) C. M. Che, H. W. Lam, W. T. Wong and T. F. Lai, unpublished work.
W. A. Nugent and J. M. Mayer, Metal Ligand Multiple Bonds, p. 186. Wiley-Interscience, New York (1988).
![Table 2. Selected bond lengths (A) and angles (“) for the [Os204Lz@- OH),]‘- anion 1637 Os-o( 1) 1.740(6) Os-O(2) 1.747(7) Os-O(3) 2.051(6) OS-N( 1) 2.022(6) 0(1)--0s--o(2) O(l)-Os-O(3) 0(3)-Os--o(3’) 0(3)-Os-N(1) 0(4)_-S(ltiN(l) O](https://thumb-ap.123doks.com/thumbv2/9libinfo/8873732.249333/3.780.151.597.84.377/table-selected-bond-lengths-angles-anion-os-ltin.webp)

