Determination of small phosphorus-containing
compounds by capillary electrophoresis
Sarah Y. Chang
a,∗, Wei-Lung Tseng
b, Sreedhar Mallipattu
b, Huan-Tsung Chang
b,∗∗aDepartment of Applied Chemistry, Chaoyang Univeristy of Technology, 168, Jifong E. Road, Wufong, Taichung County 41349, Taiwan bDepartment of Chemistry, National Taiwan University, Taipei, Taiwan
Received 8 July 2004; received in revised form 29 October 2004; accepted 29 October 2004 Available online 23 March 2005
Abstract
The review focuses on the analysis of small phosphorus-containing compounds by capillary electrophoresis (CE) with different detection modes including UV absorption, laser-induced fluorescence, conductometry, amperometry, atomic spectrometry, and mass spectrometry. Determinations of phosphates, organophosphate, and chemical warfare agents in environmental samples such as water, soil and grains are emphasized. To achieve better sensitivity, high-resolving power, and reproducibility, the optimum separation conditions for various analytes (samples) are different. We compare the merits and demerits of the different detection modes for the detection of the analytes. Although the present methods are successful in many cases, there is still a need to develop high-throughput CE techniques for screening numerous environmental samples and sample stacking techniques in CE for the analysis of trace analytes.
© 2005 Elsevier B.V. All rights reserved.
Keywords: Capillary electrophoresis; Phosphate; Organophosphate; Chemical warfare agents; Pesticides
1. Introduction
A large number of small organic and inorganic compounds containing phosphorus are extremely important in biological and chemical profiles, including nucleosides, organophos-phates (OP), and phosphate ion. Among them, phosphate ion is one of the limiting nutrients and its analysis is of great im-portance in biological and environmental chemistry[1]. OP compounds and related phosphonates are among the most toxic compounds and are widely used as pesticides in agri-cultural and also as chemical warfare (CW) agents. Unfor-tunately, their determinations by chromatographic methods such as paper chromatography and ion chromatography (IC) are not easy, mainly because of difficulty in derivatization, low sensitivity, and slow separation[2,3]. For example, IC
∗Corresponding author. Tel.: +886 423323000x4297; fax: +886 423742341.
∗∗Co-corresponding author. Tel.: +886 223621963; fax: +886 223621963. E-mail addresses: [email protected] (S.Y. Chang),
[email protected] (H.-T. Chang).
generally takes hours to complete separation and column equilibrium for one injection of a sample of inorganic phos-phate sample[4].
Over the last decade, capillary electrophoresis (CE) has proved powerful for separating analytes ranging from small molecules to macromolecules as a result of its extremely high separation efficiency, very short analysis time, and a requirement for only small volumes of sample. Among dif-ferent CE approaches, capillary zone electrophoresis (CZE) with indirect UV detection is the most universal and ef-ficient method for determination of nonabsorbing analyte compounds [5–15]. Detection of migrating sample ions is generally achieved by the displacement of the probe ions (co-ions; chromophores) in the running buffer. This causes a negative peak when such anions pass through the detector and the peak area of the resulting negative peak is used for quantification of the analytes. Despite its popularity, indirect UV absorbance suffers mainly from a lack of selectivity and detection limits are commonly in the submilli to micromo-lar range (10−4to 10−6M)[5–15]. Several other detection methods have been reported for the determination of small
0039-9140/$ – see front matter © 2005 Elsevier B.V. All rights reserved. doi:10.1016/j.talanta.2004.10.014
phosphorus-containing compounds, such as laser-induced fluorescence (LIF), indirect LIF, electrochemical, and direct UV detection modes. LIF is considered to be the most sen-sitive detection mode even to the single-molecule limit since the sensitivity does not suffer with reduced path length. How-ever, LIF detection requires derivatization of analytes with a suitable reagent to form highly fluorescent products, which often makes the analysis more time-consuming, more diffi-cult (by products and high fluorescence background)[16]. An alternative is to employ indirect LIF detection, whose sensi-tivity is at least one order of magnitude lower compared with indirect UV absorbance[17]. Yet, indirect LIF detection has seen very limited use due to the lack of appropriate fluorescent probe, although it was firstly reported in 1988 by Kuhr and Yeung[18,19]. The major advantages of CE with amperom-etry detection are more universality, remarkable sensitivity and less restriction of the detection zone when compared to LIF[20,21]. Although CE with conductivity detection is not as sensitive as that of amperometry, it is more popular for the determination of inorganic species because of their high conductivity[22,23].
There are several excellent reviews dealing with the deter-mination of small phosphorus-containing compounds in vari-ous matrices by CE with different detection methods[24–28]. The separation and quantification of various forms of inor-ganic phosphorus oxo anions have been reviewed by Stover
[24]. Stalikas and Konidari also reviewed chromatographic methods and emerging techniques for the analyses of her-bicides, such as glyphosate (GLYP), glufosinate (GLUF), bialaphos and their metabolites[26]. In addition, Hooijschuur
et al. reviewed chromatographic and mass spectrometric tech-niques for the determination of CW compounds in environ-mental samples[27]. In this review article, we cover the area of small phosphorus-containing compounds analysis using CE with different detection modes.
2. Phosphorus anions
In natural systems phosphorus exists in different forms of organic and inorganic compounds; phosphorus oxo anions that contains phosphorus oxygen linkages are the most com-mon form. Phosphorus oxo anions form different oligomeric species by existing in several oxidation states and also un-dergo multiple protonation, metal ligand reactions. Con-densed phosphates of one-dimensional chains and rings are named polyphosphate and orthophosphate, respectively.
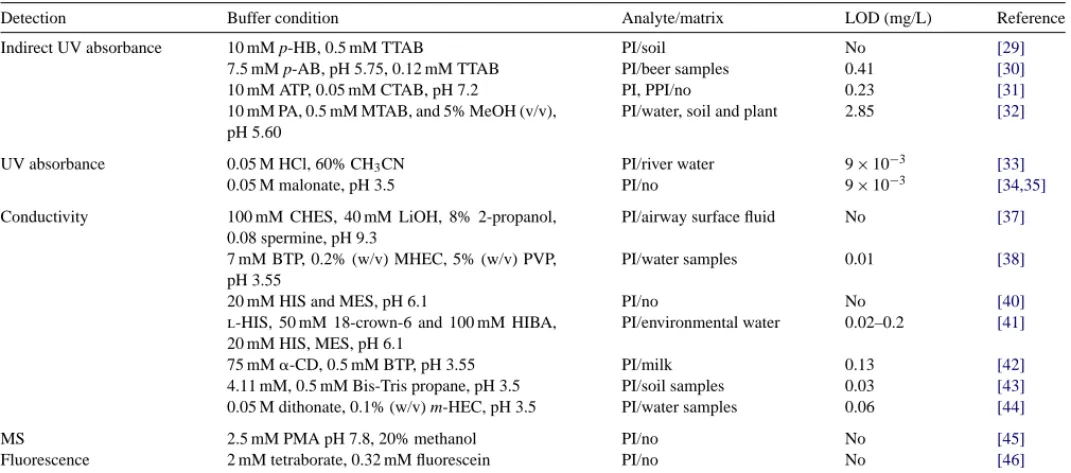
Table 1lists the conditions and compares the detection limits for phosphate ions by various detection methods in CE.
2.1. Indirect UV detection
Organic acids and phosphate ion in aqueous soil ex-tracted samples were determined by CE using a supporting electrolyte solution of 10 mM p-hydroxybenzoic acid con-taining 0.5 mM tetradecyltrimethylammonium bromide[29]. Tetradecyltrimethylammonium bromide in the background electrolyte caused reverse electroosmotic flow in order to optimize resolution and speed. Klampfl and Christian de-termined several anions including phosphate ion in different
Table 1
Representative CE methods for the analysis of phosphorus anions
Detection Buffer condition Analyte/matrix LOD (mg/L) Reference
Indirect UV absorbance 10 mM p-HB, 0.5 mM TTAB PI/soil No [29]
7.5 mM p-AB, pH 5.75, 0.12 mM TTAB PI/beer samples 0.41 [30]
10 mM ATP, 0.05 mM CTAB, pH 7.2 PI, PPI/no 0.23 [31]
10 mM PA, 0.5 mM MTAB, and 5% MeOH (v/v), pH 5.60
PI/water, soil and plant 2.85 [32]
UV absorbance 0.05 M HCl, 60% CH3CN PI/river water 9× 10−3 [33]
0.05 M malonate, pH 3.5 PI/no 9× 10−3 [34,35]
Conductivity 100 mM CHES, 40 mM LiOH, 8% 2-propanol, 0.08 spermine, pH 9.3
PI/airway surface fluid No [37]
7 mM BTP, 0.2% (w/v) MHEC, 5% (w/v) PVP, pH 3.55
PI/water samples 0.01 [38]
20 mM HIS and MES, pH 6.1 PI/no No [40]
l-HIS, 50 mM 18-crown-6 and 100 mM HIBA,
20 mM HIS, MES, pH 6.1
PI/environmental water 0.02–0.2 [41]
75 mM␣-CD, 0.5 mM BTP, pH 3.55 PI/milk 0.13 [42]
4.11 mM, 0.5 mM Bis-Tris propane, pH 3.5 PI/soil samples 0.03 [43]
0.05 M dithonate, 0.1% (w/v) m-HEC, pH 3.5 PI/water samples 0.06 [44]
MS 2.5 mM PMA pH 7.8, 20% methanol PI/no No [45]
Fluorescence 2 mM tetraborate, 0.32 mM fluorescein PI/no No [46]
p-HB: p-hydroxybenzoic acid; p-AB: p-aminobenzoic acid; TTAB: tetradecyltrimethylammonium bromide; PI: phosphate ion; PPI: pyrophosphate ion; CTAB: cetyltrimethyammonium bromide; PA: phthalic acid; MTAB: myristyltrimethylammonium bromide; CHES: Cyclohexylethenesulfonic acid; BTP: 1,3-bis[tris(hydroxymethyl)methylamino]propane; MHEC: methylhydroxyethyl cellulose; PVP: polyvinylpyrrolidone; HIS: histidine; MES: 2-(N-morpholino)ethanesulfonic acid, HIBA:␣-hydroxyisobutyric acid; CD: Cyclodextrin; m-HEC: hydroxyethylcelluose; PMA: pyromellitic acid; LOD: limit of detection.
beer samples by employing CZE with indirect UV detection and conductivity mode with the limit of detection (LOD) of 0.41 mg/L for phosphate ion[30]. Simultaneous separa-tion and determinasepara-tion of phosphate and pyrophosphate ions has been achieved by using adenosine triphosphate as elec-trophoretic chromophore and cetyltrimethylammonium bro-mide as electroosmotic flow modifier[31]. This method pro-vides the advantages of rapidity, less sample volume, and a good linear response. Simultaneous determination of phos-phate ion, organic acids, and other anions in soil, natural water, and plant materials has been achieved with the LODs over a range of 0.48–2.85 mg/L when using various aromatic acids (benzoic, phthalic, trimellitic and pyromellitic acids) as background electrolytes[32].
2.2. Direct UV detection
Nakashima et al. reported an LOD for phosphate ion of 9g/L (as PO43−) via the formation of a keggin-type
[PMo12O40]3− complex that is only stable in the presence
of 60% CH3CN solution[33]. In comparison of indirect UV
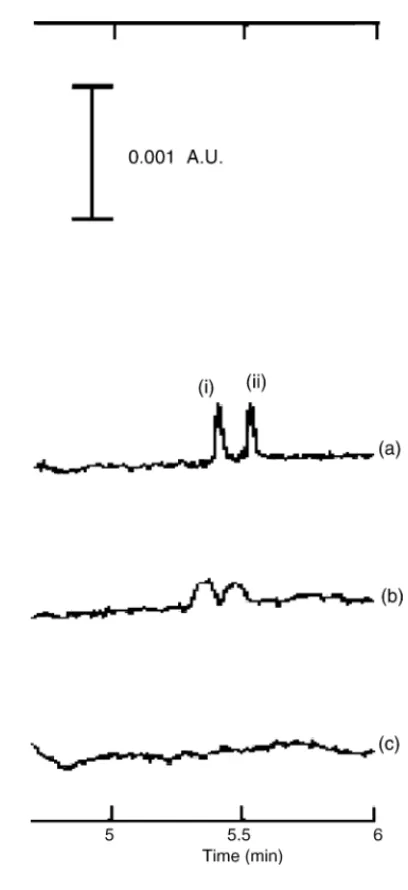
detection, the direct CE method is specific for phosphate ion with a 10-fold sensitivity improvement. Himeno et al. de-veloped a simultaneous determination method for phosphate and arsenic ion using Mo(VI)-ascorbic acid reagent as shown inFig. 1 [34,35]. The heteropoly-blue complexes formed by the reaction of phosphate or arsenic ion with Mo(VI)-ascorbic acid possess UV absorbance at 220 nm between pH 2 and 5. By detecting the complexes at 220 nm, the LOD level amount-ing to 9g/L can be reached.
2.3. Conductometric detection
Several inorganic and organic anions, organic surfactants, alkalimetals, alkaline earths, transition metals, and organic amines were separated by CE in conjunction with a con-ductivity mode[36]. Govindaraju et al. developed conduc-tivity detection method for airway surface fluid ion analysis by CE[37]. This method allows one to separate and quan-tify a variety of biologically related anions, such as nitrite, nitrate, sulfate, phosphate, bicarbonate, and chloride, with better sensitivity than indirect UV detection method. A car-rier electrolyte system for CZE with 300m inner diameter (i.d.) capillary tubes made of fluorinated ethylene–propylene copolymer, with an alternating current conductivity detector has been reported for the separation and determination of sev-eral anions[38]. The LODs in the range of 3–10g/L for the anions were achieved for 200-nL sample volumes. A series of water samples (drinking, river, rain) were tested to assess the practical applicability of this method.
Contactless conductivity detection systems offer the ad-vantages of easy alignment of the electrodes with the cap-illary, better electrical isolation, and long-term robustness over contact ones and have been employed in CE for de-terminations of cations and anions[39]. A disposable, low cost microfluidic device with contactless conductivity
detec-Fig. 1. Effect of buffer matrix: (a) without NaCl, (b) 10 mM NaCl and (c) 50 mM NaCl, on the detection of P(V) and As(V) using 3 mM K2CrO4and 0.2 mM tetradecyltrimethylammonium bromide, pH 10.0. Peak identifica-tion: (i) P(V) and (ii) As(V). Reprinted from Ref.[34], with permission from Elsevier.
tion system was developed for the separation of cations and anions including phosphate ion by Pumera et al.[40]. This system involves measurements of conductance between two aluminum film electrodes, which are placed on the outer side of poly(methyl methylacrylate) and of the impedance of the solutions in the separation channel. Flow injection–CE sys-tem with contactless conductivity detection syssys-tem was de-veloped for the determination of anions including phosphate ion and cations in drainage water system with the LODs be-tween 0.02 and 0.2 mg/L[41]. CZE with contactless conduc-tivity detection method has been utilized for the separation of phosphate ion and other inorganic anions and also based on their host–guest complexes with␣-cyclodextrin. This method provided the LODs in the range of 28–130g/L when using a 300m i.d. capillary tube made with polytetrafluoroethy-lene[42,43]. Two dimensional ITP-zone electrophoresis in
poly(methyl methacrylate) chip with a contactless conductiv-ity mode has been developed for determination of anions[44]. Prior to separation in second channel (zone electrophoresis), analytes were concentrated and interferences were removed from the first channel. The LOD value as attained under the optimal conditions on the microfluidic device for phosphate ion (0.06 mg/L) for a 960 nL sample volume is remarkable.
2.4. Other detection modes
Mixtures of inorganic species, including phosphate ion, have been separated by CE and ion exchange chromatogra-phy, then detected by mass spectrometry with an ion spray at-mospheric pressure ionization source[45]. The selectable de-gree of ion-adduct declustering and molecular fragmentation in the MS interface region allows the system to be operated as an elemental analyzer or as a molecular detector suitable for oxidation state determinations. Dasgupta and Liu introduced a simple and versatile falling drop sample injection mode with a confocal fluorescence detector for the determination of phosphate ion and other anions by CE with indirect LIF detection[46]. The peak broadening for the late peaks is due to the large difference between the electrophoretic mobility (EPM) of the analytes and the EPM of fluoresceinate ion, especially in phosphate ion.
3. Organophosphates
3.1. Chemical warfare agents
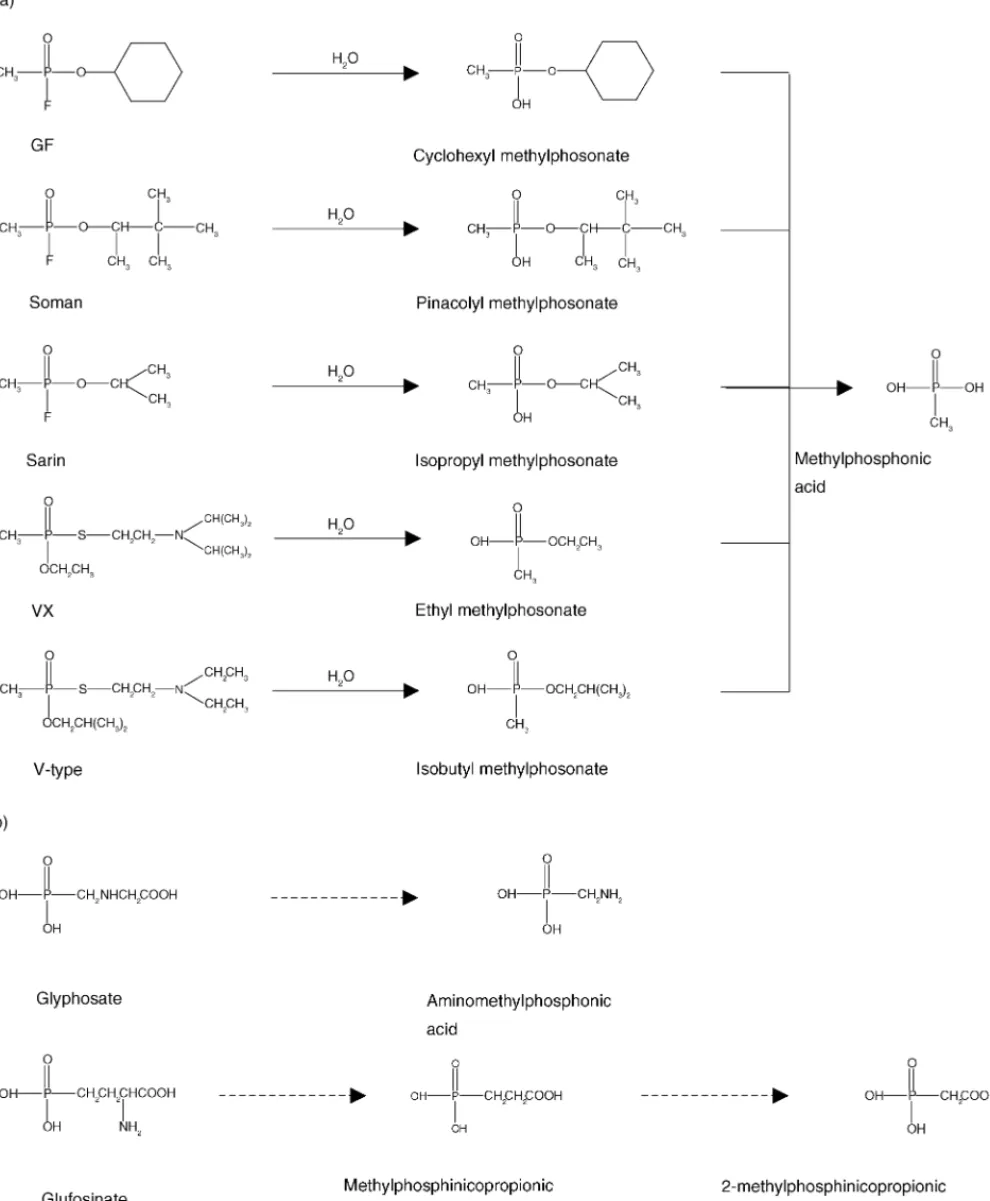
CW agents are divided into three categories: nerve, vesicant, and blood-borne agents. The nerve agents – Sarin (GB), Soman (GD), Tabun (GA), and O-Ethyl S-(2-diisopropylaminoethyl) methylphosphonothiolate (VX) – all disrupt neurological regulation within biological systems through the inhibition of acetyl cholinesterase. The G-type nerve agents, including GB, GD, and GA, rapidly hydrolyze to form various alkylphosphonic acids (AA), whereas V-type or VX nerve agents degrade to form AA, phosphonothioic acids, and various alkylaminoethanol compounds (Fig. 2a). Rochu and Masson summarizes the use of CZE methods for the investigation of the interactions between proteins and OP [47]. Two different soil bacteria, Pseudomonas
dimin-uta and Flavobacterium sp. are highly efficient catalyst for
degrading OP, including GB and GD, and the insecticides paraoxon, coumaphos and diazinon. The CW compounds an-alyzed by CE with various detection methods are summarized inTable 2, showing the optimum conditions and LODs.
3.1.1. Indirect UV detection
Mercier et al. demonstrated that AA and their monoester derivatives were separated in 15 min by using a background electrolyte of 5 mM sorbic acid and 0.1 M decamethonium bromide [48]. In order to achieve the high speed separa-tion, the capillary was dynamically coated with 0.2%
poly-bren prior to each analysis. The separation of AA mo-noesters was less in 6 min when the electrolyte consisted of 1.6 mM hydroxylamine and 5 mM sorbate acid at pH 6.0[49]. The nerve agent degradation products were detected within 5 min in the presence of a proprietary EOF modifier [50]. Melanson et al. reported that CW agent degradation prod-ucts, which are methylphosphonic acid (MPA), ethylphos-phonate (EPA), isopropylmethylphosethylphos-phonate (IMPA), and pinacolylmethylphosphonate (PMPA), were detected using phenylphosphonic acid as the indirect probe[51]. A LOD of 1.9g/L was obtained, showing that electrokinetic injection provided up to 100-fold improvement in LOD when com-pared to that using hydrodynamic injection. In a recent series of papers, Nassar et al. reported the use of a cationic sur-factant didodecyldimethylammonium hydroxide to reverse EOF for the separation of OP agent degradation products
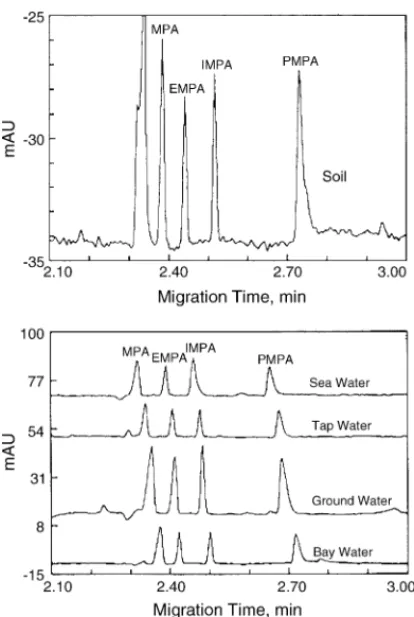
[52–54]. The concentrations of MPA, IMPA, and pinacoly-methylphosphonate from a military area were 27.4, 24.2 and 2.5 mg/L (soil) and 8.8, 9.9, and 0.3 mg/L (surface wipes), respectively. They also have shown that the validation of CE combined with indirect UV or conductivity detection to quan-titatively determine the AA compounds from the nerve agents GB and GD in the water/MPA destruction system[55]. In ad-dition, LODs are as low as 1–2 mg/L in environmental water samples and 25–50 mg/L in aqueous leachates of soil sam-ples using optimal electrokinetic injection (10 s at−10 kV) (Fig. 3). Compared to pressure injection, electrokinetic in-jection represents up to a 100-fold sensitivity improvement
[56]. Meng and Qin demonstrated for the first time extrac-tion of CW agent breakdown products from human serum by solid phase extraction using molecularly imprinted polymer
[57]. Following the extraction, the breakdown products were detected using CE without derivatization procedure. LODs of 0.1 and 0.05 mg/L were obtained from human serum and a rice sample, respectively.
3.1.2. Direct UV detection
In order to eliminate the problem of non-selectivity of indi-rect UV and fluorescence detection, Robins and Wright devel-oped the direct UV detection of AA based on the formation of borate-phosphonic acid esters[58]. Complexation occurred in 10 mM sodium tetraborate solution at pH 7.0 or higher and nanogram concentrations of AA were detected but no real-samples were analyzed. Sulfur-containing, neutral degrada-tion products of mustard and related vesicants were analyzed by micellar electrokinetic chromatography (MEKC)[59,60], and LODs for these compounds ranged from 1 to 10 mg/L using UV direct detection. Six compounds were separated in less than 10 min with an electrolyte containing 10 mM borate and 100 mM sodium dodecyl sulfate at pH 9.0.
3.1.3. Conductometric detection
Carbonate interference is one of the major drawbacks for the indirect UV detection of MPA by CE. To avoid the prob-lem, conductivity detection was used in CE for detecting the nerve agent degradation products from GB, GD, and VX
Fig. 2. Hydrolysis pathways of nerve agents (a) and herbicides (b).
because they are strong acids and fully dissociate in an aque-ous environment [61]. The application of this method for screening of MPA is demonstrated, with the detection limits around 6 mg/L in surface water, ground water, and soil
ex-tracts. Wang et al. reported a microfluidic device for screening of OP nerve agent degradation products using a contactless conductivity detector[62]. The suitability of the microfluidic device for measuring low levels of nerve agent breakdown
Table 2
Representative CE methods for the determination of CW degradation products
Detection Buffer condition Analyte/matrix LOD (mg/L) Reference
Indirect UV absorbance 5 mM sorbic acid, 0.1 M decamethonium bromide, pH 6.0
AA, AA monoesters/soil No [48]
1.6 mM hydroxylamine, 5 mM sorbate acid, pH 6.0
AA monoesters/no No [49]
4.5 mM chromate, 0.5 mM osmotic flow modifier, 1.0 mM sodium bicarbonate
AA, AA monesters/groundwater <1 [50]
10 mM glutamic acid, 1 mM phenylphos-phonic acid, 1 mM CAS-U
AA, AA monesters/no 1.9× 10−3 [51]
200 mM borate, 10 mM phenylphospho-nic acid, 0.35 mM DDAOH, 0.03 wt% Tri-ton X-100, pH 6.0
AA, AA monoesters/soil and surface wipes from military area
0.1 [52–54]
200 mM borate, 10 mM phenylphospho-nic acid, 0.35 mM DDAB, 0.03 wt% Tri-ton X-100, pH 4.0
AA monoesters/environmental water and leachates of soil
1–2 [55]
200 mM borate, 10 mM phenylphospho-nic acid, 0.03 wt% Triton X-100, pH 4.0
AA, AA monoesters/water samples and soil leachates
0.01–0.02 [56]
200 mM borate, 5 mM phenylphosphonic acid, 0.1 mM DDAB, 0.2% Triton X-100, pH 3.55
AA, AA monoesters/human serum, rice and soil
0.1, 0.05 [57]
UV absorbance 10 mM sodium tetraborate, pH 7.0 AA, AA monoesters/no Nanogram [58]
10 mM borate, 100 mM SDS, pH 9.0 AA, AA monesters, MA/water 1–10 [59,60]
Conductivity 30 mM histidine, 30 mM MES, 0.03 wt% Triton X-100, 0.35 mM CTAOH
AA monoesters/no 0.075 [52]
30 mM l-histidine, 30 mM MES, 0.7 mM TTAOH,0.03% Triton X-100, pH 6.5
AA, AA monoesters/soil, surface wa-ter and groundwawa-ter
6 [61]
5 mM MES, 5 mM l-histidine, pH 6.1 AA, AA monoesters/river water 0.048–0.086 [62]
LIF 50 mM cholate, 50 mM borate, 40% ACN, labeling with PA
AA/no 0.012–0.017 [63]
10 mM borate, pH 9.5, labeling with o-phthalaldehyde
Thiols, cyanide (hydrolysis products of CW)/no
0.002–0.089 [64]
Indirect LIF 5 mM Bis-Tris, pH 7.2, 50M TSPP as indirect probe
AA monoesters/no 9× 10−3 [65]
FPD 20 or 50 mM ammonium acetate, pH 9.0 AA, AA monoesters, MA/soil, rubber, paint and water
0.1–0.5 [66,67]
MS 5 mM sorbic acid, ammonia, pH 6.5 AA, AA monoesters/tape water 0.1 [68]
AA: alkylphosphonic acids; CAS-U: coco(amidopropyl)ammoniumdimethylsulfobetaine; DDAOH: didodecyldimethylammonium hydroxide; DDAB: dido-decyldimethylammonium bromide; CTAOH: cetyltrimethylammonium hydroxide; SDS: sodium dodecyl sulfate; MA: methylphosphonothioic acids; MES: 2-(N-morpholino)ethanesulfonic acid; TTAOH: tetradecyltrimethylammonium hydroxide; ACN: acetonitrile; PA: panacyl bromide; Bis-Tris acetonitrile bis(2-hydroxyethyl)imino-tris(hydroxymethyl)methane; TSPP: tetrakis(4-sulfophenyl)porphine.
products in the river water sample was demonstrated. Detec-tion processes have been optimized to yield high sensitivity (with 48–86g/L detection limits) and good linearity (over the 0.3–100 mg/L range).
3.1.4. LIF detection
Jiang and Lucy proposed use of MEKC with LIF detec-tion for determinadetec-tion of AA compounds [63]. A number of linear AA, including MPA, EPA, and propylphosphonic acids (PPA), were derivatized with panacyl bromide in dry
N,N-dimethylformamide. The LODs for MPA, EPA, and PPA
by high-salt stacking, using 400 mM NaCl in the dilution buffer, were 12, 14, and 17g/L, respectively. The fluores-cence derivatizing agent, o-phthalaldehyde, has also been applied to detect the hydrolysis product or its simulants of the CW agents, including VX, R-VX, and GA[64]. Unlike other CE approaches, this method uniquely targeted the thi-ols degradation product from V-type nerve agent, as well as
the cyanide degradation product from GA. The LODs for cyanide, 2-diethylaminoethanethiol, 2-mercaptoethanol by sample stacking, using a 30-s injection time at 10-cm height, were estimated to be 9.3, 1.8, and 89g/L, respectively.
3.1.5. Indirect LIF detection
Melanson et al. reported that CW agent degradation prod-ucts, including MPA, IMPA, and PMPA, were detected using the porphyrrin tetrakis(4-sulfophenyl)porphine as the indi-rect probe[65]. Compared with 325-nm HeCd laser, the use of 415-nm diode laser provided a 10-fold sensitivity improve-ment. This is because that the baseline stability achieved with the violet (415 nm) diode laser was excellent.
3.1.6. Other detection modes
Flame photometric detection (FPD) combined with CE was reported for detection of AA[66,67]. The CE-FPD is shown to be very selective and sufficiently sensitivity for
Fig. 3. Analysis of phosphonic compounds at 200–400g/L extracted from soil, sandy loam (upper trace), and water (lower trace) by CE. Reprinted from Ref.[56], with permission from ACS.
screening of CW agent breakdown products in water or soil. The same interface principle, which was derived from LC-FPD, proved to be suitable for CE-FPD. The LODs in water were ranged from 0.1 to 0.5 mg/L when the injected sample volume was 900 nL. Mercier et al. developed on-line CE-UV spectrometry—MS methods to investigate AA in different matrices[68]. MS–MS provided the LOD of 0.1 mg/L for AA in spiked tap water sample, which is higher than that (5 mg/L) by selective ion monitoring MS detection.
3.2. Pesticides
Pesticides can be classified based on functional group in their structures, such as inorganic, organophosphate, organonitrogen, and organosulfur compounds, or their spe-cific biological activity on target species, such as insecti-cides, fungiinsecti-cides, herbiinsecti-cides, and acariinsecti-cides, etc. Although pesticides are often provided to control insects, weeds, and diseases, they can leach through the soil to the ground and surface water, or drift out of the target area to the neigh-boring land. Among these pesticides, GLYP and GLUF are non-selective herbicides for control of long grasses and broad-leaved weeds. They were rapidly broken down to soil to produce aminomethylphosphonic acids (AMPA) and methylphosphinicopropionic acid (MPP) (Fig 2B). The deter-minations of GLYP, GLUF, and other phosphorus-containing pesticides by CE with different detection modes are listed
in Table 3, emphasizing the separation conditions and LODs.
3.2.1. Indirect UV detection
GLYP and GLUF, which are not chromophores in the UV–vis range, cannot be determined by simple UV detec-tion. CE with the indirect UV detection can be used to analyze Roundup®herbicide solution using the high molar absorptiv-ity of ribonucleotide electrolytes[69]. The LODs of AMPA and GLYP were 0.2 and 0.1 mg/L, respectively. In order to improve the LOD of phosphorus-containing herbicides with indirect detection, Cikalo et al. showed that field-amplified sample injection could give sensitivity improvements by a factor of up to 1000[70]. As a result, 2g/L GLYP in aque-ous sample could be detected.
3.2.2. Direct UV detection
Analysis of GLYP and AMPA in serum was achieved us-ing 0.1 M boric acid-sodium hydroxide (pH 9.6) containus-ing 10% methanol [71]. The two compounds, after derivatiza-tion with p-toluenesulphonyl chloride, were detected in the UV region with high sensitivity. The LOD for both was 0.1 mg/L in spiked sera and the recoveries of GLYP and AMPA were 87.9–88.8 and 78.4–86.9%, respectively. The anionic herbicide, chlorpyrifos, which has a dermal LD50
in rats of 202 mg/kg is commonly encountered in agricul-ture[72]. Determination of anionic herbicide has tradition-ally been carried out using MEKC methods, in connection with UV detection[73–75]. It is difficult to separate highly hydrophobic chlorpyriphos based on MEKC because it com-pletely associates with the micelles. To overcome the dis-advantages, a variety of means have been shown to be ef-fective in enhancing the separation performance, including changing surfactant system and adding various organic sol-vents[73,74]. Guardino et al. reported the determination of chlorpyrifos in air, leaves, and soil from greenhouses by gas chromatography, high-performance liquid chromatogra-phy, and CE [75]. Using a cationic additive tetraoctylam-monium bromide, the CE method allows the determination of diazinon(O,O-diethyl-O-2-isopropyl-6-methylpyrimidin-4-yl phosphorothioate) that is widely used for eradication of harmful insects but with lethal concentrations ranged from 300 to 400 mg/kg for mammals. Thuringiensin derived from the bacterium Bacillus thuringiensis is especially effective as a fly control. Diazinon and thuringiensin are readily dis-solved in MEKC buffer containing 100 mM SDS and 20% acetonitrile[76,77]. The amounts of thuringiensin in fermen-tation broth were estimated by using tryptophan as internal standard, with results of 1.641 g/L[77]. The sodium salt of octakis (2,3-diacetyl-6-sulfo)-␥-cyclodextrin has been used as a resolving agent for CE separation of the enantiomers of organophosphate triesters, which constitute one of the largest classes of agricultural insecticides [78]. The degree of toxicity for chiral OP is dependent on the seterochemistry at the phosphorus center. L¨ammerhofer et al. demonstrated the simultaneous separation of all eight N-2,4-dinitrophenyl
S.Y . Chang et al. / T alanta 66 (2005) 411–421 Table 3
Representative CE methods for the measurement of phosphorus-containing pesticides
Detection Analyte Electrolyte Martix LOD (mg/L) Reference
Indirect UV absorbance GLYP, AMPA 5 mM adenosine monophosphates, 100 mM borate, pH 7.10 Roundup herbicide 0.1, 0.2 [69]
GLYP 10 mM phthalate, 0.5 mM TTAB Wheat <2× 10−3 [70]
UV absorbance GLYP, AMPA 0.1 M boric acid, 10% methanol, pH 9.6, labeling with
p-toluenesulphonyl chloride
Sera 0.1 [71]
Chlorpyrifos-methyl and -ethyl 20 mM borate, 50 mM SDS, 40% methanol, pH 9 No 0.21, 0.25 [73]
Chlorpyrifos-methyl and -ethyl, chlorpyrifos 10 mM phosphate, 6 mM borate, 50 mM SDS or NaDCh, 25% ACN, pH 9.3
Air, leaves No [74,75]
Chlorpyrifos-methyl and -ethyl, chlorpyrifos 10 mM phosphate, 6 mM borate, 10 mM TOAB, or 50% CAN
Soil No [74,75]
Diazinon, thuringiensin 20 mM boric acid, 20 mM phosphate, 100 mM SDS, 10% ACN, pH 9.2
Fermentation broth No [76,77]
Ethyl methyl p-nitrophenyl phosphate 10% methanol, 20 mM ODAS-␥CD, 25 mM MES, 12.5 mM sodium hydroxide
No No [78]
1-amino-2-hydroxy and
2-amino-1-hydroxyprop–ane phosphonic acids
10 mM O-(tert-butylcarbamoyl)quilidine, 100 mM glacial acetic acid, 12.5 mM triethylamine
No No [79]
Conductivity GLYP, GLUF, AMPA, MPP 2-(N-Morpholino)ethanesulfonic acid as TE, chloride as LE No < 0.025 [80]
Paraoxon, fenitrothione,methyl parathion, ethyl parathion
20 mM MES, 7.5 or 10 mM SDS, pH 5.0 River water 0.21–4.48 [81–83]
LIF GLYP, GLUF, AMPA 20 mM boric acid, 15 mM Brij-35, pH 9.5, labeling with DTAF No 0.16× 10−3, 0.08× 10−3, 0.06× 10−3 [84–86] GLYP No No 1.67 [87]
Indirect LIF GLYP, GLUF, AMPA 1 mM fluorescein solution, pH 9.5 Herbicide, ground water 1.29, 0.45,1.75 [89]
MS GLYP, GLUF, AMPA, MPP 1, 2, 5 mM ammonium acetate, in methanol-water (50:50, v/v)
Wheat 0.17, 0.42 [88]
TTAB: tetradecyltrimethylammonium bromide; GLYP: glyphosate; GLUF: glufosinate; AMPA; aminomethylphosphonic acids; MPP: methylphosphinicopropionic acid; SDS: sodium dodecyl sulfate; NaDCh: sodium deoxycholate; ACN: acetonitrile; TOAB: tetraoctylammonium bromide; ODAS-␥CD: octakis (2,3-diacetyl-6-sulfo)-␥-cyclodextrin; MES: 2-(N-morpholino)ethanesulfonic acid; DTAF: 5-(4, 6-dichloro-s-triazin-2-ylamino)fluorescein; TE: terminating electrolyte; LE: leading electrolyte.
isomeric derivatives of 1-amino-2-hydroxy and 2-amino-1-hydroxypropane phosphonic acids using a partial filling tech-nique to avoid high background signal from quinine carba-mate chiral selector[79]. Interestingly, pseudo-enantiomeric corresponds to O-(tert-butylcarbamoyl)quilidine selector ex-hibited reversed migration order and nearly identical resolu-tion.
3.2.3. Conductometric and electrochemical detection
Isotachophoresis (ITP) preconcentration of GYLP, GLUF, AMPA, and MPP with conductivity detection was inves-tigated [80]. This method provides lower LODs around 25g/L for the analytes, which are around 60-fold lower than those by previous CE methods. In a recent paper, Wang et al. reported a microfluidic device incorporated into a thick-film carbon or a boron-doped diamond electrode-based elec-trochemical detector for the separation and detection of four OP nerve compounds, including paraoxon, methyl parathion, fenitrothione, and ethyl parathione[81,82]. The microfluidic device offers rapid separation and high sensitivity (micromo-lar). They demonstrated the applicability of the microfluidic device to separation of three organophosphate compounds in spiked river water samples. They also have shown that the ability to use a single-channel microfluidic device man-ifold for measurements of the nitroarmatic explosives and OP nerve agents[83]. Assay rates of 360 and 30 h−1can be realized for the total screening and individual measurement, respectively.
3.2.4. LIF detection
Three fluorescein analogues, including isothiocyanate iso-mer (FITC), 5-(4, 6-dichloro-s-triazin-2-ylamino)fluorescein (DTAF) and 5(6)-carboxyfluorescein N-succinimidyl ester (CFSE), were used as labeling reagents for the determination of GYLP, GLUF, and AMPA by MEKC with LIF[84–86]. The use of nonionic surfactants Triton X-100 and Brij-35 improved the sensitivity as a result of preventing the interfer-ence from the excess labeling reagent by shifting the corre-sponding peaks from those for derivative analytes. DTAF is superior to FITC and CFSE on the account of 15-fold sensi-tivity improvement and shorter reaction time. The LOD for these herbicides with DTAF labeling reagent ranged from 0.06 to 0.16g/L. These results indicate that this method provides great potential for its application to the environ-mental samples without an enrichment step. Rassi et al. also reported that GLYP was derivatized through the car-boxylic group with 7-amionaphthalene-1,3-disulfonic acid (ANDSA). The ANDSA-GLYP derivative has a maximum absorption at 251 nm and fluorescence intensity at 382 nm when excited at 300 nm. In comparison with UV absorption detection, the LOD was further decreased by one order of magnitude to 1.67 mg/L using fluorescence detection[87].
3.2.5. Other detection modes
Goodwin et al. coupled CE system with MS using a sheath-less microelectrospray interface for the determination of the
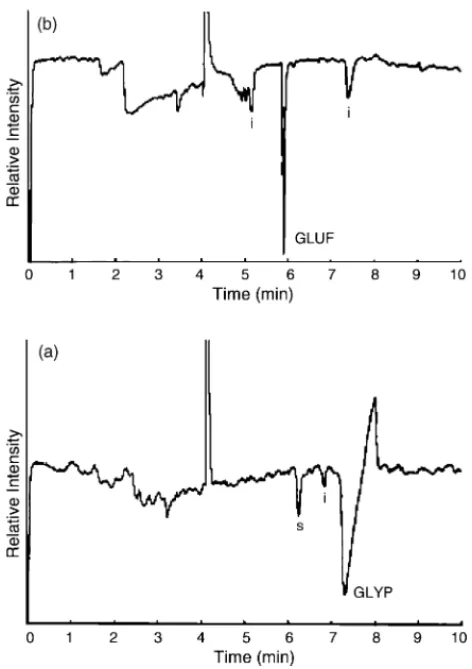
Fig. 4. Electropherograms of (a) Ninninchun herbicide solution and (b) Basida herbicide solution. Reprinted from Ref.[89], with permission from Elsevier.
same analytes [88]. A linear polyacrylamide coated capil-lary gave best reproducibility in terms of migration time and peak area due to zero electroosmotic flow. The observed LOD for GLYP in water is 0.17 mg/L and that for water–acetone extract of wheat is 0.42 mg/L. In a recent report from our lab-oratory, we have shown the potential of a CE-indirect fluo-rescence detection method in determining GLYP, GLUF, and AMPA using 1 mM fluorescein solution at pH 9.5 as indirect probe[89]. The capillary was carefully treated with methanol for 10 min, water for 5 min, and separation buffer for 30 min in order to create constant fluorescence background. The LOD values of GLYP, GLUF, and AMPA were 1.29, 0.45 and 1.75 mg/L, respectively. Quantification of these com-pounds in commercial herbicides, such as Ninninchun and Basida was demonstrated (Fig. 4). However, application of this method to the direct detection of GLYP in the ground water is still problematic.
4. Conclusions
This review article has addressed recent trends in the sepa-ration and determination of different phosphorus-containing compounds in different matrices with different detection modes. CE is a simple, attractive alternative method to that
of chromatographic techniques in the analysis of different phosphorus-containing compounds. A wide variety of de-tection methods such as UV, fluorescence, LIF, conducto-metric and amperoconducto-metric methods have been used in CE to determine different phosphorus compounds in biological and environmental matrices. Although numerous successful examples have been demonstrated, CE methods providing high-throughput, excellent reproducibility, and high resolv-ing power are still desired. In order to screen numerous sam-ples, on-line sample pretreatment (preconcentration and in-terference removal) and CE separation in microchip formats require further development.
References
[1] D.E.C. Corbridge, Phosphorus—an outline of its chemistry, in: Bio-chemistry and Technology, fourth ed., Elsevier, Amsterdam, 1990. [2] J. Weiss, Ion Chromatography, second ed., VCH, Weinheim, 1995. [3] M. Baudler, M. Mengel, Fresenus J. Anal. Chem. 211 (1965) 42. [4] F. Stover, J.A. Bulmahn, J.K. Gard, J. Chromatogr. A 688 (1994)
89.
[5] O. H´enin, B. Barbier, A. Brack, Anal. Biochem. 270 (1999) 181. [6] J. Ren, X. Huang, Anal. Chem. 73 (2001) 2663.
[7] J. He, S.-W. Chen, L.-F. Ruan, L.-L. Cao, J. Yao, Z.-N. Yu, J. Agric. Food Chem. 51 (2003) 7523.
[8] N.J. Munro, D.N. Finegold, J.P. Landers, Z. Huang, Anal. Chem. 72 (2000) 2765.
[9] K.E. Andersen, C. Bjergegaard, P. Moller, J.C. Sorensen, H. Sorensen, J. Agric. Food Chem. 51 (2003) 6391.
[10] P. Doble, M. Macka, P.R. Haddad, J. Chromatogr. A 804 (1998) 327. [11] T.-C. Chiu, M.-F. Huang, C.-C. Huang, M.-M. Hsieh, H.-T. Chang,
Electrophoresis 23 (2002) 449.
[12] E. Rudzinska, P. Wieczorek, P. Kafarski, Electrophoresis 24 (2003) 2693.
[13] S.A. Shamsi, N.D. Danielson, Anal. Chem. 66 (1994) 3757. [14] M.A. van den Hoop, J.J. van Staden, J. Chromatogr A 770 (1997)
321.
[15] M.-J. Chen, H.-S. Chen, C.-Y. Lin, H.-T. Chang, J. Chromatogr. A 171 (1999) 171.
[16] L. Hernandez, S. Tucci, N. Guzman, X. Paez, J. Chromatogr. 652 (1993) 393.
[17] P.L. Desbene, C.J. Morin, M.A.M. Desbene, R.S. Groult, J. Chro-matogr. A 689 (1995) 135.
[18] W.G. Kuhr, E.S. Yeung, Anal. Chem. 60 (1998) 1832. [19] W.G. Kuhr, E.S. Yeung, Anal. Chem. 60 (1998) 2642.
[20] Y. Peng, Q. Chu, F. Liu, J. Ye, J. Agric. Food Chem. 52 (2004) 153. [21] D.M. Osbourn, C.E. Lunte, Anal. Chem. 73 (2001) 5961. [22] Y. Zeng, H. Chen, D.-W. Pang, Z.-L. Wang, J.-K. Cheng, Anal.
Chem. 74 (2002) 2441.
[23] Y. Liu, J.A. Vickers, C.S. Henry, Anal. Chem. 76 (2004) 1513. [24] F.S. Stover, J. Chromatogr. A 834 (1999) 243.
[25] T. Wang, S.F.Y. Li, J. Chromatogr. A 834 (1999) 233. [26] C.D. Stalikas, C.N. Konidari, J. Chromatogr. A 907 (2001) 1. [27] E.W.J. Hooijschuur, Ch.E. Kientz, U.A.Th. Brinkman, J. Chromatogr.
A 982 (2002) 177.
[28] J. S´adeck´a, J. Polonsk´y, J. Chromatogr. A 834 (1999) 18. [29] I. Ahumada, J. Mendoza, P. Escudero, K. Mossert, L. Ascar, J.
AOAC Int. 84 (2001) 1057.
[30] W. Christian, J. Klampfl, J. Agric. Food Chem. 47 (1999) 987. [31] H.O. Barbier, B.B. Andr´e, Anal. Biochm. 270 (1999) 181. [32] Z. Chen, C. Tang, J.C. Yu, J. High Resol. Chromatogr. 22 (1999)
379.
[33] Y. Nakashima, T. Goto, I. Kitazum, S. Himeno, Electrophoresis 22 (2001) 3377.
[34] S. Himeno, K. Sano, Y. Nakashima, J. Chromatogr. A 966 (2002) 213.
[35] I. Kitazumi, Y. Nakashima, S. Himeno, J. Chromatogr. A 939 (2001) 123.
[36] C. Haber, W.R. Jones, J. Soglia, M.A. Surve, M. McGlynn, A. Ca-plan, J.R. Reineck, C. Krstanovic, J. Cap. Elec. 3 (1996) 1. [37] K. Govindaraju, E.A. Cowley, D.H. Eidelman, D.K. Lloyd, Anal.
Chem. 69 (1997) 2793.
[38] D. Kaniansky, V. Zelenska, D. Baluchova, Electrophoresis 17 (1996) 1890.
[39] F. Laugere, R.M. Guijt, J. Bastemeijer, G. van der Steen, A. Berthold, E. Baltussen, P. Sarro, G.W.K. van Dedem, M. Vellekoop, A. Boss-che, Anal. Chem. 75 (2003) 306.
[40] M. Pumera, J. Wang, F. Opekar, I. Jel´ınek, J. Feldman, H. L¨owe, S. Hardt, Anal. Chem. 74 (2002) 1968.
[41] P. Kub´aˇn, M. Reinhardt, B. M¨uller, P.C. Hauser, J. Environ. Monit. 6 (2004) 169.
[42] M. Mari´an, B. R´obert, K. Duˇsan, J. Chromatogr. A 834 (1999) 179.
[43] D. Kaniansky, V. Zelensk´a, M. Mas´ar, F. Iv´anyi, ˇS. Gazd´ıkov´a, J. Chromatogr. A 844 (1999) 349.
[44] R. Bodor, V. Madajova, D. Kaniansky, M. Masar, M. Johnck, B.C.W. Stanislawski, J. Chromatogr. A 916 (2001) 155.
[45] J.J. Corr, J.F. Anacleto, Anal. Chem. 68 (1996) 2155. [46] H. Liu, P.K. Dasgupta, Anal. Chem. 69 (1997) 1211. [47] D. Rochu, P. Masson, Electrophoresis 23 (2002) 189.
[48] J.-P. Mercier, Ph. Morin, M. Dreux, A. Tambute, J. Chromatogr. A 741 (1996) 279.
[49] J.-P. Mercier, Ph. Morin, M. Dreux, A. Tambute, J. Chromatogr. A 779 (1997) 245.
[50] S.A. Oehrle, P.C. Bossle, J. Chromatogr. A 692 (1995) 247. [51] J.E. Melanson, B.L.-Y. Wong, C.A. Boulet, C.A. Lucy, J.
Chro-matogr. A 920 (2001) 359.
[52] A.-E.F. Nassar, S.V. Lucas, W.R. Jones, L.D. Hoffland, Anal. Chem. 70 (1998) 1085.
[53] A.-E.F. Nassar, A. Emery, L.D. Hoffland, Proc. Am. Chem. Soc., Div. Environ. Chem. 37 (1997) 39.
[54] L.D. Hoffland, R. Calloway, A. Emery, A.-E.F. Nassar, Proc. Am. Chem. Soc., Div. Environ. Chem. 37 (1997) 41.
[55] A.-E.F. Nassar, S.V. Lucas, C.A. Myler, W.R. Jones, M. Campisano, L.D. Hoffland, Anal. Chem. 70 (1998) 3598.
[56] A.-E.F. Nassar, S.V. Lucas, L.D. Hoffland, Anal. Chem. 71 (1999) 1285.
[57] Z.-H. Meng, L. Qin, Anal. Chim. Acta 435 (2001) 121. [58] W.H. Robins, B.W. Wright, J. Chromatogr. A 680 (1994) 667. [59] R.L. Cheicante, J.R. Stuff, H.D. Durst, J. Cap. Elec. 2 (1995)
157.
[60] R.L. Cheicante, J.R. Stuff, H.D. Durst, J. Chromatogr. A 711 (1995) 347.
[61] T.E. Rosso, P.C. Bossle, J. Chromatogr. A 824 (1998) 125. [62] J. Wang, M. Pumera, G.E. Collins, A. Mulchandani, Anal. Chem.
74 (2002) 6121.
[63] J. Jiang, C.A. Lucy, J. Chromatogr. A 966 (2002) 239. [64] C.L. Copper, G..E. Collins, Electrophoresis 25 (2004) 897. [65] J.E. Melanson, C.A. Boulet, C.A. Lucy, Anal. Chem. 73 (2001)
1809.
[66] Ch.E. Kientz, E.W. Hooijschuur, U.A.Th. Brinkman, J. Microcol. Sep. 9 (1997) 253.
[67] E.W.J. Hooijschuur, Ch.E. Kinetz, U.A.Th. Brinkman, J. Chromatogr. A 928 (2001) 187.
[68] J.-P. Mercier, P. Chaimbault, Ph. Morin, M. Dreux, A. Tambut´e, J. Chromatogr. A 825 (1998) 71.
[69] S.A. Shamsi, N.D. Danieison, Anal. Chem. 67 (1995) 1845. [70] M.G. Cikalo, D.M. Goodall, W. Matthews, J. Chromatogr. A 745
[71] M. Tomita, T. Okuyama, J. Chromatogr. A 571 (1991) 324. [72] T.B. Gain, Appl. Pharmacol. 14 (1969) 515.
[73] A. Farran, S. Ruiz, C. Serra, M. Aguilar, J. Chromatogr. A 737 (1996) 109.
[74] M. Aguilar, A. Farran, C. Serra, M.J. Sepaniak, K.W. Whitaker, J. Chromatogr. A 778 (1997) 201.
[75] X. Guardino, J. Obiols, M.G. Rosell, A. Farran, C. Serra, J. Chro-matogr. A 823 (1998) 91.
[76] C.-M. Liu, C.-S. Tong, Y.-M. Tzeng, J. Liq. Chrom. Rel. Technol. 25 (2002) 3213.
[77] C.-M. Liu, Y.-M. Tzeng, J. Chromatogr. A 809 (1998) 258. [78] W. Zhu, F. Wu, F.M. Raushel, G. Vigh, J. Chromatogr. A 895 (2000)
247.
[79] M. L¨ammerhofer, E. Zarbl, W. Lindner, B.P. Simov, F. Hammer-schmidt, Electrophoresis 22 (2001) 1182.
[80] L. Goodwin, M. Hanna, J.R. Startin, B.J. Keely, D.M. Goodall, An-alyst 127 (2002) 204.
[81] J. Wang, M.P. Chatrathi, A. Mulchandani, W. Chen, Anal. Chem. 73 (2001) 1804.
[82] J. Wang, G. Chen, M.P. Chatrathi, A. Fujishima, D.A. Tryk, D. Shin, Anal. Chem. 75 (2003) 935.
[83] J. Wang, M. Pumera, M.P. Chatrathi, A. Escarpa, M. Musameh, G. Collins, A. Mulchandani, Y. Lin, K. Olsen, Anal. Chem. 74 (2002) 1187.
[84] M. Molina, M. Silva, Electrophoresis 23 (2002) 2333. [85] M. Molina, M. Silva, Electrophoresis 23 (2002) 1096. [86] M. Molina, M. Silva, Electrophoresis 22 (2001) 1175.
[87] Z. El Rassi, in: R. Clement, B. Burk (Eds.), Enviro Analysis, Pro-ceedings of the Second Biennial International Conference on Chemi-cal Measurement and Monitoring of the Environment, Ottawa, 1998, pp. 265–272.
[88] L. Goodwin, M. Hanna, J.R. Startin, B.J. Keely, D.M. Goodall, J. Chromatogr. A 1004 (2003) 107.