White spot syndrome virus protein ICP11:
A histone-binding DNA mimic that disrupts
nucleosome assembly
Hao-Ching Wang
a,b,1, Han-Ching Wang
c,d,1, Tzu-Ping Ko
b,1, Yu-May Lee
a,b, Jiann-Horng Leu
c,e, Chun-Han Ho
a,b,
Wei-Pang Huang
c,e, Chu-Fang Lo
c,e,2, and Andrew H.-J. Wang
a,b,e,2aInstitute of Biochemical Sciences,eDepartment of Life Sciences, andcInstitute of Zoology, National Taiwan University, Taipei 106, Taiwan;bInstitute of Biological Chemistry, Academia Sinica, Taipei 115, Taiwan; anddInstitute of Biotechnology, National Cheng Kung University, Tainan 701, Taiwan Communicated by James C. Wang, Harvard University, Cambridge, MA, November 6, 2008 (received for review April 18, 2008)
White spot syndrome virus (WSSV) is a large (
⬇300 kbp),
double-stranded DNA eukaryotic virus that has caused serious disease in
crustaceans worldwide. ICP11 is the most highly expressed WSSV
nonstructural gene/protein, which strongly suggests its importance in
WSSV infection; but until now, its function has remained obscure. We
show here that ICP11 acts as a DNA mimic. In crystal, ICP11 formed a
polymer of dimers with 2 rows of negatively charged spots that
approximated the duplex arrangement of the phosphate groups in
DNA. Functionally, ICP11 prevented DNA from binding to histone
proteins H2A, H2B, H3, and H2A.x, and in hemocytes from
WSSV-infected shrimp, ICP11 colocalized with histone H3 and
activated-H2A.x. These observations together suggest that ICP11 might
inter-fere with nucleosome assembly and prevent H2A.x from fulfilling its
critical function of repairing DNA double strand breaks. Therefore,
ICP11 possesses a functionality that is unique among the handful of
presently known DNA mimic proteins.
apoptosis兩 DNase enhancer 兩 crystal structure 兩 shrimp aquaculture
T
he white spot syndrome virus (WSSV) is an enveloped DNA
virus that infects crustaceans and threatens shrimp aquaculture
(1–4). The white spot disease caused by WSSV can result in 100%
cumulative mortality in farmed shrimps in 2–10 days. Based on
studies of individual genes and analysis of the complete genome
sequence, the ellipsoid-shaped WSSV has been erected as the type
species of a new genus (Whispovirus) of a new virus family
Nima-viridae (5). Because of the large size of the viral genome (
⬇300 kb)
and the uniqueness of the encoded proteins, WSSV has not yet been
fully characterized.
In previous studies, both transcriptomic (WSSV-infected EST
database and WSSV DNA microarray) and proteomic (2D
elec-trophoresis) approaches identified ICP11 as a highly expressed
WSSV gene/protein (6, 7). The high expression levels of this protein
strongly suggest its importance to WSSV infection; but until now,
its function has remained unknown. In the present article, we
determine the crystal structure of ICP11 and use Far Western
assays and indirect immunofluorescence to investigate its function
and the factors with which it interacts. We found that ICP11 acts as
a DNA mimic that prevents DNA from binding to histone proteins
and, thus, disrupts nucleosome assembly.
Results
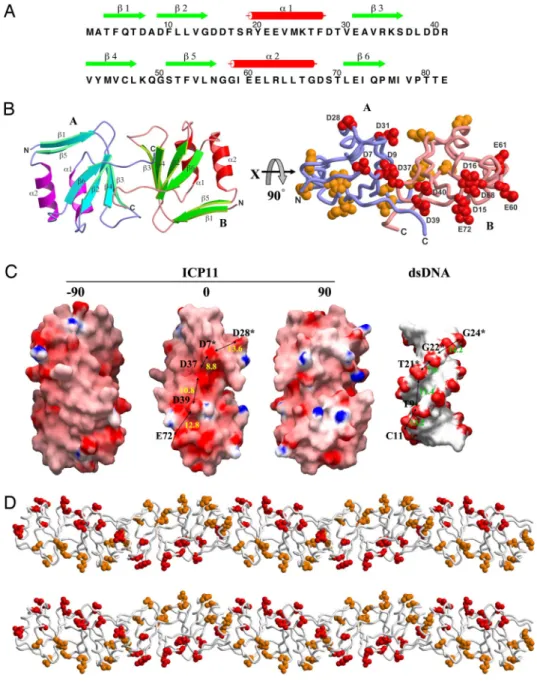
ICP11 Crystal Structure.
The protein model of ICP11 was built
manually into a clear electron density map [
supporting
infor-mation (SI) Fig. S1 A] derived from MAD X-ray diffraction data.
The refined structure contains 2 ICP11 molecules as a dimer per
asymmetric unit (Fig. 1). The refinement statistics are listed in
Table S1. Each monomer consists of a 4-stranded anti-parallel
-sheet, a 2-stranded -ribbon, and 2 flanking ␣-helices (Fig. 1
A and B). In the center of the monomer, 16-aa side chains are
associated into a hydrophobic core. Every secondary structural
element contributes to this core structure; thus, forming a stable
compact globular fold. The nature of the dimer interface is
largely nonpolar, involving Leu-11, Met-44, and Met-76. The
C-terminal segment of the ‘‘A’’ monomer docks to a groove on
the surface of the ‘‘B’’ monomer, where the side chains of Ile-77
and Val-78 (A) interact with those of Leu-38, Val-42, Ile-77 and
Pro-79 (B), and vice versa. For both the A and B monomers, at
the rim of the interface, the charged side chains of Asp-9 and
Glu-31 form salt bridges with Arg-34 of the other monomer.
Near the molecular dyad of the dimer, the 2 side chains of Cys-46
are at a distance of 6.0 Å from each other, too far to form a
disulfide bridge. In the crystal, the dimers are involved in 2 types
of lattice contact. The crystal packing shows layers of filaments
arranged in alternating directions, parallel to the a and b axes.
The layers stack along the c axis. This arrangement resulted in
a large solvent content of 78% in the crystal (Fig. S1B).
The isoelectric point of ICP11 protein is 4.2, indicating that it is
acidic and negatively charged under physiological conditions. The
electrostatic surface of the ICP11 dimer contains patches of
neg-atively charged amino acids arranged into 2 rows, separated by
⬇25–30 Å (Fig. 1B Right and Fig. 1C), which is comparable with the
interphosphate distance in the opposite strands of dsDNA (22–26
Å). The negative charge distribution of ICP11 is shown in Fig. 1C,
with dsDNA (PDB 102d) shown at the same scale for comparison.
The distances between the ICP11 carboxyl groups Asp-28, Asp-7
(monomer A), Asp-37, Asp-39, and Glu-72 (monomer B) and the
dsDNA phosphate groups G24, G22 and T21 (chain B), and T9 and
C11 (chain A) are similar, with an rmsd fit of 2.4 Å. Also, ICP11
forms a helical polymer in crystal (Fig. 1D and
Fig. S1B). These
results suggest that ICP11 mimics the negative charge distribution
of dsDNA. A recent study on another crystal structure of VP9
(another name for ICP11) reported a different dimeric form, in
which these 2 intertwined rows of negatively charged patches were
not seen (Fig. S2) (9).
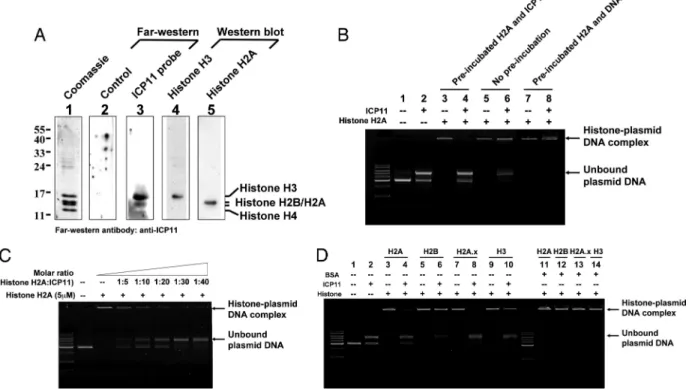
ICP11 Binds To Host Histone Proteins.
If ICP11 is a DNA mimic; then,
at least part of its function would likely involve direct interaction
with host cellular factor(s). In a screening by Far Western assay, 4
candidate shrimp proteins were identified: Kazal-type serine
pro-Author contributions: Hao-Ching Wang, Han-Ching Wang, T.-P.K., C.-F.L., and A.H.-J.W. designed research; Hao-Ching Wang, Han-Ching Wang, T.-P.K., and C.-H.H. performed research; Y.-M.L., J.-H.L., and W.-P.H. contributed new reagents/analytic tools; Hao-Ching Wang, Han-Ching Wang, T.-P.K., C.-F.L., and A.H.-J.W. analyzed data; and Hao-Ching Wang, Han-Ching Wang, T.-P.K., C.-F.L., and A.H.-J.W. wrote the paper.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2ZUG).
1Hao-Ching Wang, Han-Ching Wang, and T.-P.K contributed equally to this work. 2To whom correspondence may be addressed. E-mail: [email protected] or
This article contains supporting information online atwww.pnas.org/cgi/content/full/ 0811233106/DCSupplemental.
teinase inhibitor,
␣-macroglobulin, and 2 histone proteins, histone
H2A and H2B. In the present study, only the histone proteins were
selected for further investigation. A follow-up GST pull-down
experiment confirmed the interaction between histone H2A and
ICP11 (data not shown), and a Far Western assay of purified shrimp
core histone proteins with a ICP11 probe (Fig. 2A) showed that
histone proteins H3 and H2A/H2B both produced strong signals. In
the absence of ICP11, histone H2A formed a histone-DNA
com-plex (Fig. 2B, lanes 3, 5, and 7), and the bound DNA could not be
displaced by the subsequent addition of ICP11 (Fig. 2B, lane 8).
However, if the histone H2A was preincubated (lane 4) or
coin-cubated (lane 6) with ICP11, then some of the plasmid DNA
became unbound. This preincubation effect was dose-dependent
(Fig. 2C). These results suggest that ICP11 will only target unbound
histone H2A, but will not bind with histone that is already in a
histone-DNA complex, consistent with the hypothesis that ICP11
targets the DNA binding site of the histone. Preincubation of other
histone proteins for 1 h with ICP11 also resulted in some unbound
plasmid DNA, and in the cases of histone H2A, H2A.x, and H3, the
amount of histone-plasmid DNA complex was visibly reduced (Fig.
2D, lanes 4, 6, 8, and 10). Neither of the 2 acidic protein negative
controls (BSA, BSA, pI 4.7; soybean trypsin inhibitor, pI 4.55) were
able to prevent histone-DNA binding in this assay (Fig. 2D, lanes
11–14; soybean trypsin inhibitor, data not shown). Also, we tested
whether ICP11 would nonspecifically block DNA from binding to
another basic DNA binding protein. For this test, we used archaeal
nucleoprotein Sso7d (pI 9.66) from Sulfolobus solfataricus to
re-place histone proteins in the reaction (10). Our results showed that
ICP11 did not block DNA from binding to Sso7d (
Fig. S3). These
results suggest that the interactions seen in Fig. 2 B–D were specific
and biologically meaningful, not just the result of a nonspecific
charge-driven interaction.
ICP11 Colocalizes with Histone H3 and
␥histone H2A.x in Vivo.
ICP11
binds to at least 3 of the core histone proteins (H2A, H2B, and H3;
Fig. 2 A), and it also prevents plasmid DNA from forming a complex
with these same histones (Fig. 2D). Therefore, it seems likely that
at least part of the function of this DNA mimic protein is to disrupt
the assembly of the host cell nucleosome. We used
immunofluo-rescence to investigate this possibility in vivo, and found that in
hemocytes of WSSV-infected shrimp, ICP11 and histone H3 were
colocalized in both the nucleus and the cytoplasm (compare the
Fig. 1. The negative charge distribution of ICP11 resembles the B-form DNA helix. (A) The secondary structural elements are shown above the amino-acid sequence, with green arrows and red cylinders representing -strands and ␣-helices. (B) A ribbon diagram of the ICP11 dimer (Left). The strands and heli-ces of monomer A on the right side are colored as in A; those of monomer B on the left side are colored in cyan and magenta. (Right) A dia-gram of the ICP11 dimer, in which each nega-tively-charged amino acid is shown as a cluster of spheres (except for E21 and E22, which are omitted). (C) The molecular surfaces of the ICP11 dimer and dsDNA. Color-coding is by GRASP (8), with red to blue representing the electrostatic potential from⫺15 kBT to⫹15 kBT. The ICP11 dimer is shown in 3 orthogonal views normal to the dyad axis. Labels indicate the ICP11 acidic residues that correspond to the negatively charged spots on the dsDNA. Aster-isks indicate the ICP11 monomer B or the com-plementary strand of the dsDNA. The approx-imate distances between spots are also shown. (D) Distribution of negatively charged amino acids in the ICP11 helical filament. The diagram is shown in stereo and colored as in B.CELL
merged images of Fig. 3 A and B). In most infected cells, histone H3
was mainly located outside the nucleus, suggesting that the histone
H3 failed to translocate into the nucleus to participate in
nucleo-some assembly. A second immunofluorescence study used a
spe-cific antibody to show that high accumulations of Ser 139
phos-phorylated histone H2A.x (
␥histone H2A.x) was also colocalized
Fig. 2. ICP11 binds to host histone proteins. (A) Histone H3 and H2A/H2B bind to ICP11. After separation by SDS/PAGE and Coomassie blue staining, the acid-extracted histone proteins from P. monodon hemocyte (lane 1) were identified by LC-MS/MS spectrometry (Table S2). The 2 positive histone protein bands on the Far Western assay (lane 3) matched the Western blotting signals detected by histone H3 antibody (lane 4) and histone H2A antibody (lane 5). (B) ICP11 blocked DNA-histone binding but did not displace DNA from the DNA-histone complex. Preincubation (1 h) of ICP11 and histone H2A prevented the histone from subsequently binding to DNA, as shown by the large quantity of unbound plasmid DNA (lane 4). When ICP11 and plasmid DNA were introduced at the same time, much less plasmid DNA remained unbound (lane 6). After preincubation (1 h) of the histone H2A and plasmid DNA, the addition of 30g of ICP11 (200 M) was unable to displace the plasmid DNA from the histone H2A-plasmid complex (lane 8). (C) The inhibition of histone H2A-DNA complex formation by ICP11 was dose-dependent. Reactions with a gradient series of ICP11 show dose-dependent prevention of the formation of the histone H2A-plasmid DNA complex. (D) ICP11 blocked DNA from binding to histone proteins. When ICP11 was preincubated with histone proteins, the histone proteins were unable to bind all of the subsequently added plasmid DNA (lanes 4, 6, 8, and 10). ICP11 did not bind to DNA (lane 2), and preincubation with BSA had no effect (lanes 11–14).
Fig. 3. ICP11 colocalizes with histone H3 and␥histone H2A.x in vivo. Control and WSSV-infected (72 hpi) shrimp hemocytes were fixed with paraformaldehyde, stained for ICP11 (FITC, green) and with TRITC (red) for histone H3 (A and B) and␥histone H2A.x (C and D). The yellow areas in merged images show colocalized histone proteins and ICP11.
with ICP11 (Fig. 3 C and D). These results are consistent with our
earlier binding assay, which showed that ICP11 prevented plasmid
DNA from binding to the histone H2A variant H2A.x (Fig. 2D, lane 8).
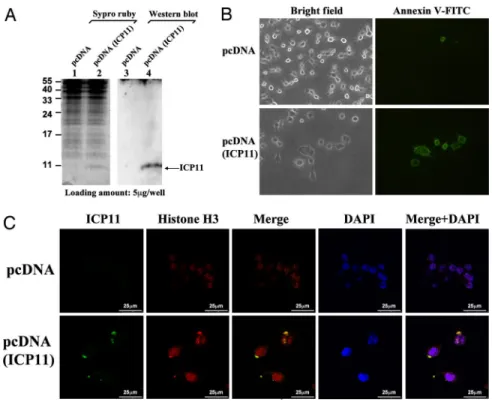
ICP11 Expression in HeLa Cells.
To reconfirm that our findings are
biologically meaningful, to begin to explore the potential
applica-tion of ICP11 to mammalian studies, we expressed the protein in
HeLa cells. At 24 h post transfection, ICP11 expression was
confirmed by Western blotting with a specific anti-ICP11 antibody
(Fig. 4A). ICP11 induced morphological changes in the HeLa cells
(Fig. 4B), and cell attachment was also weaker than in the control
cells. The loss of plasma asymmetry, a key feature of apoptosis, was
detected by FITC-conjugated Annexin V: clear Annexin V signals
were observed in the HeLa cells at 24 h post transfection (Fig. 4B).
Another sign of apoptosis, DNA fragmentation, was also observed
by using a TUNEL assay (data not shown). These results suggest
that ICP11 expression may induce death in HeLa cells. We noted
above that ICP11 was colocalized with histone H3 in shrimp
hemocytes (Fig. 3B), and we found a similar colocalization here: in
the transfected HeLa cells, large amounts of the human histone H3
were seen outside the nucleus and were colocated with ICP11 (Fig.
4C). We infer that ICP11 is the cause of the effect in both cell types,
and further hypothesize that ICP11 also induces nucleosome
dis-order in the HeLa cells. Last, in the surviving HeLa cells at 48 h
after transfection, ICP11 expression levels were low and the histone
H3 was mostly localized in the nucleus (data not shown).
ICP11 Enhances DNase Activity.
In addition to showing that ICP11
could prevent the formation of histone-DNA complex, we also
observed in our earlier experiments that the unbound plasmid
was digested (Fig. 2D, lanes 2, 4, 6, 8, and 10). Improving the
purity of ICP11 reduced the extent of digestion; and in a highly
purified preparation of ICP11-n (
Fig. S4), no DNase activity was
detectable at a protein concentration of 0.2
M. These results
indicated that the DNase activity was most likely due to the
presence of a contaminant. However, it is plausible that ICP11
may stimulate the activity of a DNase present in trace amounts.
Fig. 5A indicates that 1
M of ICP11-n increased the activity of
bovine DNase I by
⬇1.6-fold, as assayed by monitoring the rate
of absorbance increase of herring genomic DNA at 260 nm (Fig.
S5). Fig. 5B further shows that ICP11-n also increased the DNase
activity of protein extracted from shrimp stomach tissue, the
main tissue expressing ICP11. However, the functional
signifi-cance of this stimulation is presently unclear.
Discussion
DNA mimic proteins have previously been reported in
pro-karyotes (Haemophilus influenzae, Escherichia coli,
Mycobac-Fig. 4. ICP11 expression induced HeLa cell death and translocation of human histone H3 protein. (A) The ex-pression of ICP11 in HeLa cells was confirmed by Western blotting. (B) ICP11 expression induced morphological changes and death of the HeLa cells. Annexin V (FITC-conjugated, green) was used to detect cell death. (C) ICP11 colocalizes with human histone H3 in HeLa cells at 24 h post transfection. Control and transfected HeLa cells were fixed with paraformaldehyde, and stained for ICP11 (FITC, green) and for histone H3 (TRITC, red). The yellow areas in the merged images indicate colocaliza-tion of the histone protein and ICP11.Fig. 5. DNase activity was increased by ICP11. (A) Reactions (37 °C, 1 h) with different concentrations (0, 0.05, 0.1, and 0.2M)ofICP11showadose-dependentincrease of the DNA digestion activity of bovine DNase I (0, 10, 25, and 50 nM). (B) The effect of ICP11 on the DNase activity of protein extracted from shrimp stomach.
CELL
terium tuberculosis), prokaryotic viruses (Bacillus subtilis
bac-teriophage PBS2, bacbac-teriophage T7), and a eukar yote
(Drosophila melanogaster). They usually function through
sim-ple competitive inhibition, by occupying the DNA binding sites
of DNA binding proteins; thus, denying their access to the
DNA (11). They often target DNA regulatory proteins such as
restriction enzymes, DNA gyrase, TATA-box binding protein,
and prokaryotic nucleoid-associated protein. The DNA mimic
proteins from the bacteriophages act to knock out a defense
mechanism of the host bacterium. For example, B. subtilis
bacteriophage PBS2 has uracil instead of thymine in its
genome, and the phage uses the DNA mimic protein uracil
glycosylase inhibitor (UGI) to bind to host uracil-DNA
gly-cosylase, so that it is prevented from cleaving this uracil base
from the DNA (12). Another example is the T7 bacteriophage
Ocr protein, which dimerizes to form a long DNA-shaped rod
with acid groups arranged on the surface, and it binds to type
I restriction enzymes to block their protective function (13). In
contrast, the H. influenzae DNA mimic protein HI1450, which
has homologs in 10 different bacterial species, binds to the
DNA binding site of the nucleoid-associated protein HU-␣,
and it may have a role in controlling the chromosome-like
structure in prokaryotes (14, 15).
Compared with these previously identified DNA mimic
proteins, WSSV ICP11 is unique in that it binds directly to the
DNA binding site of histone proteins (Fig. 2D). Our data also
suggest this binding is specific, at least in so far as ICP11 does
not prevent Sso7d from binding to DNA (
Fig. S3). Our
colocalization data further suggest that ICP11 binds to the
nucleosome-forming histone H3 protein (Fig. 3 A and B, and
Fig. 4C), as well as to
␥histone H2A.x (Figs. 3 C and D).
Histone H2A.x represents 2–25% of the total histone H2A
expressed by cells, and it is involved in the maintenance of
genomic stability. Histone H2A.x is rapidly phosphorylated at
Ser-139 by ATM kinase in response to DNA damage. Once
activated, it accumulates in the nucleus at the sites of double
strand breaks (DSBs) and has a critical role in DSB repair
(16 –19). The high accumulation of
␥histone H2A.x after
WSSV infection (Fig. 3D) is presumably in response to the
DNA damage. The fact that most of the
␥histone H2A.x was
primarily located in the cytoplasm is further evidence that
ICP11 has acted to block its DSB repair function.
A model for this direct binding suggests that the negatively
charged carboxyl groups of Asp-28, Asp-37, Asp-39, and Glu-72 are
close to the positively charged amino acids on the DNA binding
region of the histone H2A/H2B heterodimer (Fig. S6). This direct
binding is unusual and it has implications for the functionality of
ICP11. In contrast to ICP11, most other pathogens that target
nucleosomes do so indirectly. For example, the HSV-1 tegument
protein VP22 interacts with template-activating factor 1 (TAF-1, a
histone chaperone) to inhibit nucleosome assembly, whereas
pro-teins from other viruses use strategies such as direct or indirect
binding to the histone-modifying enzymes to affect the
posttrans-lational modification of histone proteins (20–22). Conversely, our
HeLa cell data showed not only that ICP11 was colocalized with
histone H3, but also provided evidence of apoptosis, as well as
detecting human histone H3 outside the nucleus. Clearly, these
results suggest that ICP11 has a biologically crucial role. One
hypothesis is that ICP11 directly induces nucleosome disorder; thus,
leading to cell death. Another possibility is that ICP11 may cause
cell death by means of a more indirect mechanism: it has recently
been shown that nucleosome destabilization is very important for
the epigenetic regulation of gene expression (23), and by binding to
histone in the cytoplasm, ICP11 may impair this regulatory
mech-anism by preventing the bound histone from translocating to the
nucleus. In this hypothesis, cell death is only an incidental
conse-quence of ICP11 being expressed in the cell.
In summary, we conclude that an abundance of ICP11 in the
host cell deprives the cell nucleus of histone proteins, makes
the host DNA in cell vulnerable to damage, and finally leads
to a disruption of the genetic machinery in the nucleus. The
HeLa cell studies also suggest the existence of an
ICP11-histone mechanism that leads to cell death. Because ICP11-histone
proteins are highly conserved (the histone H3 amino acid
sequences of shrimp and human are 100% identical), these
findings suggest that ICP11 has potential application in
mam-malian studies (e.g., as an apoptotic agent).
Materials and Methods
Preparation and Purification of Recombinant ICP11. The full length ICP11 gene
(residues 1– 82) was amplified by PCR and cloned into pET 21b expression vector (Novagen). The recombinant ICP11 protein contained a C-terminal His6-tag (90
aa). After induction with IPTG, it was expressed by E. coli BL21 (DE3) cells at 20 °C for 16 h. Soluble ICP11 was purified by immobilized metal-ion chromatography with a Ni-NTA column, followed by gel filtration by using Superdex 200pg (Amersham Biosciences). For the histone-DNA binding and DNase assays, non-His-tagged ICP11 was used. To produce this protein, the ICP11 gene was cloned into pET16b expression vector. Similar procedures were used to produce another recombinant ICP11 protein that contained a N-terminal His10-tag. The His10-tag
was then removed by FactorXa cleavage (Amersham Biosciences).
For the DNase enhancer assays, newly prepared ICP11 with improved purity (ICP11-n) was used. The ICP11-n protein was produced by denaturing the His-tagged ICP11 by using 6M GuHCl, repurification by using a NiNTA column, and protein refolding with gel filtration, followed by removal of the His-tag by factor Xa cleavage. The resulting ICP11-n is very pure, because no other band could be seen even when stained with the highly-sensitive sypro ruby (Fig. S4).
Recombinant ICP11 Crystallization and Data Collection.
Selenomethionine-labeled C-terminal His6-tagged ICP11 (SeMet-ICP11) was produced by
re-placing the LB medium with selenium-Met minimal medium (24). Purified seMet-ICP11 was dialyzed overnight at 4 °C against crystallization buffer (100 mM NaCl/5 mM DTT/50 mM Tris pH 8.0), and the purified seMet-ICP11 was concentrated to 80 mg/mL. For crystallization, 2 L of the ICP11 solution was mixed with 2L of a reservoir containing 0.2 M sodium acetate trihydrate and 2.2 M ammonium sulfate (pH 6.5) as a precipitant, and equilibrated with the reservoir by the sitting drop method. Before flash-cooling, crystals were rinsed with a cryoprotectant solution of 30% glycerol and 70% reservoir. Both native and 3-wavelength multiwavelength anom-alous dispersion (MAD) X-ray diffraction data from the ICP11 crystals were collected on beamline BL13B1 at the National Synchrotron Radiation Re-search Center (NSRRC) in Hsinchu, Taiwan. Data were processed by using HKL2000 (25). The space group of the seMet-ICP11 crystals was P4x2x2, with
typical unit cell dimensions of a⫽ b ⫽ 91 Å and c ⫽ 98 Å.
Structure Determination and Refinement. The ICP11 structure was determined
by using the MAD phasing method and the programs SOLVE and RESOLVE (26, 27). Three datasets in the range of 15- to 3.1-Å resolution collected at the wavelengths of 0.9790 Å (edge), 0.9788 Å (peak), and 0.9636 Å (high-energy remote) were used. The space group was identified to be P41212,, and 5 Se sites
were located for an asymmetric unit. After solvent flattening and phase exten-sion to 2.72 Å by the Crystallography and NMR System (CNS) (28), by using the native dataset, the resulting electron density map was clear (Fig. S1 A). This map allowed manual building of the model with the program O (29), and revealed that each asymmetric unit contained 2 ICP11 molecules. Therefore, noncrystal-lographic symmetry restraints were imposed on equivalent parts of the 2 mono-mers, and the model was refined by using CNS. The rms deviations between the 2 monomers, excluding the C-terminal segment after Pro-79, were 0.24 Å for 308 pairs of the backbone atoms and 1.34 Å for 296 pairs of the side-chain atoms. The molecular surface areas were 5480 Å2and 5280 Å2for monomer A and B when
they were separated. On formation of the dimer, the interface covers 910 Å2and
920 Å2on monomers A and B, respectively.
Statistics for the MAD phasing and refinement are shown inTable S1. The CCP4 package (30), ALSCRIPT (31), MOLSCRIPT (32), RASTER3D (33), and GRASP (8) were used for the structural analyses to produce the figures.
Far Western Assay To Identify Proteins That Bind To ICP11. For the Far Western
assay (34), the shrimp core histone proteins were extracted by an acid extraction method (35). Briefly, after centrifugation of hemolymph at 800⫻ g, hemocyte pellets were collected and washed twice with cold PBS
buffer. The hemocyte cells were resuspended in 0.2 N HCl and incubated at 4 °C overnight. After centrifugation (5000⫻ g, 30 min), the acid-soluble histone proteins in the supernatant were dialyzed overnight at 4 °C against deionised water. The extracted shrimp histone proteins (5g) were sepa-rated by 17.5% SDS/PAGE and transferred to a nitrocellulose membrane. The membrane was incubated overnight at 4 °C with purified C-terminal His6-tagged ICP11 protein (20g/mL) in binding buffer (10 mM Tris䡠HCl, pH 6.5/5 mM CaCl2/10 mM MgCl2), containing 0.02% skim milk and 1% Triton X-100. After washing, the membrane-bound ICP11 proteins were detected by anti-ICP11 antibody. LC-MS/MS and Western blotting were used to identify the positive signals that appeared in this assay.
Histone-DNA Binding Assays. For each reaction, 1g of histone H2A or H2B from calf thymus (Roche) or recombinant human histone H2A.x (1g) or H3 (20g) (Millipore) was mixed with 30 g of non-His-tagged ICP11. The reaction mixtures were adjusted to a volume of 14L in binding buffer (20 mM Tris, pH 8.0/200 mM NaCl), and preincubation (if any) proceeded at 37 °C for 1 h. Last, 1L of binding buffer containing 300 ng of plasmid DNA (pGEX5T-1) was added, and incubation continued for 1 h. The reaction products were run on a 0.8% agarose gel and stained with ethidium bromide. For 1 of the control protocols, 30g of ICP11 was added to 300 ng of plasmid DNA without histone proteins, and for the other controls, 30 g of BSA was used in place of ICP11.
Indirect Immunofluorescence Assay of WSSV ICP11 in Shrimp Hemocytes.
He-molymph was collected from healthy Penaeus monodon shrimp and from WSSV-infected shrimp at 72 hpi by using a syringe that contained cold MAS (modified Alsever solution) (36). Hemocytes were placed on glass coverslips, washed with PBS, and fixed in 4% paraformadehyde for 10 min at 4 °C. After acetone treat-ment (3 min on ice), the hemocytes were incubated with 3% normal goat serum and 5% BSA (Sigma) for 16 h at 4 °C. After this blocking, the hemocytes were incubated for 3– 4 h at room temperature with a 1:200 dilution of ICP11-specific mouse antiserum and either histone H3 or Ser-139 phosphorylated histone H2A.x rabbit polyclonal antibody (Millipore). The cells were then washed twice (15 min each) in PBST (containing 0.3% Tween-20) and incubated for 1 h with a 1:200 dilution of FITC-conjugated polyclonal goat anti-mouse IgG and TRITC-conjugated polyclonal goat anti-rabbit IgG. After further extensive washing with
PBST, the cells were mounted and viewed by using a Leica TCS SP5 Confocal Spectral Microscope Imaging System. DAPI was used to counterstain the nucleus.
HeLa Cell Transfection. HeLa cells (American Tissue Cell Collection) were cultured
in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and main-tained at 37 °C in a humidified atmosphere with 5% CO2. The ICP11 expression vector pcDNA(ICP11) was constructed by inserting the full-length ICP11 gene into pcDNA3.1⫹ myc-his vector (Invitrogen). Transient transfections were carried out with Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen).
Detection of Cell Apoptosis (Annexin V). After transfection of the HeLa cells with
pcDNA (ICP11) or the empty vector (pcDNA3.1⫹ myc-his), an Annexin V-FITC fluorescence microscopy kit (BD Bioscience) was used to detect apoptosis. At 24-h post transfection, the cells were washed twice in PBS and incubated for 15 min with FITC conjugated Annexin V in binding buffer (10 mM Hepes, pH 7.4/140 mM NaCl/2.5 mM CaCl2). After extensive washing in binding buffer, the cells were observed by fluorescence microscopy.
DNase Enhancer Assays. DNase activity was determined by using an assay
de-scribed previously (37), with minor modifications. Plasmid DNA (pGEX5T-1) was purified and used as the DNA substrate. The plasmid DNA (200 ng) was incubated with different concentrations of bovine DNase I (0, 10, 25, and 50 nM, Sigma) and ICP11-n (0, 0.05, 0.1, and 0.2M) in 20 l of reaction buffer (20 mM Tris䡠HCl, pH 8.0/200 mM NaCl) with 1 mM MgCl2at 37 °C. To test whether ICP11-n also increases the DNase activity of shrimp protein, the bovine DNase I was replaced by 250 ng of protein extracted from shrimp stomach. After 1 h of incubation, the total reaction solutions were analyzed on a 0.8% agarose gel and stained with ethidium bromide.
ACKNOWLEDGMENTS. We thank Paul Barlow for helpful comments on the
manuscript. X-ray diffraction data were collected on beamline BL13B1 at the National Synchrotron Radiation Research Center in Hsinchu, Taiwan. Proteomic mass spectrometry analyses were performed by the Core Facil-ities for Proteomics Research located at the Institute of Biological Chem-istry, Academia Sinica. This work was supported by National Science Coun-cil Grants NSC96 –2317-B-002-020, NSC95–3112-B-001-018, and NSC96 – 3112-B-001-014.
1. Wang CH, et al. (1995) Purification and genomic analysis of baculovirus associated with white spot syndrome (WSBV) of Penaeus monodon. Dis Aquat Organ 23:239 –242. 2. Lo CF, et al. (1997) Detection and tissue tropism of white spot syndrome baculovirus
(WSBV) in captured brooders of Penaeus monodon with a special emphasis on reproduc-tive organs. Dis Aquat Organ 30:53–72.
3. Yang F, et al. (2001) Complete genome sequence of the shrimp white spot bacilliform virus.
J Virol 75:11811–11820.
4. Tsai JM, et al. (2004) Genomic and proteomic analysis of thirty-nine structural proteins of shrimp white spot syndrome virus. J Virol 78:11360 –11370.
5. Vlak JM, et al. (2004) Nimaviridae (VIIIth Report of the International Committee on Taxonomy of Viruses), eds Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (Elsevier, Amsterdam), pp 187.
6. Wang HC, Wang HC, Kou GH, Lo CF, Huang WP (2007) Identification of icp11, the most highly expressed gene of shrimp white spot syndrome virus (WSSV). Dis Aquat Organ 74:179–189. 7. Wang HC, et al. (2007) Protein expression profiling of the shrimp cellular response to white
spot syndrome virus infection. Dev Comp Immunol 31:672– 686.
8. Nicholls A, Honig B (1992) GRASP: A Manual (Columbia Univ Press, New York, NY), 1st Ed. 9. Liu Y, Wu J, Song J, Sivaraman J, Hew CL (2006) Identification of a novel nonstructural protein, VP9, from white spot syndrome virus: Its structure reveals a ferredoxin fold with specific metal binding sites. J Virol 80:10419 –10427.
10. Gao YG, et al. (1998) The crystal structure of the hyperthermophile chromosomal Protein Sso7d bound to DNA. Nat Struct Biol 5:782–786.
11. Dryden DT (2006) DNA mimicry by proteins and the control of enzymatic activity on DNA.
Trends Biotechnol 4:378 –382.
12. Putnam CD, Tainer JA (2005) Protein mimicry of DNA and pathway regulation. DNA Repair 4:1410 –1420.
13. Walkinshaw MD, et al. (2002) Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol Cell 9:187–194.
14. Parsons LM, Yeh DC, Orban J (2004) Solution structure of the highly acidic protein HI1450 from
Haemophilus influenzae, a putative double-stranded DNA mimic. Proteins 54:375–383.
15. Parsons LM, Liu F, Orban J (2005) HU-alpha binds to the putative double-stranded DNA mimic HI1450 from Haemophilus influenzae. Protein Sci 14:1684 –1687.
16. Fillingham J, Keogh MC, Krogan NJ (2006) GammaH2AX and its role in DNA double-strand break repair. Biochem Cell Biol 84:568 –577.
17. Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004) H2AX: The histone guardian of the genome. DNA Repair 3:959 –967.
18. Downs JA, Lowndes NF, Jackson SP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408:1001–1004.
19. Foster ER, Downs JA (2005) Histone H2A phosphorylation in DNA double-strand break repair. FEBS J 272:3231–3240.
20. Ren X, Harms JS, Splitter GA (2001) Bovine herpesvirus 1 tegument protein VP22 interacts with histones, and the carboxyl terminus of VP22 is required for nuclear localization. J Virol 75:8251– 8258.
21. Pumfery A, et al. (2003) Chromatin remodeling and modification during HIV-1 Tat-activated transcription. Curr HIV Res 1:343–362.
22. Hamon MA, et al. (2007) Histone modifications induced by a family of bacterial toxins. Proc
Natl Acad Sci USA 14:13467–13472.
23. Henikoff S (2008) Nucleosome destabilization in the epigenetic regulation of gene ex-pression. Nat Rev Genet 9:15–26.
24. Guerrero SA, et al. (2001) Production of selenomethionyl-labelled proteins using simpli-fied culture conditions and generally applicable host/vector systems. Appl Microbiol
Biotechnol 56:718 –723.
25. Otwinowski Z, Minor W (1997) Processing of X-ray Diffraction Data Collected in Oscillation
Mode. Methods in Enzymology: Macromolecular Crystallography, part A, eds Carter CW,
Jr, Sweet RM (Academic, New York), Vol 276, pp 307.
26. Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta
Crystallogr 55:849 – 861.
27. Terwilliger TC (2000) Maximum likelihood density modification. Acta Crystallogr 56:965– 972.
28. Bru¨nger AT, et al. (1998) Crystallography & NMR System: A new software suite for structure determination. Acta Crystallogr 54:905–921.
29. Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta
Crystal-logr 47:110 –119.
30. CCP4 (1994) Collaborative Computational Project Number 4. Acta Crystallogr 50:760 – 763.
31. Barton GJ (1993) Alscript-a tool to format multiple sequence alignments. Protein Eng 6:37– 40.
32. Kraulis PJ (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structure. J Appl Crystallogr 24:946 –950.
33. Merritt EA, Murphy MEP (1994) Raster3D version 2.0: A program for photorealistic mo-lecular graphics. Acta Crystallogr 50:869 – 873.
34. Sritunyalucksana K, Wannapapho W, Lo CF, Flegel TW (2006) PmRab7 is a VP28-binding protein involved in white spot syndrome virus infection in shrimp. J Virol 80:10734 – 10742.
35. Fujitani H, Holoubek V (1974) Recovery of histones by acid extraction from chromatin and from artificial DNA-histone complex. Tex Rep Biol Med 32:461– 478.
36. Lin ST, et al. (2002) Ribonucleotide reductase of shrimp white spot syndrome virus (WSSV): Expression and enzymatic activity in a baculovirus/insect cell system and WSSV-infected shrimp. Virology 304:282–290.
37. Hou MH, Wang AHJ (2005) Mithramycin forms a stable dimeric complex by chelating with Fe(II): DNA-interacting characteristics, cellular permeation and cytotoxicity. Nucleic Acids
Res 33:1352–1361.
CELL