奈米氧化鈰構形控制對乙醇轉氫效率研
究
Morphology Control of Cerium Oxide
Nanocrystal and It Effect to Ethanol
Reforming Reaction
研 究 生 : 蕭偉印 Student : WeiYing Shiao
指導教授 : 李積琛 博士 Advisior: ChiShen Lee
國立交通大學 應用化學研究所
碩士論文
A Dissertation
Submitted to Institute of Applied Chemistry National Chiao Tung University
in Partial Fulfillment of the Requirements for the Degree of
Master of Science In
Applied Chemistry June 2006
Hsinchu, Taiwan, Republic of China 中華民國九十五年六月
奈米氧化鈰構形控制對乙醇轉氫效率研究 學生:蕭偉印 指導教授:李積琛 博士 國立交通大學應用化學研究所 摘要 本實驗利用共沉澱法合成出氧化鈰奈米纖維,以及水熱合成法合 成出氧化鈰奈米粒子,奈米柱及奈米方塊,並且使用掃描及穿透式電 子顯微鏡鑑定其奈米結構以及晶體成長方向,其中氧化鈰奈米纖維為 多晶結構,氧化鈰奈米柱是由兩組{ 1 1 0 }及一組{ 1 0 0 }晶面組成之 柱狀體,而氧化鈰奈米方塊是由三組{ 1 0 0 }晶面組成之正方形方塊。 經過使用上述合成之奈米氧化鈰晶體,在添加重量百分比 5%的銠 金屬,經過測試乙醇轉換裂解產氫實驗,發現與不加控制構形及所暴 露晶面的氧化鈰比較,明顯有較佳的轉換效率,在相同情況下,利用傳 統涵浸法所得催化劑之氫氣選擇率約 116%,而利用氧化鈰奈米柱所製 成催化劑可提高 10%氫氣選擇率。而上述奈米構形催化劑在長時間催 化反應後由於結構穩定性不佳,使得催化效率不能維持。
Morphology Control of Cerium Oxide Nanocrystal and It Effect to Ethanol Reforming Reaction
Student: WeiYing Shiao Advisor: Dr.Chishen Lee
Institute of Applied Chemistry
National Chiao Tung University
Abstract
In this study , two methods, including (1) coprecipritation and (2) hydrothermal processes, were carried out to generate CeO2 nanocrystals with various shapes. On the basis of on the SEM study, the first method only produced polycrystalline CeO2 whereas the second method can generate rod and cube shape of CeO2. According to the TEM images , we found that the predominantly exposed planes are {001} in cerium oxide nanocubes . The predominantly exposed planes are {001} and {110} in cerium oxide nanorods .
The catalytic activity for the conversion of ethanol to hydrogen have been carried out using the 5% wt Rh/CeO2 as the catalysts with as-synthesized CeO2 rods and cubes. The results from H2 selectivity, the effect of C/O ratio and stability test indicate that the cerium oxide which exposed planes under controlled more active than catalyst with irregular shape of CeO2 particles . Under the same condition , hydrogen selectivity of the cerium oxide nanorod catalyst is 10% higher than that of the catalyst prepared by impregnation method whose hydrogen selectivity is about 116% . However , the H2 selectivity for catalyst with rod and cube CeO2
gradually decrease after 24 hours, indicating the {100} and {110} surface are not stable at high temperature .
誌謝 首先我想感謝的是我的指導老師,李積琛老師,老師在就讀研究 所的兩年當中,對於學生的研究都是支持並細心指導,不會因為做錯 便有所苛責,而老師讓學生自己去找答案並在一旁提點的指導方式, 讓我們學到的是尋找答案的方式以及思考模式更是受益良多。 再來我要感謝實驗室的博班學長:王明芳,陳奎伯,黃文亨以及吳 明誠,因為我的個性比較怪,容易鑽牛角尖,胡思亂想,有他們的包 容以及細心開導,在實驗室裡面過的很自在,也很開心。 再來在實驗上,也感謝所有幫過忙的人,其中陳月枝老師以及林 亞玄學長在整個實驗系統的架設與分析上給予莫大的幫助,如果沒有 陳老師的幫助,也許就不會有這篇論文的寫成。 最後感謝的是大毛,每天跟我一起吃飯,還幫我買便當,陪我聊 天,聽我發牢騷,如果沒有他,可能每天都會臭著一張臉做實驗,謝 謝你,謝謝大家。
目錄 中文摘要 i 英文摘要 ii 誌謝 iv 目錄 v 表目錄 viii 表目錄 ix 第一章 緒論 1 1-1 實驗緣起 1 1-2 乙醇催化產氫原理 3 1-3 氧化鈰簡介 4 1-4 已知氧化鈰奈米晶體 6 1-4-1 二氧化鈰奈米粒子 6 1-4-2 二氧化鈰奈米柱 7 1-4-3 二氧化鈰奈米方塊 9 第二章 奈米結構分析法及轉換效率計算法介紹 10 2-1 奈米結構分析法 10 2-1-1 掃描式及穿透式電子顯微鏡 10 2-2 轉換效率計算法 12
2-2-1 最大氫氣選擇率( Hydrogen Selectivity ) 12 2-2-2 定量測量方式 12 2-2-3 轉換效率計算法 15 第三章 實驗方法 17 3-1 實驗藥品 17 3-2 儀器設備 18 3-3 實驗步驟 19 3-3-1 利用 IA IIA 鹵化鹽共沉澱合成氧化鈰奈米纖維 19 3-3-2 利用水熱法合成二氧化鈰奈米柱及奈米方塊 19 3-3-3 粉末分析 19 3-3-4 掃描式電子顯微鏡分析 19 3-4-5 穿透式電子顯微鏡分析 20 3-4-6 催化劑製備 20 3-4-7 轉換效率測試系統 20 3-4-8 氣相層析儀分析設置 22 第四章 結果討論 23 4-1 利用 IA IIA 鹽類共沉澱法合成奈米氧化鈰 23 4-1-1 實驗簡介 23 4-1-2 不同鹽類對氧化鈰晶體構形的影響 22
4-2 利用水熱法合成氧化鈰奈米柱 28 4-2-1 實驗步驟 28 4-2-1 氫氧化納濃度對合成之影響 28 4-3 利用水熱法合成氧化式奈米方塊 33 4-3-1 實驗簡介 33 4-3-2 氫氧化鈉濃度對合成之影響 33 4-3-3 穿透式顯微鏡分析 35 第五章 乙醇轉氫催化效率測試 37 5-1 催化測試系統 37 5-2 反應氣體與催化劑接觸時間 39 5-3 碳氧比值對催化效率影響 40 5-3-1 實驗原理 40 5-3-2 實驗結果 40 5-4 結果與討論 44 5-5 結論 45 參考文獻 46 附錄(1)固態反應列表 48
表目錄 表 2-2-1 熱導偵測器訊號對真實訊號誤差 14 表 2-2-2 熱導偵測器訊號對真實訊號誤差 15 表 3-1-1 實驗藥品 17 表 4-1-1 反應列表 23 表 4-2-1 氫氧化鈉濃度與所得產物 28 表 4-2-2 氧化鈰層間距列表 32 表 4-3-1 氫氧化鈉濃度對反應結果 33 表 5-1-1 分析氣體滯留時間 37
圖目錄 圖 1-3-1 氧化鈰晶體結構圖 4 圖 1-4-1 二氧化鈰奈米粒子穿透視顯微鏡照片 6 圖 1-4-2 二氧化鈰晶體成長形成八面體 7 圖 1-4-3 二氧化鈰奈米柱 8 圖 1-4-4 二氧化鈰奈米柱穿透視電子顯微鏡圖及晶面示意圖 8 圖 1-4-5 二氧化鈰奈米方塊穿透式電子顯微鏡圖 9 圖 2-1-1 掃描式電子顯微鏡構照示意圖 11 圖 2-1-2 穿透式電子顯微鏡構造示意圖 11 圖 2-2-1 氫氣濃度對熱導偵測器訊號圖 13 圖 2-2-2 氮氣濃度對熱導偵測器訊號圖 13 圖 2-2-3 氫氣濃度對熱導偵測器訊號圖 14 圖 2-2-4 氮氣濃度對熱導偵測器訊號圖 15 圖 3-4-1 汽化及反應系統示意圖 21 圖 4-1-1 a 、b、c、d 分別為表 4-1-1 所列編號 a 、b、c、d 之產物掃描 式電子顯微鏡觀測結果 24 圖 4-1-2 e、f、g、h 分別為表 4-1-1 所列編號 e、f、g、h 之產物掃描式 電子顯微鏡觀測結果 25 圖 4-1-3 使用氯化鈉鹽類共沉澱所得氧化鈰粉末繞射圖 26
圖 4-1-4 使用氯化鎂共沉澱所得粉末繞射圖 26 圖 4-1-5 使用氯化鈉鹽類共沉澱所得氧化鈰穿透式電子顯微鏡圖 27 圖 4-2-1 a、b、c、d 分別為 1M 、2M 、3M 、4M 氫氧化納濃度下, 所得產物經掃描式電子顯微鏡觀測圖形 29 圖 4-2-2 a、b、c、d 分別為 5M 、10M 、15M 、15M 氫氧化納濃度 下,所得產物經掃描式電子顯微鏡觀測圖形 30 圖 4-2-3 氧化鈰奈米柱穿透式顯微鏡圖 31 圖 4-2-4 氧化鈰奈米柱粉末繞射圖 31 圖 4-2-5 氧化鈰奈米柱製成催化劑之圖 32 圖 4-3-1 a、b、c、d、e、f 分別表示 1M、2M、3M、5M、10M、15M 電子顯微鏡圖 34 圖 4-3-2 氧化鈰奈米方塊穿透視顯微鏡圖暨選區繞射圖 35 圖 4-3-3 奈米氧化鈰方塊粉末繞射圖 36 圖 4-3-4 氧化鈰奈米方塊製成催化劑之掃描式電子顯微鏡圖 36 圖 5-1-1 測試及分析系統圖 38 圖 5-2-1 改變流速對氫氣選擇率之影響 39 圖 5-3-1 不同碳氧比值對轉換效率之影響 41 圖 5-3-2 不同催化劑在 C/O 值 0.7 下之氫氣選擇率 41 圖 5-3-3 催化劑穩定性測試結果 41
圖 5-3-4 奈米方塊催化劑經過 24 小時反應之掃描式電子顯微鏡圖 42 圖 5-3-5 奈米柱催化劑經過 24 小時反應之掃描式電子顯微鏡圖 42
第一章緒論
1-1 實驗源起
在石油短缺的今天,再生能源的研究已成為熱門議題,再加上京都 議定書所提到對於地球暖化現象的控制,未來新能源必須能控制並減 少二氧化碳的產生,目前已有不少研究提及利用生質酒精作為未來可 再生能源,由於生質酒精來自於植物光合作用,即使燃燒亦不會如化 石燃料般排放多餘二氧化碳於空氣中,加速地球暖化,合於京都議定 書規範,再者,乙醇可由自然界植物醱酵生成,來源不至於匱乏,更 是符合再生能源的需求。 目前已有不少研究針對由乙醇熱裂解或是蒸氣改質方式得到氫 氣,其中多數反應皆需要催化劑催化反應進行,催化劑的選擇更是多 樣化,由銅1,鈷2,鎳3到貴重金屬4如鉑,銠, 鈀,釕…等,但如果單 獨使用金屬催化劑容易因為反應過程產生積碳或是一氧化碳氣體造成 催化劑毒化而失效,目前傾向添加氧化物如氧化鋁(Al2O3)4,氧化鋯(ZrO2)3,氧化鈰(CeO2)3, 5,氧化鑭(La2O3)6…等,以降低催化劑毒化現
象,而在以上不同金屬以及氧化物搭配下,催化方向,活性及反應產 物亦不相同7,如何使反應專一化,使氫氣產率提升等研究目前都在進 行中。其中最具突破性的發現,莫過於L.D. Schmidt在 2004 年Science 期刊8中所提及利用銠金屬(Rh)及氧化鈰(CeO2),在有水的情況下,可 以進行自身放熱的乙醇產氫反應,亦即表示在反應過程中僅需提供少 量額外能量來使乙醇裂解產氫,更具突破性的發現乃是,此反應過程 中由於水的加入反應,除了乙醇分子中的氫原子可以被轉化成氫氣, 甚至連水分子中的氫原子亦可以轉換成氫氣而有額外的氫氣生成。 然而,在Schmidt的論文8中提到,其所使用的催化劑製造方式為傳 統的涵浸法,並未針對催化劑的構形或大小進行研究,有鑒於近年來
顯微觀測技術的進步,各種奈米材料競相研發出現,由於奈米材料表 面積增加,表面活性亦隨材料大小尺度鉅幅減少而加強,奈米材料將 會為催化劑研究帶來衝擊性的影響,如果能針對奈米材料對催化活性 的影響研究,也許是一項可行的研究。 不少理論計算指出9-11曾經提出在氧化鈰結構中,{1 0 0}、{1 1 0} 以及{1 1 1}三個晶面中以{1 0 0}表面能最高,{1 1 1}表面能最低,而 的確實驗結果12得到在自然不加干擾的長晶環境下,由於{1 1 1}表面能 最低最穩定,易生成由四組{1 1 1}晶面構成的八面體。 而表面能的高低是否影響催化效率?Hao-Xin Mai在他的著作13中 提到他利用水熱法合成出僅暴露出{1 0 0}晶面的氧化鈰奈米方塊以及 暴露出 {1 0 0}及{1 1 0}晶面的氧化鈰奈米柱,在經過針對氧氣儲存能 力(Oxygen Storage Capacity)的實驗,發現相對於未加控制晶面及晶粒 大小的氧化鈰粉末,氧化鈰奈米方塊氧氣儲存能力高出約三倍,而氧 化鈰奈米柱更高出氧化鈰奈米方塊約一點五倍,而氧氣儲存能力可視 為氧化鈰在做為催化劑時,調節催化劑表面氧氣濃度能力的指標。 在Kebin Zhou的著作14中更提到利用氧化鈰奈米粒子(為不控制其 所暴露晶面之粉末)以及氧化鈰奈米柱(與上述氧化鈰奈米柱相同構形 且暴露相同晶面)進行對一氧化碳氧化反應(CO Oxidation)氧化鈰奈米 柱在比氧化鈰奈米粒子在比正常溫度攝氏 350 度低約攝氏一百度之溫 度(225oC),即可發生反應,由此可見其催化活性遠高於氧化鈰奈米粒 子。 有鑑於此,在本項研究中,企圖將氧化鈰顆粒奈米化,檢測其是 否能增加在乙醇催化產氫反應中的催化活性,進一步,更嘗試利用控 制其不同奈米構形,暴露出不同的氧化鈰晶面,來測試不同晶面對催 化能力的影響。
1-2 乙醇催化產氫原理
15-17 在正常情況下,乙醇直接和氧氣反應燃燒 : (1) C2H5OH + 3 O2 → 3 H2O + 2 CO2 △HR≒ -1280 KJ/mol 但是D. K. Liguras18 提到將乙醇汽化混和氧氣通過加熱到攝氏 500 度 含 鎳 的 氧 化 鑭(La2O3)表面時,乙醇進行將部份氧化反應(partial oxidation reaction): (2) C2H5OH + 1/2 O2 → 2 CO + 3 H2 △HR≒ +20 KJ/mol 產生的一氧化碳在有水的情況下加上適當的催化環境可進行 Water-Gas shift reaction 但此反應並非完全反應,其反應視所使用催化 劑以及催化環境不同而有程度上不同 : (3) CO + H2O →CO2 + H2 △HR≒ -40 KJ/mol L. D. Schmidt8 提到利用氧化鈰加入百分之五的銠金屬為催化劑 可將總反應導向一自身放熱反應,合併上列反應式(2) 、(3)可得到 : (4) C2H5OH + 2 H2O + 1/2 O2 → 2 CO2 + 5H2 △HR≒ -50 KJ/mol 總反應為放熱反應,因此催化此反應僅需在反應起始時預熱催 化劑,再汽化乙醇混和空氣及水蒸氣通過催化劑,其中不僅可得到一 莫耳乙醇分子中所含三莫耳氫氣,更可將水分子中的氫還原得到額外 還原來自水分子中的氫氣,理論上一莫耳乙醇在有水及氧氣反應將可 以得到五莫耳氫氣。1-3 氧化鈰簡介
19 氧化鈰特殊的化學性質,使得其用途十分廣泛。由於其對一氧化 碳及硫化物的氧化催化活性極高,更可催化甲醇裂解以及WGS反應, 因此經常被用來當作催化劑。更因為其具有氧氣儲存,釋放的性質, 通常利用它來控制整體催化劑表面的氧氣濃度而使用在汽車排氣管裡 的三向催化劑(Three-Way Catalysts ),因其可用來同時將引擎內燃燒未 完全產生的一氧化碳(CO),碳氫化合物,以及氮氧化物(NOx)催化使之 氧化所以稱之。此外也可製成化學機械研磨( Chemical Mechanical Polishing)用的漿料(原理為利用含氧化鈰之漿料將欲研磨物表面不平 處發生化學反應,使之鬆散易脫落,再進一步將其磨除)20。 氧化鈰晶體結構如圖 1-3-1 所示,為CaF2類型的Fluorite結構,鈰原 子為面心立方堆積,而氧原子則填滿其中的八個八面體空隙,三軸長 各為5.41Å,空間群為Fm3m。 圖1-3-1 氧化鈰晶體結構圖鈰的電子組態為 [Xe]4f15d16s2,在Ce4+時電子組態與Xe相同,Ce3+
時電子組態為 [Xe]6s1(因 6s軌域半填滿)亦可穩定存在,因此Ce可以輕
易在三價/四價轉換,所以氧化鈰可視作二氧化鈰以及三氧化二鈰固態
傾向為CeO2,表面部分則為Ce2O3。 當氧化鈰在適當條件下,與一氧化碳接觸則行下列反應 : (1) CO + 2 CeO2 → CO2 + Ce2O3 而Ce2O3再和空氣中氧氣反應 : (2) O2 + 2 Ce2O3 → 4 CeO2 由上列二式可知,氧化鈰可儲存及釋放氧,因此氧化鈰對一氧化 碳的氧化具有極大活性,可利用來減輕汽機車排放廢氣中的一氧化碳 含量,而值得注意的是( 2 )式可逆向進行,利用此性質,在固態氧化物 燃料電池中(Solid State Fuel Cell)可以擔任利用氧原子傳遞電流的材料
1-4 已知氧化鈰奈米晶體
1-4-1 二氧化鈰奈米粒子 Z. L. Wang 在 2003 年的文獻12提出,在二氧化鈰結構中,經過理 論計算得知{1 0 0}表面能最高,{1 1 1}表面能最低,因此在自然不加 干擾的長晶環境下,易生成由四組{1 1 1 }晶面構成的八面體,如圖 1-4-1 所示,在晶體開始成長時,{1 1 1 }及{1 0 0}共同構成變形之八面 體球狀粒子,但晶體持續成長至接近微米尺度時,由於{1 1 1 }晶面成 長速度較快,便形成八面體形狀的奈米粒子如圖1-4-2。 圖 1-4-1 二氧化鈰奈米粒子穿透視顯微鏡照片12圖1-4-2 二氧化鈰晶體成長形成八面體12 1-4-2 二氧化鈰奈米柱 Aurelien Vantomme22 在 2005 年提出利用氨水使氯化鈰變成氫氧 化鈰沉澱,並加入界面活性劑(溴化十六烷基三甲基銨)在水熱法環境下 可合成長度 150-400 奈米,寬度 10-25 奈米的二氧化鈰奈米柱,如圖 1-4-3 所示,其晶體成長方向為[1 1 0 ]。 其優點在於奈米柱大小分佈均勻,且可大量合成,但是缺點在於 界面活性劑難以去除,因此Kebin Zhou提出使用氫氧化鈉14使硝酸鈰沉 澱析出為氫氧化鈰,在高濃度氫氧化鈉(2M)的水熱環境下即可合成類 似大小的二氧化鈰奈米柱,如圖1-4-4 所示,且此奈米柱為一由兩組( 1 1 0 )及一組( 1 0 0 )晶面構成之短柱體。
圖1-4-3 二氧化鈰奈米柱22
1-4-3 二氧化鈰奈米方塊
Hao-Xin Mai 在 2005 年的著作13中提出,利用與 1-4-2 類似水熱環
境,加長反應時間,可使產物變成二氧化鈰奈米方塊,其中邊長為 40
奈米,其方塊六個面都是{ 1 0 0}晶面。
第二章奈米結構分析法及轉換效率計算法介紹
2-1 奈米結構分析法
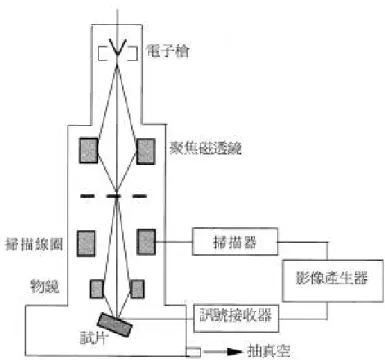
23 在本實驗中奈米結構直接觀測均採用掃描式及穿透視電子顯微 鏡。 2-1-1 掃描式及穿透式電子顯微鏡 掃描式電子顯微鏡利用 de Broglie 波動理論: λe= h / mv = 12.2 / (V)1/2 (Å) 或 (1x10-10m) 在 1 萬伏特的加速電壓下將可得到波長 0.12Å亦即是波長為 1.2 x 10-11公尺的電子束,再將此電子束經過聚焦投射到所需觀測樣品上,經 過反射後,再利用置於樣品上側方的訊號接受器接收二次電子等訊號 再經電腦分析即可得到影像訊號(圖 2-1-1),而穿透式電子顯微鏡所不 同的是在於電子束是直接穿透樣品將影像投射在下方的螢光板上(圖 2-1-2),因此可得到原子排列的干涉影像,並利用計算原子層間距,得 到樣品的晶體成長方向,或是可利用已經聚焦的電子束,直接照射樣 品,進行選區電子繞射(SAED),利用所得繞射圖形,亦可得到相關晶 體資訊。圖2-1-1 掃描式電子顯微鏡構照示意圖23
2-2 轉換效率計算法
8 2-2-1 最大氫氣選擇率( Hydrogen Selectivity ) 由前述乙醇轉換氫氣的反應式: C2H5OH + 2 H2O + 1/2 O2 → 2 CO2 + 5 H2 △HR≒ -50 KJ/mol 假設一莫耳的乙醇經過催化,將有五莫耳的氫氣生成,但是三莫 耳氫氣是來自乙醇分子提供,兩莫耳由水分子提供,最大氫氣選擇率 為:(乙醇提供之三莫耳氫氣+水提供之兩莫耳氫氣) / (乙醇提供三莫耳 氫氣)= 5 / 3 = 166%。 2-2-2 定量測量方式 本實驗反應後氣體分析測量採用氣相層析儀,利用氣相層析儀首 先針對欲分析之氫氣、氮氣,製作檢量線,得到儀器訊號對氣體莫耳 數之公式,藉此分析並定量氣體。定量方式為利用兩組氣體質量流量控制器(Mass Flow Controller)分 別控制氫氣及氮氣之質量流速(Mass Flow Rate)並流經相同管線混和得 到所需特定氫氣及氮氣莫耳比例之氣體再經由氣相層析儀自動進樣分 析,分別對氫氣及氮氣訊號峰面積積分,經過變動兩者氣體組成比例, 即可得到氫氣及氮氣濃度對氣相層析儀之熱導偵測器訊號反應關係 圖,再畫出檢量線得到兩者相關公式,即可利用所得到公式(圖 2-2-1 , 圖2-2-2)來分析欲分析氣體中氫氣及氮氣所佔比例。 在首次分析時,氫氣及氮氣濃度變化範圍為 0%到 100%,假使氫 氣佔 X%則氮氣佔(1-X)%在此範圍內取十點作分析而得到下列兩校正 曲線圖。
y = 416097x + 760968 R2 = 0.9987 0.00 10.00 20.00 30.00 40.00 50.00 0 20 40 60 80 100 120 百 萬 氫氣濃度(%) 熱導偵測器訊號 圖2-2-1 氫氣濃度對熱導偵測器訊號圖 y = 40639x - 117126 R2 = 0.997 -0.50 0.00 0.50 1.00 1.50 2.00 2.50 3.00 3.50 4.00 4.50 0 20 40 60 80 100 120 百 萬 氮氣濃度(%) 熱導 偵測 器 訊 號 圖2-2-2 氮氣濃度對熱導偵測器訊號圖 但是若依上圖所得公式換算之特定濃度下熱導偵測器訊號,則發 現將有 10%左右的誤差值出現,以下為儀器真實訊號及經公式換算訊 號誤差列表 :
表2-2-1 熱導偵測器訊號對真實訊號誤差 氣體莫耳濃度(%) H2誤差(%) N2誤差(%) 0 0 0 10 12.3 -23.5 20 12.2 -17.8 30 10.6 -14.9 40 9.6 -12.1 50 9.7 -8.7 60 8.5 -5.1 70 7.5 -4.1 80 6.1 -2.3 90 5.1 1.7 100 3.8 1.5 而在真實實驗中發現,氫氣及氮氣在產物氣體中所佔比例各約在 40% 上下波動,故重新選定校正範圍,在40%到 60%之間,取五點作校正, 得到下列校正曲線圖 y = 398164x + 2E+06 R2 = 0.9991 15.00 17.00 19.00 21.00 23.00 25.00 40 45 50 55 60 百 萬 氫氣濃度(%) 熱導偵測器訊號 圖2-2-3 氫氣濃度對熱導偵測器訊號圖
y = 40737x - 237649 R2 = 0.9992 1.00 1.20 1.40 1.60 1.80 2.00 2.20 2.40 40 45 50 55 60 百 萬 氮氣濃度(%) 熱導偵測器訊號 圖2-2-4 氮氣濃度對熱導偵測器訊號圖 而由檢量線公式換算之訊號值和真實訊號值比較發現其誤差範圍 下降至百分之一以下,由此可知,當進行氣體濃度對儀器訊號校正時 其檢量線公式之R2應高於0.999 方可利用此公式進行氣體組成分析。 表2-2-2 熱導偵測器訊號對真實訊號誤差 氣體濃度(%) H2誤差(%) N2誤差(%) 40 -1.3 2.3 45 -0.3 2.0 50 -0.2 2.1 55 -0.4 1.2 60 -0.1 2.3 而經由趨勢線所得校正公式如下: (1) 氫氣莫耳比例 = ( 熱導偵測器訊號 - 2*106 ) / ( 398164 ) (2) 氮氣莫耳比例 = (熱導偵測器訊號 + 237649 ) / (40737 ) 2-2-3 轉換效率計算法 假設由上述所得公式計算反應後氣體中含 A % 氫氣,B % 氮氣, 而乙醇進樣為C(莫耳/分鐘),空氣中所含氮氣比例為 D%,則轉換效率 為: ( A / 3*C ) * ( D / B ),其中 ( A / 3*C ) 表示氫氣每分鐘產生莫耳數
除以乙醇毎分鐘進樣莫耳數之三倍(因一莫耳乙醇可取得三莫耳氫 氣),而再乘上 ( D / B ) 是因為氣相層析儀僅針對產物氣體濃度及組成 分析,無法進行產物氣體體積測量(因反應過後氣體膨脹),所以 A 不 能代表氫氣真正產生量,需利用氮氣進行體積校正。
試舉一例說明,假設通入之氣體其所含氣體組成為 79%氮氣,21%
氧氣其流速為0.5 SLPM( Standard Liter Per Minute 標準狀態下每分鐘 流量一公升 將其值除以 22.4 將可得到氣體莫耳數 ),固定碳氧比值 (C/O ratio)為 0.7,而產物氣體中經過分析其氮氣訊號值為通入氣體之 2.03 倍,即可認定為出口流速為入口流速之 2.03 倍,因氮氣濃度被稀 釋2.03 倍,換算出出口流速為每分鐘 1.025 SLPM,藉由檢量線公式可 得到產物氣體中氫氣所佔比例為37.15%,兩者相乘再除以 22.4 換算成 每分鐘輸出之氫氣莫耳數之後再除以每分鐘通入之乙醇莫耳數之三倍 即是氫轉換選擇率為111%。
第三章 實驗方法
3-1 實驗藥品
表3-1-1 實驗藥品
編號 藥品 英文藥名 分子式 純度 廠商
1 硝酸氨鈰 Cerium ammonium nitrate (NH4)2Ce(NO3)6 99.5% Alfa Aesar 2 硝酸鈰 Cerium nitrate(III) Ce(NO3)3.6H2O 99.5% Alfa Aesar 3 草酸鈰 Cerium oxalate(III) Ce2(C2O4)3.XH2O 99.9% Alfa Aesar
4 氧化鈰 Cerium oxide(IV) CeO2 99.5% Alfa Aesar
5 氯化鈰 Cerium chloride(III) CeCl3 99.5% Alfa Aesar
6 氫氧化鈉 Sodium hydroxide NaOH 99% J.T.Baker
7 硝酸銠 Rhodium nitrate(III) Rh(NO3)3.2H2O 99.9% Alfa Aesar
8 氯化銠 Rhodium chloride(III) RhCl3 99.9% Alfa Aesar
9 氯化鋰 Lithium chloride(I) LiCl 99% Alfa Aesar
10 氯化鈉 Sodium chloride NaCl 99% Alfa Aesar
11 氯化鉀 Potassium chloride KCl 99% Alfa Aesar
12 氯化鎂 Magnesium chloride MgCl2 99% Alfa Aesar
13 氯化鈣 Calcium chloride CaCl2 93% Alfa Aesar
14 氯化鍶 Strontium chloride SrCl2 95% Alfa Aesar
15 氯化鋇 Barium chloride BaCl2 99.95% Alfa Aesar
16 乙醇 Ethanol C2H5OH 99.9% J.T.Baker
3-2 儀器設備
1.粉末繞射儀 : Bruker AXS D8 Advance (Leipzig Germany) 2.掃描式電子顯微鏡 : Hitachi S-4700 (Tokyo Japan)
3.穿透式電子顯微鏡 : Philips TECNAI 20 4.高溫爐 : Lindberg /Blue 5.水熱罐 :造奕企業社 6.氣體流量計 : Alicat Scientific: MC-500SCCM-D,MC-5SLPM-D, MC-10SLPM-D 7.氣相層析儀 : Thermo Trace GC 2000
8.質譜儀 : Thermo Trace DSQ Mass Spectrometer 9.注射式幫浦 : Core Parmer 60061
10.熱電偶溫度計 : YSC Thermometer YS-1300K 11.酸鹼度計 : Suntex Micprocessor PH Meter SP2200
12.超音波震碎機 : Misonix Ultrasonic Cell disruptor XL2000 13.離心機 : Hettich Zentrifugen EBA20
3-3 實驗步驟
3-3-1 利用 IA, IIA 鹵化鹽共沉澱合成氧化鈰奈米纖維 本實驗首先利用去離子水將不同比例之 IA 與 IIA 鹵化鹽與草酸 鈰混和溶解,再利用高溫爐加熱去水使鹵化鹽及草酸鈰沉澱,之後加 以攝氏四百度高溫分解草酸鈰為二氧化鈰,再用去離子水洗去鹵化 鹽,便可得到產物。 3-3-2 利用水熱法合成二氧化鈰奈米柱及奈米方塊14 本實驗首先利用去離子水調配不同濃度之氫氧化鈉水溶液,以及 硝酸(Ce(NO3)3.6H2O)鈰水溶液,在固定攪拌子轉速(500RPM)情況下, 將硝酸鈰水溶液倒入氫氧化鈉水溶液中,經過十分鐘攪拌,倒入水熱 罐之鐵弗龍內罐密封,置於高溫爐以不同條件加熱,反應過後,利用 離心機以及去離子水反覆清洗至溶液呈中性,乾燥之後便可得到產物。 3-3-3 粉末分析 將反應後所的之粉末,經過研磨,利用粉末繞射儀進行繞射,繞 射角度(2θ) 5。-60。,所得繞射圖譜以EVA軟體處理,並從 Joint Committee Powder Diffraction Standards (JCPDS)資料庫中比對是否為二氧化鈰,經 過確認後,可利用謝樂公式( Scherrer Formula ),針對( 1 1 1)繞射峰之 半高寬進行初步的產物顆粒大小鑑定。謝樂公式( Scherrer Formula) : D = ( 0.89*λ ) / ( B*Cosθ ) D:粉末顆粒大小 λ:繞射光源波長(本實驗使用銅靶 Kα,為 1.54Å) B:繞射峰半高寬(弧度) θ:繞射角度 3-3-4 掃描式電子顯微鏡分析 將碳膠帶黏於樣品銅座,再將產物粉末塗佈在碳膠上,利用攝氏
50 度烘箱烘乾,且用真空系統抽去剩餘水氣,即可進入機台觀測。 3-4-5 穿透式電子顯微鏡分析 取少量產物粉末置於 10 毫升樣品瓶中,加入 8 毫升乙醇,置於超 音波震盪器中震盪一小時,再將液體滴在銅網上,待乾燥後以真空系 統抽真空至少四小時,方可進入機台觀測。 3-4-6 催化劑製備8 將所得之奈米二氧化鈰粉末0.2 公克,置於 20 毫升樣品瓶中,加 入15 毫升乙醇及 0.0157 公克硝酸銠(熱解後可得 0.01g銠金屬為氧化鈰 5%重量),利用超音波粉碎機連續粉碎一小時,再放入比表面積( 300 m2/g),顆粒大小約 1.4~1.0 公厘之氧化鋁顆粒,利用攝氏 50 度烘箱烘 乾,放入高溫爐以攝氏500 度加熱一小時使硝酸銠分解即可。 3-4-7 轉換效率測試系統 在轉換效率測試系統方面,最大之困難在於如何將乙醇以及水的 混合物完全汽化並通過催化劑,經過數次嘗試,設計出如圖3-4-1 的反 應系統,圖中的汽化瓶為不繡鋼製,其上方有三個連接口由左至右分 別為:催化反應管,進氣噴頭,以及溫度探測器。 左方的反應管為石英所製,以承受反應時高達攝氏七百度以上的 高溫,管內填充有 1.5 公分的催化劑,兩端再加以石英棉固定,中間的 進氣噴頭連接經由氣體流量控制器輸出之空氣以及注射式幫浦輸出之 乙醇與水,三者混合物,將其均勻噴灑於罐內,右邊溫度探測器用來 監測汽化瓶內溫度,控制汽化瓶外的高溫爐加熱溫度,使罐內溫度維 持攝氏兩百度,當罐內液體汽化通過左邊反應管內催化劑,此時氣體 溫度約兩百度,通過催化劑時,反應迅速起始,由於反應生成氣體量 遠大於通入氣體量,所以出氣流速遠大於進氣流速,在石英管後方為 一冷凝器,將反應後產生之水蒸氣冷凝之後再由氣相層析儀進行分析。
3-4-8 氣相層析儀分析設置
氣相層析所用管柱為 CARBOXEN 1010 PLOT Capillary Column
長度 30 公尺,內徑 0.53 毫米,分析時溫度條件為管柱所在烘箱攝氏 45 度,樣品注射口溫度攝氏 200 度,熱導偵測計(TCD Detector)燈絲溫 度攝氏 350 度,熱導偵測計本體溫度攝氏 200 度,攜帶氣體為氬氣, 每次注入樣品氣體為 10 毫升,攜帶氣體流速 72 毫升每分鐘,混合後 取分流 1/24 注入管柱,即注入管柱氣體為 3 毫升,單次分析時間為十 分鐘,每次測試結束需經過一分鐘溫度平衡,方可進行下一次測試
第四章 結果討論
4-1 利用 IA IIA 鹽類共沉澱法合成奈米氧化鈰
4-1-1 實驗簡介 本實驗最初之構想在於將可熱分解之鈰化合物與 IA, IIA 鹽類共 溶於水,再將水蒸乾使其沉澱,期望藉結晶析出時,IA ,IIA 鹽類對 鈰化合物的晶體成長方向產生影響,加上 IA, IIA 鹽類可加水溶解去 除,便可得到產物。 4-1-2 不同鹽類對氧化鈰晶體構形的影響 在不同鹽類對氧化鈰晶體構形的影響共嘗試了如表 4-1-1 所列八 種,其中以氯化鋰,氯化鈉及氯化鎂的實驗出現特殊構形的氧化鈰。 表4-1-1 反應列表 編號 鹽類 結構 a LiCl cubic b NaCl cubic c KCl cubic d MgCl2 hexagonal e CaCl2 tetragonal f SrCl2 cubic g KOH orthorhombic h NaOH orthorhombic 表 4-1-1 所列各項實驗均以鹽類:草酸鈰=8:1(重量比),鹽類重量為 兩公克,以20 毫升去離子水溶解後,在攝氏六十五度下蒸乾再以攝氏 四百度分解草酸鈰,圖 4-1-1,4-1-2 為所得產物利用掃描式電子顯微鏡 觀測所得之結果。a) b) c) d) 圖4-1-1 a 、b、c、d 分別為表 4-1-1 所列編號 a 、b、c、d 之產物掃描式電子顯微 鏡觀測結果 (a) 如圖可見氧化鈰呈長條狀,但表面不平整估計因為鋰原子而造成 (b) 如圖可見氧化鈰呈現寬 60 奈米長度 1 到 2 微米之纖維狀 (c) 如圖可見氧化鈰雖略具長方形外觀,但不規則 (d) 如圖可見產物為六角形薄片,厚度 20 奈米但並非氧化鈰而是氫氧化鎂為主,參雜部 分氧化鈰在內
e) f)
g) h)
圖4-1-2 e、f、g、h 分別為表 4-1-1 所列編號 e、f、g、h 之產物掃描式電子顯微鏡
觀測結果
其中以使用氯化鋰,氯化鈉,以及氯化鎂出現特殊形狀之氧化鈰, 在使用氯化鋰所得的實驗結果中可以看出氧化鈰為規則性的長柱狀, 但表面並不光滑呈現凹凸不平狀,而使用氯化鈉的實驗結果,氧化鈰 呈現長條狀,其寬度大小約為60 奈米,長度在 1 到 2 微米之間,經由 穿透式電子顯微鏡觀測得知其為多晶結構,在使用氯化鎂的實驗結果 中可以看到大小約100 奈米,而厚度約在 20 到 30 奈米的六角形平板, 經過粉末繞射實驗及EDX證實其為氫氧化鎂( Mg(OH)2 )之晶體為主, 但含少量氧化鈰。 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 5 10 20 30 40 50 a.u. 圖4-1-3 使用氯化鈉鹽類共沉澱所得氧化鈰粉末繞射圖 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 5 10 20 30 40 50 a.u. 圖4-1-4 使用氯化鎂共沉澱所得粉末繞射圖,為氫氧化鎂及氧化鈰混和物
圖4-1-5 使用氯化鈉鹽類共沉澱所得氧化鈰穿透式電子顯微鏡圖 由圖中明顯看出為多晶結構
4-2 利用水熱法合成氧化鈰奈米柱
4-2-1 實驗步驟 本實驗主要利用 10 毫升氫氧化鈉溶液滴入 0.5 克硝酸鈰溶於 6 毫 升去離子水之溶液(以下反應列表所指為最後溶液氫氧化鈉濃度),生成 氫氧化鈰奈米粒子,並利用在水熱環境中,氫氧化鈰奈米粒子在鹼性 環境下自組裝成寬20-30 奈米,長 100-200 奈米之氫氧化鈰( Ce(OH)3 ) 奈米柱,經過高溫爐攝氏300 度氧化脫水生成氧化鈰奈米柱晶體。 4-2-2 氫氧化納濃度對合成之影響 本實驗主要參考 Kebin Zhou 在 2005 所發表文獻14,欲合成類似之 氧化鈰奈米柱,發現使用相同氫氧化鈉濃度下,無法得到重複實驗結 果,進而改變氫氧化鈉濃度,發現在原始文獻所提及之 2M氫氧化鈉濃 度下,無法合成,需至少在 5M濃度下,方可出現柱狀氧化鈰奈米柱, 依濃度變化之實驗結果如表4-2-1,以下反應皆相同在水熱環境下加熱 十小時,所得產物。 表4-2-1 氫氧化鈉濃度與所得產物 濃度 結果 1M 奈米粒子 2M 奈米粒子 3M 奈米粒子 4M 奈米粒子 5M 奈米粒子及奈米柱 10M 奈米柱為主 15M 奈米柱a) b)
c) d)
圖4-2-1 a、b、c、d 分別為 1M 、2M 、3M 、4M 氫氧化納濃度下,所得產物經
掃描式電子顯微鏡觀測圖形,均為不規則粒子狀 (a~d) 氧化鈰奈米粒子,顆粒大小約 20~30 奈米
a) b) c) d) 圖4-2-2 a、b、c、d 分別為 5M 、10M 、15M 、15M 氫氧化納濃度下,所得產 物經掃描式電子顯微鏡觀測圖形,5M 時開始出現柱狀物 (a) 出現部份氧化鈰奈米柱 (b)氧化鈰奈米柱成為主要產物 (c) 氧化鈰奈米柱成為主要產物但在高氫氧化鈉濃度下奈米柱寬度變寬 (d) 氧化鈰奈米柱成為主要產物但在高氫氧化鈉濃度下奈米柱寬度變寬
圖4-2-3 氧化鈰奈米柱穿透式顯微鏡圖,成長方向為[1 1 0] 0 100 200 300 400 500 600 700 5 10 20 30 40 50 800 6 a.u. 圖4-2-4 氧化鈰奈米柱粉末繞射圖 由掃描式電子顯微鏡觀測可知,當氫氧化鈉濃度低於 5M下,無法 得到柱狀產物,當氫氧化鈉濃度高於5M時,奈米柱寬度隨濃度略為增 加,氫氧化鈉濃度10M之產物其( 1 1 1)繞射峰半高寬為 1.01o,經過謝 樂公式計算大小為 7.33 奈米,而實際掃描式電子顯微鏡觀測寬度為 20-30 奈米,長度為 100-200 奈米,而穿透式顯微鏡分析可得到其成長 方向層間距為1.9Å,故其奈米柱晶體成長方向為[ 2 2 0 ],與原始論文 結果相符合,稍後將以10M濃度之產物進行催化效率測試。
圖4-2-5 氧化鈰奈米柱製成催化劑於氧化鋁顆粒上之掃描式電子顯微鏡圖 表4-2-2 氧化鈰 d spacing 表 d spacing (Å) ( h k l ) 3.1248 1 1 1 2.7062 2 0 0 1.9135 2 2 0 1.6319 3 1 1 1.5264 2 2 2 1.3531 4 0 0 1.2416 3 3 1 1.2102 4 2 0 1.1048 4 2 2
4-3 利用水熱法合成氧化式奈米方塊
4-3-1 實驗簡介 本實驗為 4-2 實驗之衍生,在合成氧化鈰奈米柱過程,發現有部分 疑似立體方塊產物,經過延長水熱反應時間為四十八小時,提高反應 溫度為攝氏150 度,可以得到邊長為 40 奈米之氧化鈰方塊。 4-3-2 氫氧化鈉濃度對合成之影響 在合成出氧化鈰奈米方塊之後,希望藉由改變反應條件以控制方 塊大小,首先改變的為氫氧化鈉濃度,以下所列之反應皆為改變最終 溶液之氫氧化鈉濃度,再置入水熱罐內以攝氏 150 度反應 48 小時所 得,所得產物利用去離子水清洗至中性。 表4-3-1 氫氧化鈉濃度對反應結果 氫氧化鈉濃度 結果 1M 奈米粒子 2M 奈米粒子 3M 奈米粒子 5M 奈米方塊逐漸成形 10M 奈米方塊 15M 奈米方塊a) b) c) d) e) f) 圖4-3-1 a、b、c、d、e、f 分別表示 1M、2M、3M、5M、10M、15M 電子顯微鏡 圖 (a) 氧化鈰奈米粒子 (b) 氧化鈰奈米粒子 (c) 氧化鈰奈米粒子
(d) 奈米方塊逐漸成形 (e) 氧化鈰方塊成為主要產物大小約為 40 奈米 (f) 氧化鈰奈米方塊但其大小較 10M 氫氧化鈉濃度時略大 由掃描式電子顯微鏡觀測結果可知在低於 5M氫氧化鈉濃度下,奈 米方塊無法形成,而高於 5M情況下,氫氧化鈉濃度對方塊大小亦無顯 著影響,其反應時間較氧化鈰奈米柱長且生成產物直接為氧化鈰並非 氫氧化鈰( Ce(OH)3 ),在經過粉末繞射後,氫氧化鈉 10M之產物其( 1 1 1 )繞射峰半高寬為 0.372o,經過謝樂公式推算,其粒子大小為24.2 奈 米,而掃描式電子顯微鏡觀測其正方體邊長為40-50 奈米,而在催化效 率轉換實驗中,以氫氧化鈉 10M下的產物為催化劑,其邊長為 40 奈米。 4-3-3 穿透式顯微鏡分析 合成氧化鈰奈米方塊之後,藉由穿透視電子顯微鏡以及選區電子 繞射來鑑定所合成之方塊的六個晶面,發現氧化鈰方塊,三軸成長方 向層間距皆為2.7Å,故可知其成長方向為[ 2 0 0 ](參照表 4-2-2),六個 晶面都是( 1 0 0 )面。 圖4-3-2 氧化鈰奈米方塊穿透視顯微鏡圖暨選區繞射圖(右下)
0 100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400 1500 5 10 20 30 40 50 60 a.u. 圖4-3-3 奈米氧化鈰方塊粉末繞射圖 圖4-3-4 氧化鈰奈米方塊製成催化劑於氧化鋁顆粒上之掃描式電子顯微鏡圖
第五章 乙醇轉氫催化效率測試
5-1 催化測試系統
催化系統整體如圖 5-1-1 所示,其中反應器部分如前文所述,而在 進樣分析部分,樣品進樣採用六向閥自動進樣裝置,可固定每次氣體 進樣體積以及樣品注入時間,利用熱導偵測計偵測之訊號經過積分, 將訊號波形面積對氣體濃度作檢量線,以此作為氣體濃度分析,而各 氣體對管柱滯留時間如表5-1-1 所示: 表5-1-1 分析氣體滯留時間 氣體 滯留時間(S) 氫氣 3.32 氧氣 3.82 氮氣 4.715-2 反應氣體與催化劑接觸時間
在Schmidt的論文8中提及,此催化反應的接觸時間約為 10 毫秒, 而可以控制接觸時間的方式為控制流速,提高流速,將可减短反應氣 體與催化劑接觸時間,在本實驗中採用內徑 1/4 英吋石英管為催化反應 管,其中催化劑填充量固定為填入石英管 1 公分長,在實驗中發現當 反應進行時,催化劑會因為高溫而發出紅色光,但並非 1 公分長的催 化劑都在發光,因此無法利用催化劑體積來計算所需氣體流速,便利 用改變流速並觀察氫氣選擇率來偵測最佳反應流速,如表 5-2-1 所示, 為涵浸法製成之催化劑,改變氣體流速由 0.4SLPM到 0.6SLPM時所得 到的氫氣選擇率變化,其中1SLPM表示流速為每分鐘通過標準狀態下 的氣體一公升,可以明顯看出在流速 0.6SLPM下可以得到最高氫氣選 擇率。 0.8 0.85 0.9 0.95 1 1.05 1.1 1.15 1.2 1.25 1.3 0.4 0.45 0.5 0.55 0.6 0.65 0.7 氣體流速(SLPM) 氫氣選擇率( % ) Nanocube Nanorod Schmidt 圖5-2-1 改變流速對氫氣選擇率影響 而利用控制氧化鈰構形之催化劑進行效率測試時,發現不同催化 劑亦需要不同之氣體流速,由上列圖表可知,不同催化劑所需之流速 亦不相同,此後的實驗將會根據本節所測試之最佳流速,來進行氫氣 選擇率測試。5-3 碳氧比值對催化效率影響
16, 24, 255-3-1 實驗原理
在進行催化反應時,通入氣體為空氣(79%氮氣+21%氧氣),水蒸
氣,以及乙醇蒸氣,其中氧氣為進行 1-2 所提到反應式(2)之部分氧化
反應(Partial Oxidation Reacton)所需氣體,而水蒸氣為 1-2 中所提到反 應式(3)的 WGS 反應(Water-Gas Shift Reaction)所需氣體之一,乙醇則為 反應所需原料,在反應過程中氧氣含量多寡對於反應結果影響十分之 大,主要是因為當氧氣含量過高,將不易進行 1-2 中之反應式(2)反而 較易進行反應式(1)的全氧化反應,使氫氣產量下降,加上若在反應後 仍有氧氣存在亦可能會與氫氣再反應,消耗產生之氫氣使催化效率下 降,因此為進行此相關實驗,定義碳氧比值(C/O ratio)為乙醇及氧氣中 所含碳原子總數除以所含氧原子總數,假設為純乙醇蒸氣則碳氧比值 為 2,若乙醇與氧氣莫耳數比為 2:1,則碳氧比值為 1,利用改變碳氧 比值測試在不同情況下的氫氣轉換效率,希望能得到最佳反應條件。 5-3-2 實驗結果 本實驗中,所有不同的碳氧比值,均測試在一小時內的氫氣轉換 效率平均值,其中碳氧比值變化由 0.5 到 1.0,共測試三項催化劑,分 別為涵浸法製成之催化劑,以及奈米氧化鈰方塊,奈米氧化鈰柱,其 中氧化鈰含量均為 0.1 克而銠金屬含量為氧化鈰重量 5%。由測試結果 可知(圖 5-3-1)在氧化鈰奈米柱及奈米方塊在碳氧比值為 0.7 時,可得到 最佳轉換效率,而涵浸法所得催化劑,在碳氧比值0.6 時,可得到最高 效率,而在三種不同催化劑中,可以看出利用奈米氧化鈰奈米柱製成 之催化劑效率最高,在碳氧比值0.7 時,可以達到 125%。
0 0.2 0.4 0.6 0.8 1 1.2 1.4 0 0.2 0.4 0.6 0.8 1 1.2 C/O值 氫氣 選擇 率 Cube Schmidt's paper Rod 圖5-3-1 不同碳氧比值對轉換效率之影響 1.1 1.12 1.14 1.16 1.18 1.2 1.22 1.24 1.26
cube schmidt rod
氫氣 選擇 率 圖5-3-2 不同催化劑在 C/O 值 0.7 下之氫氣選擇率 0 0.2 0.4 0.6 0.8 1 1.2 1.4 0 5 10 15 20 25 30 時間(小時) 氫氣選擇率( 100% ) Schmidt Rod Cube 圖5-3-3 催化劑穩定性測試結果
圖5-3-4 奈米方塊催化劑經過 24 小時反應之掃描式電子顯微鏡圖
5-4 結果與討論
在本實驗中,所測試的三種催化劑,其中氧化鈰奈米柱為由一組{ 1 0 0 }晶面以及兩組{ 1 1 0 }晶面構成之柱狀物,氧化鈰奈米方塊為由三 組{ 1 0 0 }晶面構成之正方形,氫轉換效率測試可得之奈米柱氧化鈰轉 換效率最高,氧化鈰奈米柱次之,未控制構形催化劑(涵浸法)最低,的 確控制暴露不同晶面的氧化鈰的確對轉換效率有顯著的影響,氫氣選 擇率變化可達到 10%,而相對於傳統涵浸法催化劑製法,改變氧化鈰 奈米構形的確可以提升催化效能,使氫氣選擇率提高 10%,但根據理 論計算9-11,{ 1 0 0 }面活性最高,其催化效能應該最高,實際情況並未 如此,推測與其表面積並不相同所致,此論點在Hao-Xin Mai的著作13中 亦有提到,Mai 測試氧化鈰奈米柱以及氧化鈰奈米方塊的氧氣儲存能 力(Oxygen Storage Capacity)發現氧化鈰奈米柱高於氧化鈰奈米方塊,但經過考慮表面積之後,將氧氣儲存能力除以個別的表面積(BET Surface)發現氧化鈰奈米方塊略高於氧化鈰奈米柱,與理論計算相符 合,在本實驗中氧化鈰奈米柱氫氣選擇率高於氧化鈰奈米方塊應是相 同原因。 而在長時間測試催化劑穩定度實驗中,可以發現涵浸法製成之催化 劑,其氫氣選擇率經過二十四小時催化實驗後,並未有明顯降低氫氣 選擇率情況發生,而包含{ 1 0 0 }及{ 1 1 0 },晶面之氧化鈰奈米柱則 降低至與涵浸法催化劑相同,只包含{ 1 0 0 }晶面之奈米方塊氧化鈰則 明顯降至 100%,其效率降低程度亦與其各晶面表面能高低成相對關 係,由於奈米氧化鈰方塊中{ 1 0 0 }晶面表面能最高,穩定度相對較 低,在約攝氏 800 度的高溫下逐漸轉換成其他相對較穩定的面導致氫 氣選擇率降低,而奈米氧化柱中含{ 1 1 0 },其相對來說較{ 1 0 0 }穩 定,故其氫選擇率降低程度亦較小,由掃描式電子顯微鏡觀測亦觀察
到明顯的外形改變。
由此可知,雖然改變其奈米構形可已經增進氫選擇率但是無法長時 間穩定,但是為何奈米方塊氧化鈰之氫選擇率會降至較涵浸法之催化劑 (未控制氧化鈰暴露晶面)低,則需進一步進行反應前後催化劑組成及表 面晶面方能得知。
5-5 結論
在本實驗中,合成出氧化鈰奈米柱以及奈米方塊,並將其加入 5%銠 (Rh)金屬製成催化劑,與傳統涵浸法製成之氧化鈰含 5%銠(Rh)金屬之 催化劑一起進行乙醇催化產氫效率比較。其中以氧化鈰奈米住所得到 氫選擇率最高為126%,而以傳統涵浸法得到之催化劑其氫氣選擇率為 116%,和文獻上之結果相符合,氧化鈰奈米方塊製成之催化劑氫氣選 擇率則居中,為122%。 然而由長時間催化測試結果可知,具奈米構形之催化劑,在經過 反應進行中的高溫環境下,其構形無法維持,導致效率下降,如要進 一步研究如何穩定其奈米構形,可嘗試加入氧化鋯(ZrO2),使之形成氧 化鈰氧化鋯固態溶液之奈米結構,此部分研究仍待進行中。參考文獻
1. V.S. Bergamaschi; F.M.S. Carvalho; C. Rodrigues; D.B. Fernandes,
Chem. Eng. J. 2005, 112, 153-158.
2. Jordi Llorca; Jean-Alain Dalmon; Pilar Ram´ırez de la Piscina; Narc´ıs Homs, Appl. Catal. A-Gen. 2003, 243, 261-269.
3. D. Srinivas; C.V.V. Satyanarayana; H.S. Potdar; P. Ratnasamy, Appl.
Catal. A-Gen. 2003, 246, 323-334.
4. V. Fierro; O. Akdimb; H. Provendier; C. Mirodatos, J. Power Sources 2005, 145, 659-666.
5. J.R. Salge; G.A. Deluga; L.D. Schmidt, J. Catal. 2005, 235, 69-78. 6. Athanasios N. Fatsikostas; Dimitris. I. Kondarides; Xenophon E.
Verykios, Catal. Today 2002, 75, 145-155.
7. Agus Haryanto; Sandun Fernando; Naveen Murali; Sushil Adhikari,
Energy & Fuels 2005, 19, 2098-2106.
8. G. A. Deluga; J. R. Salge; L. D. Schmidt; X. E. Verykios, Science 2004, 303, 993.
9. Zongxian Yang; Tom K. Woo, J. Chem. Phys. 2004, 120, 16.
10. Karl Sohlberg; Sokrates T. Pantelides; Stephen J. Pennycook, J. Am.
Chem Soc. 2001, 123, 21.
11. Dean C. Sayle; S. Andrada Maicaneanu; Graeme W. Watson, J. Am.
Chem Soc. 2002, 124, 11429-11439.
12. Zhong Lin Wang; Xiangdong Feng, J. Phys. Chem. B 2003, 107, 13563-13566.
13. Hao-Xin Mai; Ling-Dong Sun; Ya-Wen Zhang; Rui Si; Wei Feng; Hong-Peng Zhang; Hai-Chao Liu; Chun-Hua Yan, J. Phys. Chem. B 2005, 109, 24381.
14. Kebin Zhou; Xun Wang; Xiaoming Sun; Qing Peng; Yadong Li, J.
Catal. 2005, 229, 206-212.
2001, 851.
16. A. N. Fatsikostas; D. I. Kondarides; X. E. Verykios, Catal. Today 2003, 75, 145.
17. S. Cavallaroet, Energy Fuels 2000, 14, 1195.
18. D. K. Liguras; K. Goundani; X. E. Verykios, J. Power Sources in press. 19. D. K. Liguras; D. I. Kondarides; X. E. Verykios, Appl. Catal.
B-Environ. 2003, 43, 345-354.
20. Tetsuya Hoshino; Yasushi Kurata; Yuuki Tarasaki; Kenzo Susa, J.
Non-Cryst. Solids 2001, 283, 129-136.
21. H. Yokokawa; T. Horita, N. S.; K. Yamaji; M.E. Brito; Y.-P. Xiong; H. Kishimoto, Solid State Ion. 2004, 174, 205-221.
22. Aure´lien Vantomme; Zhong-Yong Yuan; Gaohui Du; Bao-Lian Su,
Langmuir 2005, 21, 1132-1135.
23. 謝詠芬; 何快容, 材料分析技術在積體電路製程中的應用.
24. E.C. Wanat; K. Venkataraman; L.D. Schmidt, Appl. Catal., A 2004, 276, 155-162.
附錄(1) 固態反應列表(碩一所進行之固態實驗列表)
編號 實驗比例 反應溫度(oC) 結果
1 FeAgInSe3 800 AgInSe2,FeSe Fe7Se8
2 FeAgInSe3.5 800 AgInSe2,FeSe2,Fe3Se2
3 FeAgInSe4 800 AgIn5Se8,AgInSe2 Fe7Se8
4 CoAgInSe3 800 AgInSe2,Co
5 CoAgInSe3.5 800 AgInSe2,CoSe2
6 CoAgInSe4 800 AgInSe2,In2Se3
7 ZnAgInSe3 800 AgInSe2,ZnInSe2,Se
8 ZnAgInSe4 800 AgIn5Se8,AgInSe2,ZnSe
9 ZnAgIn5Se9 800 AgIn5Se8,ZnSe
10 RuAgInSe3 800 AgIn5Se8,RuSe2
11 RuAgInSe3.5 800 AgIn5Se8,RuSe2
12 MoAgInSe4 600 AgInSe2,MoSe2
13 MoAgInSe5 600 AgInSe2,MoSe2
14 MoAgInSe6 600 AgInSe2,MoSe2
15 WAgInSe4 600 AgInSe2,WSe2
16 WAgInSe5 600 AgInSe2,WSe2
17 WAgInSe6 600 AgInSe2,WSe2
18 TaAgInSe3.5 600 AgInSe2,TaSe2
19 TaAgInSe4 600 AgInSe2,TaSe2
20 TaAgInSe4.5 600 AgInSe2,TaSe2,TaSe3
21 SrAg2In2Se4 670 AgInSe2,In2Se3,SrSe
22 SrAg2In10Se16 670 AgIn5Se8,InSe
23 SrAg2In2Se5 670 AgInSe2,SrSe, AgIn5Se8
24 SrAg2In10Se17 670 AgIn5Se8,SrSe
25 SrAgIn3Se6 670 SrSe, AgInSe2
26 SrAgIn7Se12 670 AgInSe2.SrSe
27 SrAuIn2Se4 670 In2Se3,SrSe,Au
28 SrAuIn4Se7 670 In2Se3,SrSe,Au
29 SrAu2In2Se4 670 In2Se3,SrSe,Au
編號 實驗比例 反應溫度(oC) 結果
31 MgAuIn4Se7 800 MgSe,In2Se3
32 Er4In5Se13 800 ErSe2,Er2Se3,In2Se3
33 Er3InSe6 800 ErSe2,Er2Se3,In2Se3
34 Ce4In5S13 800 CeS2,In2S3
35 Ce3InS6 800 CeS2,InS
36 CeIn3S6 800 CeS2,In2S3
37 Bi3AgIn8Se17 600 AgInSe2,In2Se3,Bi2Se3
38 Bi2Ag4In8Se17 600 AgInSe2,In2Se3,Bi2Se3
39 Bi2Ag2In6Se13 600 AgInSe2,In2Se3,Bi2Se3
40 Sb3AgIn8Se17 600 AgInSe2,In2Se3
41 Sb2Ag4In8Se17 600 AgInSe2,In2Se3
42 Sb2Ag2In6Se13 600 AgInSe2,In2Se3,AgIn5Se8
43 Er4In5S13 600 ErS2,Er2S3,In2S3
44 Er3InS6 600 ErS2,Er2S3,In2S3
45 ErIn3S6 600 ErS2,In2S3
46 SbInSe3 600 In2Se3,Sb2Se3 47 Sb3In5Se12 600 In2Se3,Sb2Se3,InSb 48 Sb2In4Se9 600 In2Se3,Sb2Se3,InSe 49 AgSnInSe4 600 In2Se3,SnSe,AgInSe2 50 AgSn2In4Se8 600 In2Se3,SnSe,AgInSe2 51 AgSn2In2Se4 600 InSe,SnSe
52 ErPbIn2Se5.5 600 PbSe,In2Se3,ErSe2
53 ErPb2In4Se9.5 600 PbSe,In2Se3,ErSe2
54 Er2PbIn2Se7 600 PbSe,In2Se3,ErSe2
55 Tb4In5Se13 1050 Tb2Se3,InSe
56 Tb3InSe6 1050 Tb2Se3,InSe
57 Tb5In3Se12 1050 Tb2Se3,InSe
58 MnPb4Ga8Se17 850 PbGa2Se4,PbSe,MnGa2Se4
59 Mn2Pb3Ga8Se17 850 PbGa2Se4,PbSe,MnGa2Se4
編號 實驗比例 反應溫度(oC) 結果
61 Mn4Pb1Ga8Se17 850 GaSe,PbGa2Se4PbSe,MnGa2Se4
62 Mn1Pb1Ga10Se17 850 GaSe,PbGa2Se4PbSe,MnGa2Se4
63 Mn5Pb1Ga8Se17 850 GaSe,PbGa2Se4PbSe,MnGa2Se4
64 AgPb4Ga8Se17 850 PbGa2Se4,Gs2Se3,PbSe
65 Ag2Pb3Ga10Se17 850 PbGa2Se4,Gs2Se3,PbSe
66 Ag3Pb2Ga8Se17 850 PbGa2Se4,Gs2Se3,PbSe
![圖 1-4-2 二氧化鈰晶體成長形成八面體 12 1-4-2 二氧化鈰奈米柱 Aurelien Vantomme 22 在 2005 年提出利用氨水使氯化鈰變成氫氧 化鈰沉澱,並加入界面活性劑(溴化十六烷基三甲基銨)在水熱法環境下 可合成長度 150-400 奈米,寬度 10-25 奈米的二氧化鈰奈米柱,如圖 1-4-3 所示,其晶體成長方向為[1 1 0 ]。 其優點在於奈米柱大小分佈均勻,且可大量合成,但是缺點在於 界面活性劑難以去除,因此Kebin Zhou提出使用氫氧化鈉 14 使硝酸鈰沉](https://thumb-ap.123doks.com/thumbv2/9libinfo/8409917.179785/19.892.251.703.99.712/八面體二氧化基銨在水熱法環境可合成長寬度向為量合成但是缺點在.webp)