LTA and LPS mediated activation of protein kinases in the regulation
of inflammatory cytokines expression in macrophages
Shih-Chi Su
a,1, Kuo-Feng Hua
a,1, Hsinyu Lee
b, Louis Kuoping Chao
b,1, Sai-Koong Tan
a,
Hsinyu Lee
c, Shun-Fa Yang
d,⁎
, Hsien-Yeh Hsu
a,⁎
aBiotechnology and Laboratory Science in Medicine, Institute of Biotechnology in Medicine, National Yang-Ming University,
155 Li-Nong Street, Shih-Pai, Taipei 112, Taiwan
bDepartment of Biological Science and Technology, Chung Hwa College of Medical Technology, Tainan 717, Taiwan cDepartment of Life Science, Institute of Zoology, National Taiwan University, Taipei 110, Taiwan
d
Institute of Medicine, Chung-Shan Medical University, Taichung 402, Taiwan Received 10 May 2006; received in revised form 27 May 2006; accepted 31 May 2006
Available online 10 June 2006
Abstract
Background: Lipoteichoic acid (LTA) and lipopolysaccharide (LPS), the toxicants from bacteria, are potent inducers of inflammatory cytokines, such as tumor necrosis factor-α (TNF) and interleukin-1β (IL-1). Although LTA is much less reported than that on LPS, LTA is regarded as the gram-positive equivalent to LPS in some aspects. We investigated the LTA-induced signal transduction and biological effects, as well as to compare the effect of LTA with that of LPS.
Methods: Kinase assay, ELISA and RT-PCR were performed to delineate LTA and LPS signaling as well as to determine the secretion and RNA expression of TNF and IL-1.
Results: Src, Lyn and MAPKs are involved in LTA and LPS signaling in murine macrophages. Additionally, blockades of PKC, PI3K and p38, respectively, caused significant inhibition of both LTA- and LPS-induced proIL-1/IL-1 and TNF expression. ERK inactivation moderately reduced LTA- and LPS-induced proIL-1/IL-1, but considerably reduced TNF expression. Inhibition of JNK engendered super-induction of IL-1 secretion, but diminished TNF secretion. Strikingly, both IL-1 and TNF protein induction were declined by overexpression of dominant negative form of JNK. Conclusions: The results clarify the similarity and difference between LTA- and LPS-mediated signal transduction and induction of inflammatory cytokines in macrophages.
© 2006 Elsevier B.V. All rights reserved.
Keywords: LTA; LPS; Cytokines; Macrophages; Protein kinases; Signal transduction
1. Introduction
LTA and LPS are the major elements of cell walls of gram-positive and gram-negative bacteria, respectively. Both LTA and LPS are antigenic and able to activate monocytes/macrophages to secrete various inflammatory cytokines. Our previous report demonstrated that LPS is one of the most potent stimuli for triggering cytokine production in these cells[1]. Although LTA
is much less reported than that on LPS, LTA is already regarded as the Gram-positive equivalent to LPS in some aspects[2,3].
Macrophages engender a wide range of inflammatory mediators and cytokines in response to infectious pathogens. A family of receptors named Toll-like receptors (TLRs), consisting of 12 members of TLRs is characterized as a premier signaling transducer for the recognition of diverse bacterial products[4,5]. Recent studies reveal that TLR2 and TLR4 have been implicated in cellular response to LTA and LPS, respectively[6,7]. Various signal transduction pathways are activated by bacterial infection in macrophages[8–10]. Signaling may converge or diverge, and often have the “cross-talk” properties, which make them com-plicated and affect each other. Generally speaking, signaling initiates with ligand-induced activation of the intrinsic kinase
⁎ Corresponding author. Tel.: +886 11 886 2 2826 7252; fax: +886 11 886 2 2826 4092.
E-mail addresses:[email protected](S.-F. Yang),[email protected]
(H.-Y. Hsu).
1
These authors have contributed equally to this work.
0009-8981/$ - see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.cca.2006.05.045
activity of receptors. Myriads of regulatory proteins, or kinases, are in turn activated by receptor-mediated phosphorylation or dephosphorylation. These proteins include protein tyrosine kinases (PTKs)/Ras/IκB kinase/Raf-1/MEKK/MEK/MAPK and NF-κB [11–15]. Subsequently, multiple genes encoding for inflammatory mediators and cytokines are triggered.
TNF, produced primarily by monocytes and macrophages, is a key mediator in a variety of diseases. The precursor of TNF is a 26 kD membrane-bound protein, which is cleaved by TNF-converting enzyme to form a 17 kD secreted cytokine [16]. Upon binding to its specific receptors TNFR1 and TNFR2, TNF will exert intracellular signaling pathways[17,18] and elicit a large number of biological effects including cytotoxicity, im-munoregulation and apoptosis, etc. IL-1 is another potent inflammatory cytokine with several biological activities regu-lating host defense and cellular responses[19]. At the beginning of IL-1 (17 kD) secretion, a non-active form of precursor termed proIL-1 (∼34 kD) is produced and sequestered in cytosol. It is enzymatically processed into an active form by IL-1 converting enzyme (ICE)[20,21]. After cleavage by ICE, the active IL-1 is released and exhibits its diverse biological functions.
Sepsis is the systemic inflammatory response to the infection of gram-negative and gram-positive bacteria. It is associated with a generalized activation and systemic expression of the host's inflammatory pathways[22]. The activation occurs via stimulation of the host immune effector cells that subsequently synthesize and release potent mediators for cell inflammation. Recently, we have started to assess the signal transduction events by which bacterial products induce the production of TNF and 1 in macrophages, since inductions of TNF and IL-1 not only achieve the antibacterial action but also concurrently trigger severe inflammatory responses. If this phenomenon is not controlled, the systemic inflammation may progress to septic shock and result in multiple-organ failure. This promoted us to investigate the mechanisms responsible for macrophage cytokine induction by LTA and LPS. Here, we present the evidence that LTA and LPS stimulate TNF and IL-1 expression via PKC, PI3K, ERK, JNK, and p38 pathways. Moreover, LTA and LPS stimulate multiple tyrosine phosphorylations, includ-ing protein tyrosine kinases, Src and Lyn. Our results may provide a basis for understanding how the induction of inflam-matory cytokines is regulated in monocyte-derived macro-phages following infections by gram-positive or gram-negative bacteria.
2. Materials and methods
2.1. Cell cultures, stimulation and transfection
Murine macrophage J774A.1 cells were from ATCC (Rock-ville, MD), propagated in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT) and 2 mmol/l L-glutamine (Invitrogen), and cultured in
37 °C, 5% CO2 incubator, unless otherwise indicated. For
individual experiments, 0.5 × 107cells were seeded in a 10-cm plate containing 6 ml of media. After incubated for 24 h, the media were replaced by fetal bovine serum-free medium 1 h
prior to LTA or LPS treatment. Both bacterial products were prepared using the phenolic extraction and nuclease digestion procedure by the manufacturer (< 1% protein contaminants), despite that putative contaminants cannot be excluded com-pletely[23]. The transient transfection (the indicated amount of plasmid DNA per 100-mm plate) was conducted by using the LipofectAMINE PLUS® reagent (Life Technologies,Inc.), according to the protocol from the manufacturer. The efficiency of both transfection and expression was monitored by the hemagglutinin tag expression.
2.2. Materials
LPS from E. coli 0111:B4 and LTA from Streptococcus pyogenes were purchased from Sigma Co. (St. Louis, MO). REZOl®C and T was from PROtech Technology Co. (Taipei,
Taiwan). GeneAmp® RNA PCR kit for RT-PCR amplification was from Perkin Elmer Inc. (Branchburg, NJ). Anti-phosphotyrosine, clone 4G10 (mouse monoclonal IgG2bκ) was from Upstate Biotechnology, Inc. (Lake Placid, NY). Monoclonal anti-MAP kinase, activated (diphosphorylated ERK) antibody, monoclonal anti-JNK kinase, activated (diphosphorylated JNK) antibody, monoclonal anti-p38 MAP kinase, activated (diphosphorylated p38) antibody, monoclonal anti-actin antibody, and mouse monoclonal IgG was from Sigma Chem Co. (St. Louis, MO). Anti-IL-1β, anti-Src, anti-IRAK-1, anti-MyD88, rabbit polyclonal IgG, anti-rabbit IgG-HRP, anti-mouse IgG-HRP were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Lyn, mouse monoclonal IgG was purchased from Transduction Laboratories (Lexington, KY). The p44/42 MAP Kinase Assay Kit, SAPK/JNK Assay Kit and p38 MAP Kinase Assay Kit were purchased from Cell Signaling Technology (Beverly, MA). Ro-31-8220, LY294002, PD98059, SP60015, and SB203580 were from Calbiochem-Novabiochem Corp. (La Jolla, CA). Primers for TNF, proIL-1/IL-1, glyceraldehyde phosphate dehydrogenase (GAPDH) were synthesized from local MD Bio. Inc. (Taipei, Taiwan). 2.3. RNA isolation, RT and PCR amplification for detecting the expression of TNF or proIL-1/IL-1, Western blotting analysis, and Enzyme-Linked Immunosorbent Assay (ELISA) for measurement of cytokines TNF and IL-1
All detailed methods and procedures were followed the previously published methods [1]. In brief, total RNA was isolated from J774A.1 cells by using REZOl®C and T as per
manufacturer's instruction. The isolated RNA was normalized and monitored by the intactness of a constitutively expressed gene, GADPH. Random hexamer was used for reverse tran-scription, and PCR was carried out by using indicated primers. For Western blotting, the protein concentrations were deter-mined using Bio-Rad protein assay; cell lysates (25 μg of protein or as indicated) were subjected to SDS-PAGE and electrotransferred to a polyvinylidene fluoride filter (Millipore Inc, Bedford, MA), and then probed with the indicated antibodies. The experimental procedure of ELISA for measur-ing the cytokine secretion was followed the instructions from manufacturer (Endogen Co., MA).
2.4. Immunoprecipitation and phosphotyrosine examination of Src and Lyn
The cell lysates (100 μg/100 μl) were pre-incubated (immunoprecipitated) with either Src or Lyn anti-body at 4 °C for 1 h and then, the mixture of lysates and antibody were incubated with protein A/G PLUS-agarose (Santa Cruz Biotechnology, CA) at 4 °C for overnight. The precipitates were subjected to SDS-PAGE, and then probed with anti-phosphotyrosine mAb (Upstate Biotechnology, Inc.). 2.5. Assay phosphorylation and activity of extracellular signal-regulated kinase (ERK), of c-Jun NH2-terminal kinase (JNK), and of p38 mitogen activated protein kinase (p38) in LTA- and LPS-treated J774A.1 cells with or without protein kinase (PK) inhibitors
Methods for assay phosphorylation and activity of these PKs including cell lysate preparation, immunoprecipitation of PK, in vitro PK reaction, analysis of PK activity and quantification of PK activity were as described previously [1]. Briefly, cells were stimulated with indicated treatment, and the indicated amount of total protein lysate (for Erk: 200 μg; for JNK: 250 μg; for p38: 200 μg) was immunoprecipitated with the indicated antibodies(for Erk: 20 μl of resuspended immobilized phospho-p44/42 MAPK; for JNK: 2 μg/20 μl of c-Jun fusion protein beads; for p38: 20μl of resuspended immobilized phospho-p38 MAPK). The precipitates were resuspended with 50 μl of kinase buffer in
the presence of the corresponding substrates (for Erk: 200 μmol/l of ATP and 2 μg of Erk-1 fusion protein; for JNK: 100 μmol/l of ATP; for p38: 200 μmol/l of ATP and 2μg of ATF-2 fusion protein at 30 °C for 30 min. The kinase activity was determined by Western blotting. Specifically, the Erk activity was measured by the phosphorylation of Elk-1 using anti-phospho-Elk-1 (Ser383) antibody; the JNK activity was measured by the phosphorylation of c-Jun using anti-phospho-c-Jun (Ser63) antibody; the p38 activity was measured by the phosphorylation of ATF-2 using anti-phospho-ATF-2 (Thr71) antibody.
2.6. Statistical analysis
Statistical differences between the experimental groups were examined by analysis of variance, and statistical significance was determined at p < 0.05. The experiments were conducted 3 times or as indicated, all data are expressed as mean ± S.E.
3. Results
3.1. LTA and LPS stimulate protein tyrosine kinases and activate Src and Lyn in murine macrophage J774A.1 cells (J774A.1 cells)
To determine whether LTA and LPS trigger specific signal transduction pathways in J774A.1 cells, we first determined the tyrosine phosphorylation in stimulated cells. Both LTA
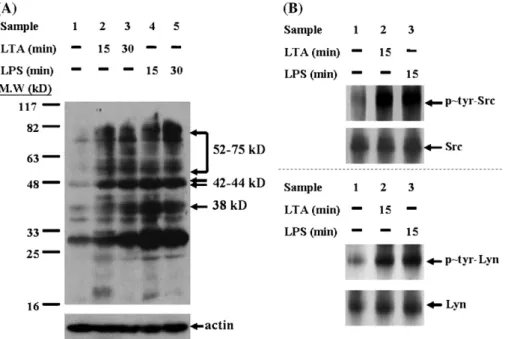
Fig. 1. LTA and LPS stimulate protein tyrosine kinase and activate Src and Lyn. (A) Comparison of protein tyrosine phosphorylation stimulated by LTA and LPS in J774A.1 cells. Cells were stimulated with LTA (1μg/ml), LPS (0.1 μg/ml) or medium. Briefly, sample 1, un-stimulated control; sample 2, LTA 15 min; sample 3, LTA 60 min; sample 4, LPS 15 min; sample 5, LPS 60 min. At the indicated time, cells were lysed and analyzed by Western blotting with anti-phosphotyrosine monoclonal antibody (anti-P∼Tyr Ab 4G10) as previously described[1]. The experiment is representative of 3 similar experiments. (B) LTA and LPS activate Src and Lyn in J774A.1 cells. Cells were treated with LTA or LPS for 15 min, and cell lysates were collected and immunoprecipitated via anti-Src antibody (upper panel) and Lyn antibody (lower panel), respectively. Then, the precipitates were subjected to SDS-PAGE and blotted with anti-phosphotyrisine mAb. For the activation of Src, J774A.1 cells were treated with LTA (1μg/ml) or LPS (100 ng/ml). For the activation of Lyn, J774A.1 cells were activated by LTA (10μg/ml) and LPS (1 μg/ml).
and LPS were found to induce the appearance of many phosphotyrosyl proteins as compared to un-treated cells (Fig. 1A). Whereas LPS appeared to be superior to LTA with respect to induction of tyrosine phosphorylation, LTA induced profound tyrosine phosphorylation 15 min after stimulation. Accordingly, we further explored the identities of these phosphotyrosyl proteins by means of examining the tyrosine phosphorylation of Src and Lyn, which are strong candidates for the primary signal transducers of LPS responses. The activation of Src (M. W.∼60 kD, pp60Src) was detected in cells treated with LTA (1μg/ml) or LPS (100 ng/ml) (Fig. 1B, upper panel); nevertheless, there was no detectable activation of Lyn in response to the same dosage of stimulus as that in Src activation. Upon increasing the concentration of LTA and LPS to 10 μg/ml and 1 μg/ml, respectively, the phosphory-lation of Lyn (M. W.∼56 kD, pp56Lyn) has been detected (Fig. 1B, lower panel).
3.2. LTA and LPS induce activation of MAPKs: ERK, JNK and p38 MAPKs were known to play a critical role in the LTA- and LPS-mediated signaling in macrophages. Accordingly, we determined the time course for phosphorylation of MAPKs in response to LTA and LPS. As shown in Fig. 2A, LTA and LPS differentially induced the phosphorylation of ERK, JNK and p38, albeit a relatively reduced level of MAPKs
phosphorylation was observed in LTA-treated cells. In addition, LTA- or LPS-induced MAPKs activities were examined by in vitro kinase assay, showing that the initial kinetic analysis at an interval of 60 min after stimulation produced the similar activated pattern as those obtained in MAPKs phosphorylation (Fig. 2B). Specifically, the activities of ERK, JNK, or p38 were analyzed by detection of the phosphorylation of Elk, c-Jun, or ATF-2, respectively. Furthermore, all TLRs trigger a common intracellular signaling pathway that involves the adaptor protein, myeloid
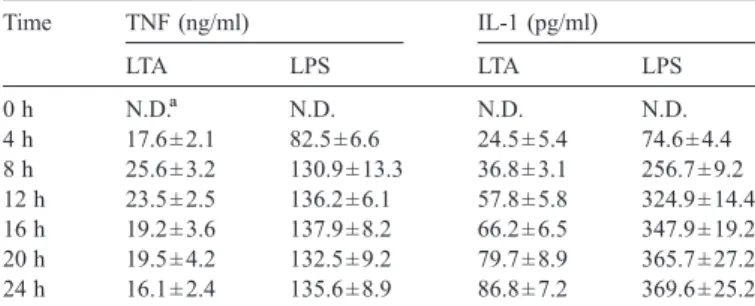
Table 1
LTA and LPS induced cytokines production
Time TNF (ng/ml) IL-1 (pg/ml) LTA LPS LTA LPS 0 h N.D.a N.D. N.D. N.D. 4 h 17.6 ± 2.1 82.5 ± 6.6 24.5 ± 5.4 74.6 ± 4.4 8 h 25.6 ± 3.2 130.9 ± 13.3 36.8 ± 3.1 256.7 ± 9.2 12 h 23.5 ± 2.5 136.2 ± 6.1 57.8 ± 5.8 324.9 ± 14.4 16 h 19.2 ± 3.6 137.9 ± 8.2 66.2 ± 6.5 347.9 ± 19.2 20 h 19.5 ± 4.2 132.5 ± 9.2 79.7 ± 8.9 365.7 ± 27.2 24 h 16.1 ± 2.4 135.6 ± 8.9 86.8 ± 7.2 369.6 ± 25.2 Cells were stimulated with LTA (1 μg/ml) and LPS (100 ng/ml), and the condition medium was collected after stimulation at the time indicated. The secreted TNF and IL-1 in condition medium were analyzed by ELISA.
a
N.D. means non-detectable.
Fig. 2. LTA and LPS induce phosphorylation and activation of MAPKs. (A) Time course analysis of phosphorylation of ERK, JNK, and p38 in LTA- or LPS-stimulated J774A.1 cells. Cells were LPS-stimulated with LTA (1 μg/ml) and LPS (100 ng/ml), and the cell lysates were collected at the time indicated. Phosphorylation of ERK, JNK, and p38 were detected by specific antibody. (B) MAPKs activities were analyzed by in vitro kinase assays. The activated ERK, JNK, and p38 were immunoprecipitated from cell lysates using specific antibodies. Recombinant Elk-1 fusion protein, c-Jun fusion protein, and ATF-2 fusion protein were used as substrates for ERK, JNK, and p38, respectively. LTA and LPS-induced ERK, JNK and p38 activities were monitored by phosphorylation of their individual substrates. These were measured by quantitative immunoblotting with phospho-Elk-1 (Ser-383) antibody, phospho-c-Jun (Ser-63) antibody, and phospho-ATF-2 (Thr-71) antibody, respectively. The experiments were conducted 3 times and a representative result is shown. (C) Effects of LTA and LPS on IRAK-1 and MyD88 expression. Cells were stimulated by LTA or LPS for the indicated time, and the expression level of IRAK-1 and MyD88 were analyzed by Western blotting.
differentiation factor 88 (MyD88) and IL-1R-associated kinase (IRAK)-1. We further examined the downstream signals upon LTA and LPS binding to TLR2 and TLR4, respectively. As shown in Fig. 2C, LPS treatment rendered a dramatic decrease in the IRAK-1 protein level around and
even before 15 min LPS stimulation. However, IRAK-1 protein level was slightly attenuated after 15–30 min LTA treatment, and significantly decreased at 60 min. Strikingly, neither LTA nor LPS affected the expression of MyD88 (Fig. 2C).
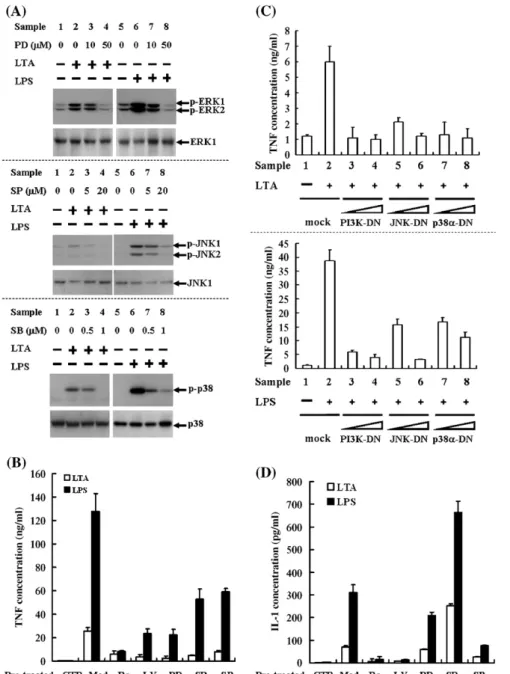
Fig. 3. Effects of protein kinase inhibitors on TNF and IL-1 secretion/proIL-1 expression in LTA- or LPS-stimulated cells. (A) Effects of PD98059, SP600125, and SB203580 on LTA- and LPS-induced phosphorylation of ERK, JNK, and p38, respectively. Cells were pre-incubated with inhibitor for 30 min, followed by LTA (1μg/ml) or LPS (100 ng/ml) stimulation for additional 15 min. Phosphorylations of ERK, JNK, and p38 were detected by Western blotting. (B) Effects of PK inhibitors on LTA- or LPS-induced TNF secretion. Cells were pre-incubated with inhibitor as indicated for 30 min, followed by LTA (1μg/ml) or LPS (100 ng/ml) stimulation for additional 8 h. Conditioned media were collected for TNF assessment using ELISA; one of 4 experiments is presented (n = 4). CTR means samples without stimulation; Med., samples with stimulation and without pre-treatment of inhibitor. (C) Effects of dominant negative forms of PI3K, JNK, or p38α on LTA-or LPS-induced TNF secretion. Cells were transiently transfected with 5- LTA-or 15-μg of plasmid DNA encoding dominant negative forms of PI3K, JNK, or p38α as described previously[1]. After incubated for 48 h, cells were stimulated with LTA or LPS for 8 h, and then TNF secretion was analyzed by ELISA. Mock, transfected with the pcDNA3.1/neo plasmid. (D) Effects of PK inhibitors on LTA- or LPS-induced IL-1 secretion. Cells were pre-incubated with inhibitor as indicated for 30 min followed by LTA (1μg/ml) or LPS (100 ng/ml) stimulation for additional 24 h. Conditioned media were harvested for IL-1 assessment using ELISA; 1 of 4 experiments is presented (n = 4). (E) Effects of PK inhibitors on LTA- or LPS-induced proIL-1 expression. Cells were pre-incubated with inhibitor as indicated for 30 min followed by LTA (1μg/ml) or LPS (100 ng/ml) stimulation for additional 6 h. ProIL-1 expression was assayed by Western blotting; 1 of 3 experiments is presented (n = 3). (F) Effects of dominant negative forms of PI3K, JNK, or p38α on LTA- or LPS-induced proIL-1 expression. Cells were transiently transfected with 5- or 15-μg of plasmid DNA encoding dominant negative forms of PI3K, JNK, or p38αas described previously[1]. After incubated for 48 h, cells were stimulated with LTA or LPS for 6 h, and proIL-1 expression was analyzed by Western blotting.
3.3. Role of protein kinases in the regulation of TNF and proIL-1/ IL-1 protein production in LTA-versus LPS-stimulated J774A.1 cells
To dissect the effects of LTA and LPS on the expression of inflammatory cytokines, we measured the TNF and IL-1 secretion with various treatments of J774A.1 cells. As shown inTable 1, the amount of TNF was elevated significantly at 4 h and reached to the maximal at 8 h upon LTA stimulation. The amount of LTA-induced TNF release gradually reduced after 24 h. The secretion of TNF was profoundly augmented at 4 h, and the amount of secreted TNF kept raising by times upon LPS stimulation. In addition, LTA-induced IL-1 secretion was detected at 4 h and peaked within 8–12 h. Similarly, LPS-induced IL-1 secretion was detected at 4 h, and the maximal amount of IL-1 secretion appeared around 12 h post-stimulation.
After determining the time course of LTA- and LPS-induced TNF and IL-1, we investigated the role of protein kinases in mediation of cytokines induction. First, the effects of protein kinase inhibitors, including PD98059 (PD, MEK inhibitor, 50 μmol/l), SP600125 (SP, JNK inhibitor, 20 μmol/l), and SB203580 (SB, p38 inhibitor, 1μmol/l) on LTA- or LPS-induced activation of ERK, JNK, and p38, respectively were confirmed (Fig. 3A). In addition, cells were preincubated with Ro-31-8220 (Ro, PKC inhibitor, 10 nmol/l), LY294002 (LY, PI3K inhibitor, 50μmol/l), SP, PD and SB, followed by LTA or LPS stimulation. For both bacterial molecules, all inhibitors resulted in differential
inhibition of TNF secretion as shown inFig. 3B. Specifically, in LTA-stimulated cells, Ro, LY, PD, SP, and SB decreased TNF release by 80%, 90%, 90%, 80% and 70%, respectively. In LPS-stimulated cells, Ro, LY, PD, SP, and SB decreased TNF release by 90%, 80%, 80%, 60%, and 50%, respectively (Fig. 3B). What is more, LTA- or LPS-induced TNF was considerably inhibited by overexpression of PI3K-DN, JNK-DN, and p38α-DN, which was consistent with the results analyzed by PK inhibitors (Fig. 3C).
Furthermore, we examined the effects of PK inhibitors on the LTA- and LPS-induced IL-1 secretion. In LTA-stimulated cells, Ro, LY, PD, and SB decreased IL-1 release by about 90%, 90%, 10% and 60%, respectively (Fig. 3D). In LPS-stimulated cells, Ro, LY, PD, and SB decreased IL-1 release by about 95%, 95%, 30% and 75%, respectively (Fig. 3D). Remarkably, SP increased LTA- and LPS-induced IL-1 secretion by 4 folds and 2 folds, respectively (Fig. 3D).
Due to the finding that conventional IL-1 ELISA may un-derestimate proIL-1 concentrations [24], we subsequently used the whole lysates of the LTA- and LPS-stimulated cells to examine the expression of proIL-1 protein, an inactive cytoplasmic precursor. As shown in the upper panel of Fig. 3E, in LTA-stimulated cells, Ro, LY, and SB, but not PD significantly inhibited the expression of proIL-1 protein; in contrast, SP dramatically increased proIL-1 expression (Fig. 3E, upper panel, sample 6). The results of proIL-1 protein are compatible with IL-1 secretion upon the treatment of PK inhibitors. In LPS-stimulated cells, Ro and LY completely abolished the proIL-1 expression, whereas
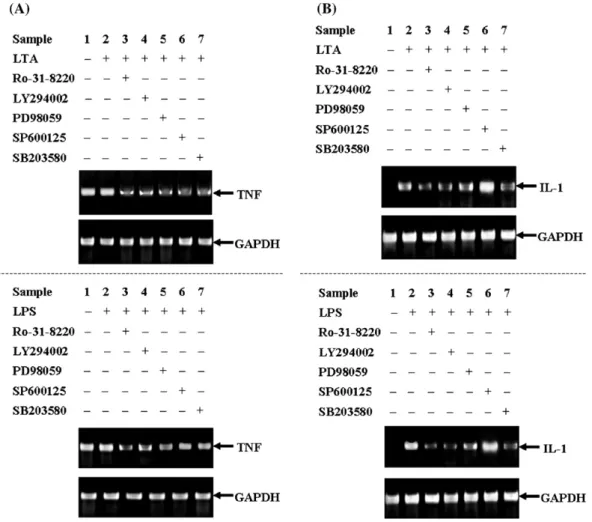
both PD and SB merely exhibited moderate inhibition of proIL-1 induction (Fig. 3E, lower panel). Similar to LTA treatment, SP dramatically increased LPS-induced proIL-1 expression (Fig. 3E, lower panel, sample 6). Moreover, we overexpressed PI3K-DN, JNK-DN, and p38α-DN to further investigates the roles of those kinases in the regulation of proIL-1 expression in LTA-versus LPS-stimulated cells. Both LTA- or LPS-induced proIL-1 expressions were significantly inhibited in the cells with over-expression of PI3K-DN, JNK-DN, and p38α-DN (Fig. 3F). 3.4. Role of protein kinases in the regulation of TNF and IL-1 mRNA expression in LTA-versus LPS-stimulated J774A.1 cells Considering the TNF mRNA expression, we were specifi-cally interested in whether the differential TNF secretion in conditioned medium was contributed by the differential up-regulation of TNF mRNA in response to LTA or LPS. By RT-PCR, we demonstrated that TNF mRNA was present in the un-treated control sample and the effect of LTA or LPS stimulation on TNF mRNA expression appeared minor (Fig. 4A). To further investigate the roles of various signaling events in TNF mRNA expression, the cells were stimulated with LTA or LPS in the
presence of Ro, LY, PD, SP, and SB, respectively. The results indicated that all PK inhibitors diminished TNF mRNA induc-tion (Fig. 4A). In addition, the role of LTA- and LPS-mediated protein kinases in the regulation of IL-1 mRNA was also inves-tigated. As shown inFig. 4B, IL-1 mRNA was barely expressed in the un-stimulated cells, but significantly induced by LTA or LPS. Whereas SP augmented the induction of IL-1 mRNA, the other 4 PK inhibitors, including Ro, LY, PD, and SB, differ-entially attenuated LTA- or LPS-induced IL-1 mRNA (Fig. 4B). 4. Discussion
LTA and LPS, the components of cell walls in gram-positive and gram-negative bacteria, respectively, stimulate macro-phages to produce TNF and IL-1. Current studies demonstrate that both LTA and LPS activate the Src and Lyn, the Src family protein tyrosine kinases (PTKs), in murine macrophages. LPS-induced signaling was mainly initiated through TLR4, but the molecular mechanism by which LTA triggers the activation of PTKs was not clear. Here, we demonstrated that LTA and LPS triggered inflammatory cytokines, TNF and IL-1 through similar intracellular pathways, yet LPS showed a tendency for
Fig. 4. Effects of protein kinase inhibitors on TNF and IL-1 mRNA expression in LTA- or LPS-stimulated cells. Effects of Ro-31-8220, LY294002, PD98059, SP600125, and SB203580 on LTA- or LPS-induced mRNA expression of TNF (A) or IL-1 (B). Cells were pre-incubated with inhibitor as indicated for 30 min followed by LTA (1μg/ml) or LPS (100 ng/ml) stimulation for additional 4 h. The isolated RNA was normalized and monitored by the intactness of a constitutively expressed gene, GADPH, and PCR was carried out by using indicated primers. One of 4 experiments is presented.
a higher level of tyrosine phosphorylation and MAPKs activation. In this paper, we further presented data showing that the phosphorylation level (Fig. 2A) and the kinase activities (Fig. 2B) of ERK, JNK, and p38 were higher in LPS-stimulated macrophages than that in LTA-stimulated macrophages. Besides, LPS not only rapidly (within 15 min) but remarkably induced degradation of IRAK-1, whereas LTA triggered relatively slow and slight IRAK-1 degradation. These results suggest that somewhat more efficiency of LPS-mediated signals was exhibited in the regulation of TNF and IL-1 expression in comparison with LTA.
The mechanisms of LTA- and LPS-induced TNF production are subjects of great importance. Considerable evidence indicated that the LPS-induced signaling leading to TNF production bifurcates to control both at transcription of TNF gene and at translation of TNF mRNA [15,25,26]. Relatively, signal trans-duction pathway and mechanism by which LTA stimulates TNF production in macrophages remain incompletely understood. Here, we revealed that LTA- and LPS-induced TNF secretions (Table 1) are independent of mRNA expression (Fig. 4A). In previous report, MAPKs activation by LPS challenge in macrophages was manifested[1]. In this study, we found that Src, Lyn, PKC, PI3K, ERK, JNK and p38 participate in LTA signaling, and made a collective comparison between LPS- and LTA-induced signaling in macrophages. Using specific inhibitors of JNK and p38 as well as transient transfection with dominant negative forms of JNK and p38, respectively, we demonstrated that JNK and p38 participate in both TNF mRNA expression and TNF secretion in LTA- and LPS-stimulated macrophages, respec-tively, which is compatible with other publications[27–30]. In addition, the PKC, PI3K, and ERK pathways are also involved in both LTA- and LPS-stimulated TNF expression, suggesting that these pathways are potential therapeutic targets for treatment of both Gram-positive and Gram-negative sepsis in which excessive TNF production occurs. Recently, the significance of PI3K and ERK for TNF production was examined in primary rat Kupffer cell cultures stimulated by LTA and LPS[31]. By using the same inhibitors as in our experiments, the authors concluded that TNF production was profoundly affected by neither PI3K nor ERK inhibitor, even at 100μM. In our experiments, a considerable reduction in TNF levels caused by PI3K and ERK inhibitor was observed. This difference may be consequent on distinct suscep-tibilities of Kupffer cells and monocyte-derived macrophages to PK inhibitors. PI3K regulates the turnover of second messenger inositol phosphates within the cell and favors tempered macro-phages to response of LPS. The studies and discussions regarding the relationship between LPS and PI3K mostly were focused on the nitric oxide induction in macrophages[32–34]. Although it was reported that inhibition of PI3K prevents induction of endotoxin tolerance [35], the role and effect on which PI3K modulates the LTA- or LPS-induced TNF expression should be examined further. Besides, PI3K participates in various signal pathways induced by a cohort of growth factors and cytokines. It has been reported that the p85 regulatory subunit of PI3K exerts the physical interaction with type I interleukin-1 receptor (IL-1R) leading to NF-κB activation upon stimulation[36]. Due to the homology of the intracellular domains between IL-1R and TLRs,
we speculate that PI3K may utilize a similar association with TLRs to regulate LTA- or LPS-induced TNF expression.
It has been reported that LPS-induced TNF expression is PKC-independent [25,37]; however, we demonstrated that PKC is important for TNF expression in J774A.1 murine macrophages, which is consistent with the results published by other investigators[38,39]. The discrepancy may be due to cell types or different inhibitors chosen in different experiments. Alternatively, we used PK inhibitors to inhibit TNF mRNA expression, but we could not rule out the effect of these inhibitors on TNF processing or protein sorting, like the acti-vity of TNF alpha converting enzyme or other protein sorting-related molecules.
Meanwhile, in addition to the study of the regulation of LTA-or LPS-induced TNF, we also examined another pro-inflam-matory cytokine, IL-1. In the process of IL-1 maturation, IL-1 is initially synthesized as an inactive 34 kD cytoplasmic precursor, proIL-1 lacking a secretion signal sequence. A single proteolysis cleavage of proIL-1 by interleukin 1 converting enzyme (ICE) or caspase 1 generates a∼17 kD mature IL-1, which is released from cells by an unknown mechanism. p38, an important mediator of stress-induced gene expression is reported to function by regulating translation processes of cytokine mRNA[40,41]. Our current results indicated that p38 plays a pivotal role in both IL-1 mRNA and proIL-1/IL-1 expression as macrophages were stimulated by LTA or LPS (Figs. 3D–F and 4B). In addition, the requirement of ERK for LTA- or LPS-induced proIL/IL-1 expression is relative minor compared to other protein kinases (Fig. 3D and E). Furthermore, transient transfection with JNK-DN attenuated proIL-1 protein expression in LTA- and LPS-stimulated macrophages, indicat-ing that JNK plays an important role in the regulation of IL-1 expression (Fig. 3F). However, SP600125, a known JNK inhibitor,[42]enhanced IL-1 mRNA (Fig. 4B) and proIL-1/IL-1 protein expression (Fig. 3D and E). The inconsistency between JNK-DN and SP600125 in the regulation of IL-1 may come from the non-specific inhibition or action of SP600125, and the detailed mechanism needs further investigation. Besides, involvement of PI3K in regulations of IL-1 expression is also clarified. Our current results showed that inhibition of PI3K by LY294002 (Fig. 3D and E) and PI3K-DN (Fig. 3F), respectively abrogated LTA- or LPS-stimulated proIL-1/IL-1 expression. This finding reveals that PI3K plays a novel and significant role in modulation of IL-1 release during bacterial infection. Next, we elucidated that both TNF and IL-1 induction by bacterial cell wall components are dependent on the activation of PKC. In the presence of Ro-31-8220, LTA- or LPS-stimulated proIL/IL-1 expression was significantly re-duced (Fig. 3D and E). These results give rise to an interesting question regarding the role of PKC in inflammatory cytokine induction by bacterial cell wall components.
In summary, currently we used a macrophage model to study in vitro the immuno-modulating responses of LTA and LPS. The results demonstrated that both LTA and LPS activate Src and Lyn, respectively, and transduces diverse signaling cascades that lead to different biochemical reactions, such as TNF and IL-1 expression. Specifically, we have further
dissected the roles of PKC, PI3K, ERK, JNK, and p38 in the regulation of TNF and IL-1 expression, respectively. We hope that our current findings could expand the therapeutic possibilities for interference with inflammatory responses of sepsis in a more efficient and specific way.
Acknowledgments
This work was supported by research grants from National Science Council, Taiwan, (NSC 94-2120-M-010-002 and NSC 93-2314-B-010-003 to H.-Y.H.; NSC 94-2313-B-273-002, 95-2313-B-273 -004 -MY3 to L.-K.C.) to H.-Y. Hsu; research grant from National Health Research Institutes, Taiwan, (NHRI-EX95-9211SI) to H.-Y. Hsu; a grant from Ministry of Education, Aim for the Top University Plan (95A-C-D01-PPG-10 to H.-Y.H.); Academia Sinica Thematic project (to H.-Y.H.); The Council of Agriculture (COA) of the Executive Yuan, Taiwan, (94AS-11.2.2-FB-e1, 95AS-12.2.2-FB-e1 to L.-K.C.).
References
[1] Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen spe-cies and signal transduction in the regulation of interleukin-1 gene ex-pression. J Biol Chem 2002;277:22131–9.
[2] Kengatharan KM, Kimpe SD, Robson C, Foster SJ, Thiemermann C. Mechanism of gram-positive shock: identification of peptidoglycan and lipoteichoic acid moieties essential in the induction of nitric oxide synthase, shock, and multiple organ failure. J Exp Med 1998;188:305–15. [3] Sriskandan S, Cohen J. Gram-positive sepsis. Mechanisms and differences
from gram-negative sepsis. Inf Dis Clin North Am 1999;13:397–412. [4] Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol
2003;21:335–76.
[5] Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol 2004;4:499–511.
[6] Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282: 2085–8.
[7] Schwadner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem 1999;274:17406–9.
[8] Lichtman SN, Wang J, Lemasters JJ. Lipopolysaccharide-stimulated TNF-alpha release from cultured rat Kupffer cells: sequence of intracellular signaling pathways. J Leukoc Biol 1998;64:368–72.
[9] Heagy W, Hansen C, Nieman K, Rodriguez JL, West MA. AAST organ injury scale: correlation of CT-graded liver injuries and operative findings. J Trauma 2000;49:806–14.
[10] West MA, Bennet T, Seatter SC, Clair L, Bellingham J. LPS pretreatment reprograms macrophage LPS-stimulated TNF and IL-1 release without protein tyrosine kinase activation. J Leukoc Biol 1997;61:88–95. [11] Meng F, Lowell CA. Lipopolysaccharide (LPS)-induced macrophage
activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J Exp Med 1997;185:1661–70.
[12] Brook M, Sully G, Clark AR, Saklatvala J. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett 2000;483:57–61.
[13] Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukoc Biol 2000;67:885–93. [14] Wang SW, Pawlowski J, Wathen ST, Kinney SD, Lichenstein HS, Manthey
CL. Cytokine mRNA decay is accelerated by an inhibitor of p38-mitogen-activated protein kinase. Inflamm Res 1999;48:533–8.
[15] van der Bruggen T, Nijenhuis S, van Raaij E, Verhoef J, van Asbeck BS. Lipopolysaccharide-induced tumor necrosis factor alpha production by
human monocytes involves the raf-1/MEK1-MEK2/ERK1-ERK2 path-way. Infect Immun 1999;67:3824–9.
[16] Moss ML, Jin SL, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997;385:733–6.
[17] Medvedev AE, Espevik T, Ranges G, Sundan A. Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lympho-toxin alpha effects. J Biol Chem 1996;271:9778–84.
[18] Kalb A, Bluethmann H, Moore MW, Lesslauer W. Tumor necrosis factor receptors (Tnfr) in mouse fibroblasts deficient in Tnfr1 or Tnfr2 are signaling competent and activate the mitogen-activated protein kinase pathway with differential kinetics. J Biol Chem 1996;271:28097–104. [19] Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev 1997;8:253–65. [20] Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992;356:768–74.
[21] Cerretti DP, Kozlosky CJ, Mosley B, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science 1992;256:97–100. [22] Matot I, Sprung CL. Definition of sepsis. Intensive Care Med 2001;27:
S3–9.
[23] Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol 2000;165:618–22. [24] Wewers MD, Winnard AV, Dare HA. Endotoxin-stimulated monocytes
release multiple forms of IL-1 beta, including a proIL-1 beta form whose detection is affected by export. J Immunol 1999;162:4858–63.
[25] Takasuka N, Matsuura K, Yamamoto S, Akagawa KS. Suppression of TNF-alpha mRNA expression in LPS-primed macrophages occurs at the level of nuclear factor-kappa B activation, but not at the level of protein kinase C or CD14 expression. J Immunol 1995;154:4803–12.
[26] Kontny E, Ziolkowska M, Ryzewska A, Maslinski W. Protein kinase c-dependent pathway is critical for the production of pro-inflammatory cytokines (TNF-alpha, IL-1beta, IL-6). Cytokine 1999;11:839–48. [27] Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the
regulation of inflammatory cytokine biosynthesis. Nature 1994;372:739–46. [28] Lee YB, Schrader JW, Kim SU. p38 map kinase regulates TNF-alpha production in human astrocytes and microglia by multiple mechanisms. Cytokine 2000;12:874–80.
[29] Swantek JL, Cobb MH, Geppert TD. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol 1997;17:6274–82.
[30] Jiang JX, Zhang Y, Ji SH, Zhu P, Wang ZG. Kinetics of mitogen-activated protein kinase family in lipopolysaccharide-stimulated mouse Kupffer cells and their role in cytokine production. Shock 2002;18:336–41. [31] Dahle MK, Overland G, Myhre AE, et al. The phosphatidylinositol
3-kinase/protein kinase B signaling pathway is activated by lipoteichoic acid and plays a role in Kupffer cell production of interleukin-6 (6) and IL-10. Infect Immun 2004;72:5704–11.
[32] Diaz-Guerra MJ, Castrillo A, Martin-Sanz P, Bosca L. Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J Immunol 1999;162:6184–90.
[33] Pahan K, Raymond JR, Singh I. Inhibition of phosphatidylinositol 3-kinase induces nitric-oxide synthase in lipopolysaccharide- or cytokine-stimulated C6 glial cells. J Biol Chem 1999;274:7528–36.
[34] Salh B, Wagey R, Marotta A, Tao JS, Pelech S. Activation of phos-phatidylinositol 3-kinase, protein kinase B, and p70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 cells: differential effects of rapa-mycin, Ly294002, and wortmannin on nitric oxide production. J Immunol 1998;161:6947–54.
[35] Bowling WM, Flye MW, Qiu YY, Callery MP. Inhibition of phosphatidy-linositol-3′-kinase prevents induction of endotoxin tolerance in vitro. J Surg Res 1996;63:287–92.
[36] Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase in interleukin 1 signaling. Physical interaction with the interleukin 1 receptor and requirement in NFkappaB and AP-1 activation. J Biol Chem 1997; 272:29167–73.
[37] Shames BD, Selzman CH, Pulido EJ, et al. LPS-Induced NF-kappaB activation and TNF-alpha release in human monocytes are protein tyrosine kinase dependent and protein kinase C independent. J Surg Res 1999;83:69–74.
[38] Meldrum DR, Meng X, Sheridan Jr BC, McIntyre RC, Harken AH, Banerjee A. Tissue-specific protein kinase C isoforms differentially mediate macrophage TNFalpha and IL-1beta production. Shock 1998; 9:256–60.
[39] St-Denis A, Chano F, Tremblay P, St-Pierre Y, Descoteaux A. Protein kinase C-alpha modulates lipopolysaccharide-induced functions in a murine macrophage cell line. J Biol Chem 1998;273:32787–92.
[40] Young P, McDonnel P, Dunnington D, Hand A, Laydon J, Lee J. Pyridinyl imidazoles inhibit IL-1 and TNF production at the protein level. Agents Actions 1993;39:C67–9.
[41] Prichett W, Hand A, Sheilds J, Dunnington D. Mechanism of action of bicyclic imidazoles defines a translational regulatory pathway for tumor necrosis factor alpha. J Inflamm 1995;45:97–105.
[42] Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A 2001;98: 13681–13686.