國
立
交
通
大
學
生物醫學研究所
碩
士
論
文
幽門螺旋桿菌之熱休克蛋白 60 誘導前發炎性細胞激素的分泌但卻
抑制單核球細胞之活性
Heat Shock Protein 60 of Helicobacter pylori Induces Proinflammatory
Cytokines Secretion but Diminishes Monocyte Activation
研 究 生:林依穎
指導教授:廖光文 教授
幽門螺旋桿菌之熱休克蛋白 60 誘導前發炎性細胞激素的分泌但卻抑制單核球細胞之活 性
Heat Shock Protein 60 of Helicobacter pylori Induces Proinflammatory Cytokines Secretion but Diminishes Monocyte Activation
研 究 生:林依穎 Student:Yi-Yin Lin
指導教授:廖光文 Advisor:
Kuang-Wen Liao, Ph.D.國 立 交 通 大 學
生物醫學研究所
碩 士 論 文
A Thesis
Submitted to Institude of Biomedical Science College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in
July 2008
Hsinchu, Taiwan, Republic of China
幽門螺旋桿菌之熱休克蛋白 60 誘導前發炎性細胞激素的分泌但卻
抑制單核球細胞之活性
研 究 生:林依穎
指導教授:廖光文
國 立 交 通 大 學
生物醫學研究所
碩 士 論 文
幽門螺旋桿菌之熱休克蛋白 60 在之前的文獻中被發現它可以誘導胃上皮細胞及人 類單核球細胞產生介白素-8 (IL-8),以及巨噬細胞產生介白素-6。在此研究中,我們將 幽門螺旋桿菌之熱休克蛋白60 (rHpHSP60) 與人類單核細胞株 THP-1 共同培養,發現除 了介白素-8 以及介白素-6 之外,腫瘤壞死因子-甲型以及介白素-1 乙型都會被 THP-1 產 生。腫瘤壞死因子-甲型與介白素 1 乙型與炎症反應的開始有很大的關連。其他生物的 熱 休 克 蛋 白 雖 然 也 會 誘 導 前 發 炎 細 胞 激 素(pro-inflammatory cytokines)的 產 生 , 但 pro-inflammatory cytokines 產生的時間卻不盡相同。在此篇研究中發現,腫瘤壞死因子-甲型在受到rHpHSP60 刺激後兩小時就開始顯著的產生而在 4 小時產量最高;而介白素 -1 乙型,介白素-6,介白素-8 會隨著時間延長而小幅增加,直到 24 小時才大量的表現。 腫瘤壞死因子-甲型在這些細胞激素中最早產,因此對單核球細胞的活化最具影響力。 我們藉由偵測單核球細胞的吞噬能力以及細胞表面抗原之表現來觀察細胞的活化。結果 顯示,細胞的吞噬活性明顯下降而且主要組織相容性複合體II (MHCII) 的表現也顯著性 的下降。而共同刺激分子CD40、CD80、CD86 會顯著性的升高,而主要組織相容性複 合體I(MHCI)表現則無改變。我們更進一步用商業化之人類腫瘤壞死因子-甲型觀察其對 單核球細胞活化的影響,結果顯示隨著商業化之人類腫瘤壞死因子-甲型的增加,單核 球細胞的吞噬能力竟然減弱了,而共同刺激分子 CD40 的表現則如我們預期的增加了。 在胃部慢性發炎的病人中發現轉化生長因子–β1 (TGF-β1)會大量表現;為了模擬胃部慢 性發炎的環境,我們將商業化之人類腫瘤壞死因子-甲型與轉化生長因子–β1同時刺激 單核球細胞,結果發現腫瘤壞死因子-甲型可協助轉化生長因子–β1造成的抑制作用, 轉化生長因子–β1則更進一步抑制由腫瘤壞死因子-甲型引起的單核球 CD40 之表現,阻 礙其成熟與分化。根據以上的結果顯示,幽門螺旋桿菌之熱休克蛋白 60 會刺激單核球 細胞產生介白素-1 乙型,介白素-6,介白素-8 以及腫瘤壞死因子-甲型;其中,腫瘤壞 死因子-甲型最早產生,但單核球細胞之活性卻仍受到抑制。在我們的研究中,腫瘤壞 死因子-甲型並非扮演活化性的角色反而是抑制單核球細胞的活性。在慢性發炎反應中, 腫瘤壞死因子-甲型結合轉化生長因子–β1 對於單核球細胞的活化造成更嚴重的影響。中華民國九十七年七月
Heat Shock Protein 60 of Helicobacter pylori Induces Proinflammatory
Cytokines Secretion but Diminishes Monocyte Activation
Student: Yi-Yin Lin Advisor: Dr. Kuang-Wen Liao Institute of Biomedical Science
National Chiao Tung University ABSTRACT
Heat shock protein 60 of Helicobacter pylori has been found that it can induce interleukin-8 (IL-8) secretion in human monocytic cells and gastric epithelium cells. In this study, we further found that the IL-6, IL-8, TNF-α and IL-1β were induced in THP-1 cells after H. pylori HSP60 stimulation. The kinetic of cytokine expression showed that TNF-α was earliest secreted at 2 h, and reached a maximum at 4 h. This result consisted with the kinetic of TNF-α mRNA expression analyzed by quantitative real-time PCR. TNF-α may have a great effect on THP-1 cells activation. Dissimilarly, IL-1β, IL-6, and IL-8 were later produced by THP-1 cells. We further examined THP-1 cells activation by detecting its enodocytosis activity and surface marker expression. Surprisingly, the endocytosis ability of THP-1 cells was weakened after rHpHSP60 stimulation. However, the co-stimulatory molecules (CD40, CD80, and CD86) were up-regulated, whereas MHC class II which plays a central in presenting the foreign antigen to T helper cells was significantly down-regulated. MHC I expression was not influenced by rHpHSP60. Interestingly, the rhTNF-α mimicry experiments indicated that the endocytosis activity of THP-1 cells was diminished by rhTNF-α in a does-dependent manner. However, it can promote CD40 expression on THP-1 cells surface. To mimic the chronic inflammation area of H. pylori-infected patients, rhTNF-α and TGF-β1 were used to treat THP-1 cells. The inhibitory effect on endocytotic activity of THP-1 cells was observed by rhTNF-α and TGF-β1 synergy treatment. TNF-α seemed to synergize with TGF-β1 to decrease the engulf ability of cells. However TGF-β1 further inhibited TNF-α-mediated CD40 expression. This study suggested rHpHSP60 induced TNF-α, IL-1β, IL-6, and IL-8 secretion in THP-1 cells. Among these cytokines, TNF-α was earliest secreted. Even through the endocytosis ability of THP-1 cells was inhibited and the MHC class II was significantly decreased after rHpHSP60 stimulation. The role of TNF-α in our study was not an “effector” on THP-1 cells activation but diminished its activity. In the chronic inflammation, the inhibition effect of TNF-α combining with TGF-β1 on monocytes activation was more critical.
Acknowledgement
我最要特別感謝的是我的指導教授廖光文博士,當初願意收留對科學一無所知的我 成為碩士班學生,在科學上,老師教導我運用強力的邏輯概念及創新的思維去設計實驗, 在學術上,老師利用生動有趣的教學方式,讓我可以快速吸收學習,在待人處世上,老 師讓我懂得要時時懷著感恩的心,去感謝給個幫助過我的人,並傳授我博班八寶,讓我 在未來在博士生涯闖蕩中,有了努力的方向,這一切我都謹記在心。再來要特別的感謝 在暑假中抽空前來幫我口試的蔡女滿老師、吳彰哲老師、林志生老師、袁俊傑老師,謝 謝你們幫我批改論文及給予我寶貴的意見,讓我更多方面的去思考我的實驗,讓我對我 的實驗有更新更深層的領悟,也讓我的論文更加完整,謝謝你們。 接下來要感謝我們這一組的大 leader 靜宜學姊,謝謝學姊在這兩年中教導我各種 實驗的技術,希望有朝一日可以像學姊一樣,樣樣精通,除此之外,謝謝你常常聽我 complain,真是我訴苦的好聽眾、購物的好團員、吃東西的好夥伴,不知道以後會不會 遇到如此志同道合的好 partner 啊!美麗氣質兼備的于鈴學姊,你坐在我旁邊也一年多 了,相當初我們也有一起努力奮戰過一小段時光,雖然你後來跑去當 LPPC 的精神領袖, 那段日子也真是有笑又有淚,之後有 BABY 要叫我回來看看嘿。白爛的 DG 學長,感覺你 越來越英明啦,雖然常常被你欺負,就先放你一馬了,Leader 真的不好當,加油加油! 謝謝我的黑師父 James,從一開始很沒耐心到後來不得不有耐心的指導我,從學術上到 生活上對我的照顧與愛護,以後還有很漫長的時間可以好好感謝。我的好酒伴李其(吉) 翰,真的要好好感謝你才行,謝謝你周末有義氣的幫我做實驗,心情不好時陪我喝個幾 杯,聽我無止盡的 murmur,真是好麻吉,祝你跟你婆兒快快生個兒子,完成打棒球的夢 想!我的好姊妹 chenyu,這兩年多虧有你,讓我在煩悶的生活中增加許多樂趣,也幸好 你永續經營你的事業,拯救即將邁向人格分裂的我。碩二四大金釵何姵姵、筑婷、小溫、 小莉以及交大第一型男阿伯,你們讓實驗室無時無刻都充滿了歡樂,感覺你們的論文題 目都相當的有趣新潮,而且很有挑戰性,加油加油,可以的話,到時候回來聽你們口試。 馬馬兒也謝謝你陪我度過兩年的光陰,想當初你剛從大學部來實驗室,我剛考到碩士班, 也算是一路走來的好夥伴,之後的日子你就好好加油啦!可愛的維瞳,從你進來的那一 刻,感覺你就會是個有為的青年,幫了我們整組很多忙,壓力太大要說出來呀,哈哈, 不要爆炸了。新進人員靜敏、小葳跟你們相處的時間不算長,但是可以感覺到你們都是 滿有趣的人,你們的碩班生涯才正要開始,祝你們未來一切都順利,特別祝靜敏桃花旺 旺。大好人上知學長,多虧有你實驗室現在才沒被昆蟲侵襲,你真是貢獻良多,以後咱 們三人要繼續努力了,加油加油,祝你快交女朋友!還有已經離開實驗室很久的阿晟、 阿 pei、子慧、廖 benwa,你們是我剛進實驗室最先認識的朋友,謝謝你們在我剛進入 實驗室時,給我很多的幫忙,感謝感謝。 最後要好好感謝的是我可愛的家人,謝謝我的爸爸媽媽無條件的支持我,鼓勵我, 沒給我壓力,回家都吃到阿嬤的愛心燉補,還有妹妹、弟弟耍寶搞笑,每次回家都是充 電的時刻,謝謝你們了。Contents
Chapter 1 Introduction
………. 11-1 Helicobacter pylori... 1
1-1.1 The morphology of Helicobacter pylori………. 2
1-1.2 H. pylori-associated diseases……….. 2
1-1.2.1 Acute gastritis... 2
1-1.2.2 Chronic gastritis... 3
1-1.2.3 Peptic ulcer disease……….... 3
1-1.2.4 Atrophic gastritis, intestinal metaplasia, and gastric cancer……….. 3
1-1.2.5 Gastric MALT lymphoma………. 4
1-1.3 The virulence factors resulting in H. pylori colonization and pathogenicity…. 4 1-1.3.1 The cag PAI and Cag A protein... 4
1-1.3.2 Vac A………. 5
1-1.3.3 Urease……… 5
1-1.3.4 NAP………... 6
1-1.3.5 Arginase………. 6
1-1.3.6 Cell wall and lipopolysaccharide………... 6
1-1.3.7 Heat shock protein60………. 7
1-1.4 Interactions between H. pylori and immune cells……….. 7
1-1.5 Immune activation and cell damage by H. pylori infection………... 10
1-1.6 Immune subversion by H. pylori……… 11
1-1.7 Chronic infection……….... 12
1-2 Human innate immunity……… 13
1-2.1 The characterizations of innate immune cells……… 13
1-2.1.1 Monocytic cells... 14
1-2.1.1.1 Human monocyte subsets………. 14
1-2.1.1.2 Monocytes differentiation……… 14 1-2.1.1.3 Bacterial infection………... 14 Abstract in Chinese i Abstract ii Acknowledgements iii Contents iv Abbreviations vii
List of table viii
1-2.1.2 Macrophage………... 15
1-2.1.3 Neutrophils granulocytes………... 15
1-2.1.4 Dendritic cells……… 15
1-2.2 Toll-like receptor (TLRs)-mediated immune activation………. 16
1-2.3 Phagocytosis of pathogens……….. 17
1-2.4 Cytokines and chemokines production………... 18
1-2.4.1 Interleukin-1β... 19
1-2.4.2 Interleukine-6………. 19
1-2.4.3 Interleukine-8………. 20
1-2.4.4 Tumor necrosis factor-α... 20
1-2.4.4.1 TNF-α associated disease………. 21
1-2.4.5 Transforming growth factor-β1………. 23
1-2.4.5.1 TGF-β regulation of infectious diseases………... 24
1-3 Heat shock proteins……… 26
1-3.1 Human heat shock proteins………. 26
1-3.1.1 Human heat shock protein 60……… 26
1-3.1.2 Human heat shock protein 70……… 27
1-3.2 Bacterial heat shock proteins……….. 28
1-3.2.1 Escherichia coli heat shock proteins: GroEL, DANK, and GroES……... 28
1-3.2.2 Chlamydia heat shock protein 60……….. 29
1-3.2.3 Mycobacterial heat shock protein 65………. 29
1-3.2.4 Helicobacter pylori heat shock protein 60………. 29
Chapter 2 Materials and Methods
………... 312-1 Materials……… 31 2-1.1 Reagent………... 31 2-1.2 Antibody………. 31 2-1.3 Kit………... 31 2-1.4 Instrument………... 32 2-1.5 Bacteria………... 32 2-1.6 Cell line………... 32 2-2 Methods………. 33
2-2.1 Recombinant DNA techniques………... 33
2-2.2 Transformation………... 33
2-2.3 Expression and purification of H. pylori hsp 60 gene in Escherichia coli……. 34
2-2.4 Protein purification………. 34
2-2.5 Cell cultures……… 35
2-2.6 Detection of cytokines production in the THP-1 cells using ELISA…………... 36
2-2.8 Surface marker detection on THP-1 cells………... 37
2-2.9 RNA isolation and cDNA synthesis………... 37
2-2.10 Quantitative Real-time PCR………. 38
Chapter 3 Results
……… 403-1 Pro-inflammatory cytokines production……… 40
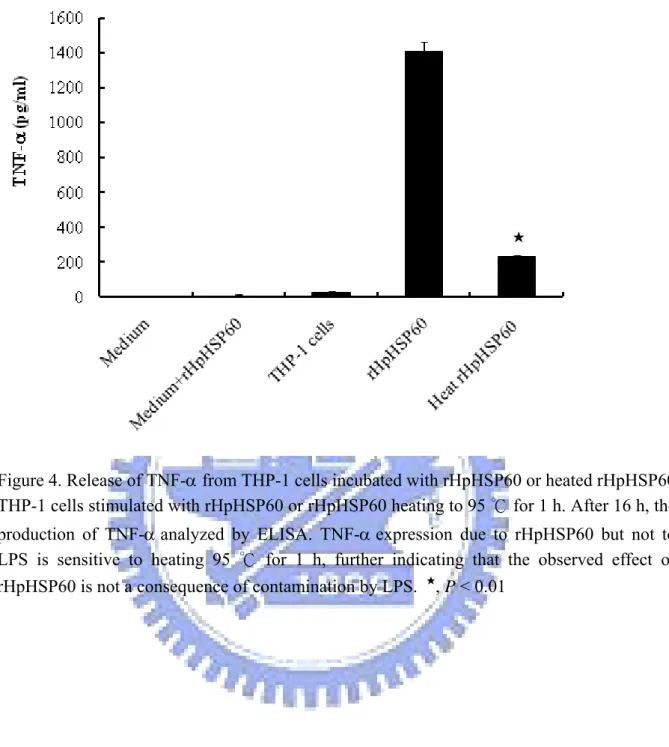
3-2 Release of TNF-α from THP-1 cells incubated with rHpHSP60 or heated rHpHSP60……… 41
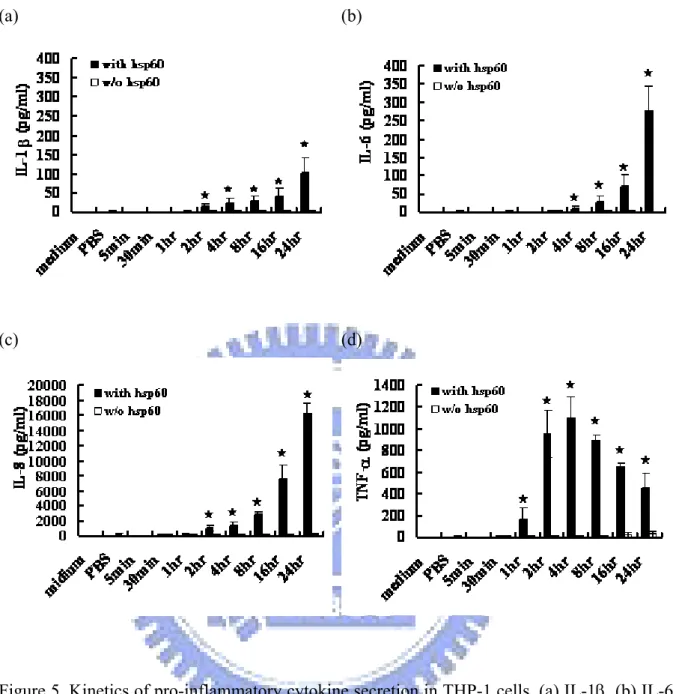
3-3 Kinetic of cytokine protein expression……….. 41
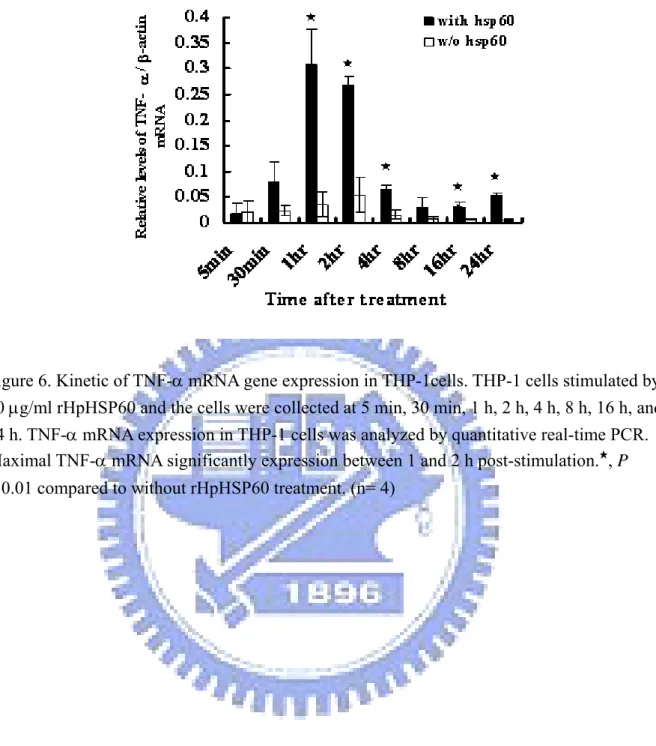
3-4 Kinetic of TNF-α mRNA expression……… 42
3-5 Detection of monocytic activation………. 43
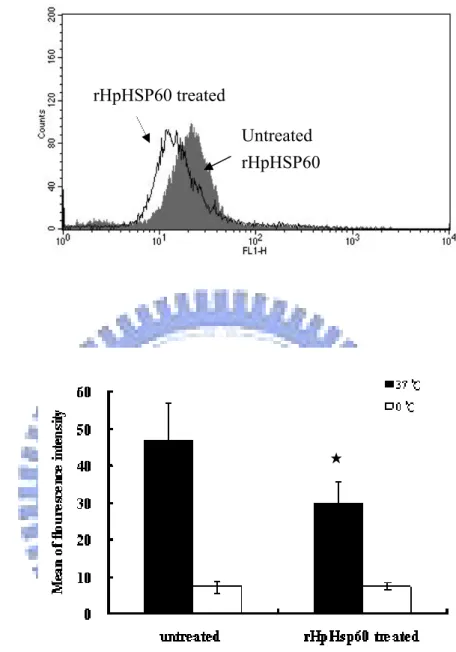
3-5.1 Endocytosis ability of THP-1 cells by rHpHSP60 treatment………. 43
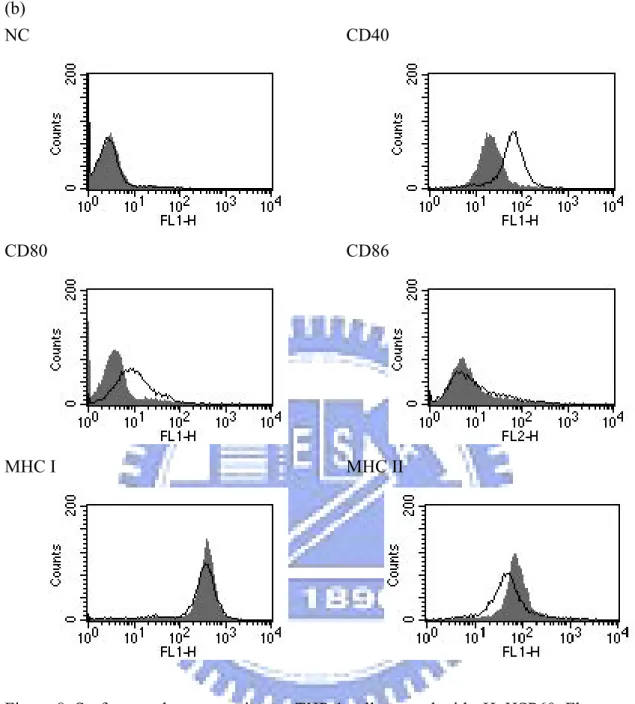
3-5.2 Surface marker expression on THP-1 cells……… 43
3-6 Effect of recombinant human TNF-α on CD40 expression and endocytotic activity of THP-1 cells………. 44
3-7 Effect of treatment with TGF-β1 and TNF-α on endocytotic activity and CD40 expression on THP-1 cells………... 45
Chapter 4 Discussion
………. 47Appendix
……….... 781-1 pET-Hp hsp60 map……….. 78
1-2 H. pylori hsp60 DNA sequence………... 79
1-3 H. pylori hsp60 protein sequence……… 80
1-4 The whole DNA sequence of H. pylori hsp60……… 81
2-1 pET-Human hsp60 map………... 85
2-2 Human hsp60 DNA sequence………. 86
2-3 Human protein sequence………. 87
2-4 Identification of recombinant human hsp60 plasmid by restriction enzyme digestion………. 88
2-5 SDS-PAGE and western blot analysis of the recombinant human HSP60 protein expressed in BL21………. 89
List of abbreviation
APCs Antigen presenting cells
Cag A Cytotoxin-associated gene A
DC Dendritic cells
HSP Heat shock protein
IFN-γ Interferon -γ
Ig Immunoglobulin IL-6 Interleukin-6 LPS Lipopolysaccharide
MALT Mucosa-associated lymphoid tissue
MDNCF Monocyte-derived neutrophil
chemotactic factor
MHC Major histocompatibility complex
NCF Neutrophil chemotactic factor
NAP-1 Neutrophil attractant/activating
peptide-1
PAI Pathogenicity island
PMN Polymorphonuclear neutrophil
leukocytes
RNIs Reactive nitrogen intermediates
TCR T cell receptor
Th T helper cells
TNF-α Tumor necrosis factor-α
TLR Toll-like receptor
List of table
List of Figure
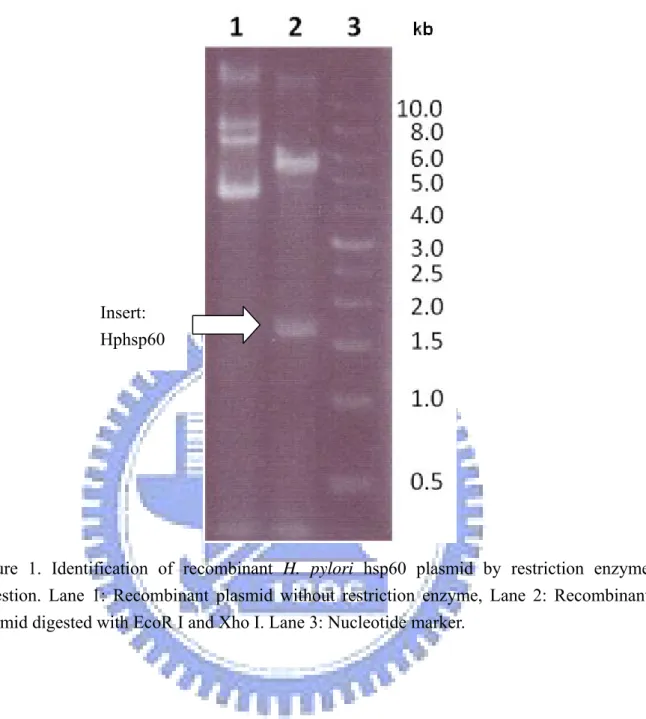
Figure 1. Identification of recombinant H. pylori hsp60 plasmid by restriction enzyme
digestion………. 57
Figure 2. SDS-PAGE and western blot analysis of the recombinant HpHSP60 protein expressed in BL21………. 58
Figure 3. Production of pro-inflammatory cytokines in THP-1 cells……… 59
Figure 4. Release of TNF-a from THP-1 cells incubated with rHpHSP60 or heated rHpHSP60……….. 60
Figure 5. Kinetics of pro-inflammatory cytokine secretion in THP-1 cells……….. 61
Figure 6. Kinetic of TNF-α mRNA gene expression in THP-1 cells……… 62
Figure 7. Endocytotic ability of THP-1 cells treated with rHpHSP60……….. 63
Figure 8. Surface marker expression on THP-1 cells treated with rHpHSP60………….. 65
Figure 9. Effect of recombinant human TNF-α on CD40 expression………... 66
Figure 10. Effect of recombinant human TNF-α on endocytotic ability………... 67
Figure 11. Immunohistochemistry of subjects infected by Helicobacter pylori………... 69
Figure 12. Effect of treatment with TGF-β1 and TNF-α on endocytotic activity of THP-1 cells……… 71
Figure 13. Effect of treatment with TGF-β1 and TNF-α on CD40 expression of THP-1 cells……… 72
Figure 14. Production of TNF-α in THP-1 cells by H. pylori hsp60 or human hsp60 stimulation……….. 73
Figure 15. Detection of human heat shock protein 60 secreted from THP-1 cells after rHpHSP60 stimulation………... 74
Figure 16. The limitation of human heat shock protein 60 detecting by the ELISA system……… 75
Chapter 1 Introduction
1-1 Helicobacter pylori
Helicobacter pylori is a well-known gastric-parasitical pathogen. Since 1982, Dr.
Marshell swallowed ten hundred million of H. pylori personally to prove that the persistence of H. pylori in stomach can result in some kind of gastric diseases, and lead a new epoch in human gastroenterology. H. pylori has been found in human in all parts of the world, with over half of the world’s population infected with H. pylori. In developing countries, 70-90 % of the population caries H. pylori. In 20-30 % of cases, the end result of the infection can be life-threatening (1). Many publishes revealed that H. pylori infection was associated with acute or chronic gastritis, peptic ulcer, gatroduodenal ulcer, and gastric cancer development, which led to H. pylori becoming classified as a classⅠcarcinogen by the World Health Organization. The complete process of H. pylori invasion, infection, and proliferation is really complex. Host immune surveillance system plays an essential role in pathogen elimination at early or late stage. However, H. pylori seems not be effectively cleared by host immune system and persist in host stomach for a half life time. Recently, scientists work hard to investigate many subversives of H. pylori and found that H. pylori can utilize multiple factors to protect them living in host stomach where a horrible environment for most pathogens is.
1-1.1 The morphology of Helicobacter pylori
H. pylori organisms are spiral, microaerophilic, gram-negative bacteria that colonizes the
gastric mucosa of humans. In gastric biopsy specimens, H. pylori organisms are 2.5 to 5.0 μm long and 0.5 to 1.0 μm wide; there are four to six unipolar sheathed flagella, which are essential for bacterial motility. Moreover, the surface of individual bacteria may be linked to gastric epithelial microvilli by thread-like extensions of the glycocalyx (2, 3). Interestingly, H.
pylori is classified as a noninvasive bacterial organism because it typically does not traverse
the epithelial barrier (4). Nevertheless, the bacterium is able to induce strong immune responses in such environment and results in some kind of gastric disease.
1-1.2 H. pylori-associated diseases
Colonization with H. pylori is not a disease in itself but a condition that affects the relative risk of developing various clinical disorders of the gastrointestinal tracts.
1-1.2.1 Acute gastritis:
Several reports showed that the acute phase of colonization with H. pylori may be associated with transient nonspecific dyspeptic symptoms, such as fullness, nausea, and vomiting, and with great inflammation of stomach mucosa. This phase is often associated with hypochlorhydria and it is unclear whether this initial colonization can be cleared spontaneously and prevents gastritis occurrence (5, 6).
1-1.2.2 Chronic gastritis:
Colonization with H. pylori always results in infiltration of the gastric mucosa with neutrophilic and mononuclear cells. H. pylori colonization relates to the chronic active gastritis, and other H. pylori-associated disorders result from this chronic inflammatory process. When colonization becomes persistent, a close correlation exists between the level of acid secretion and the distribution of gastritis. In subjects with intact acid secretion, H. pylori in particular colonizes the gastric antrum, where few acid-secretory-parietal cells are present. The pattern is associated with an antrum-predominant gastritis. Subjects with impaired acid secretion have a distribution of bacteria in antrum and corpus. The corpus bacteria in corpus are in closer contact with the mucosa, leading to a corpus-predominant pangastritis (7).
1-1.2.3 Peptic ulcer disease:
Duodenal ulcers (peptic ulcer) are defined as mucosal defects with a diameter of at least 0.5 cm penetrating through the muscularis mucosa. Duodenal ulcers usually occur in the duodenal bulb, which is the area most exposed to gastric acid. Both gastric and duodenal ulcer diseases are strongly related to H. pylori. It was showed that approximately 95 % of duodenal ulcers and 85 % of gastric ulcers occurred in the presence of H. pylori infection (8).
1-1.2.4 Atrophic gastritis, intestinal metaplasia, and gastric cancer:
Chronic H. pylori-induced inflammation can eventually lead to loss of normal mucosal architecture, with destruction of gastric glands and replacement by fibrosis and intestinal-type
epithelium. The risk atrophic gastritis depends on the distribution and pattern of chronic active inflammation (9). Patients with decreased acid output show a more rapid progression towards atrophy (10). It was reported that the risk of gastric cancer development via the sequence of atrophy and metaplasia, and the development of atrophy and cancer in the presence of H. pylori is related to host and bacterial factors, which influence the severity of the chronic inflammatory responses (6).
1-1.2.5 Gastric MALT lymphoma:
The gastric mucosa does not normally contain lymphoid tissue, after H. pylori infection, a lymphoid infiltrate appears, which constitute a chronic gastritis. In certain cases the lympoid tissue can be organized as lymphoid follicles. MALT lymphoma emerges from these lymphoid structures (11). The in vitro experiment showed that T lymphocytes sensitized for H.
pylori produce cytokines which stimulate B lymphoid proliferation. It is a B cell lymphoma
with a very unusual pathogenesis and evolution which slowly progresses and stays localized in the stomach for a long time (12).
1-1.3 The virulence factors resulting in H. pylori colonization and pathogenicity 1-1.3.1 The cag PAI and Cag A protein:
An intact cag PAI, which is associated with severe disease (13), encodes 31 proteins, which form a type IV secretion system capable of directly transferring bacterial proteins to the
cytoplasm of target cells, and it can stimulate human gastric epithelium cells to secrete IL-8, a mediator of serious gastric inflammation. (reviewed in Ref.(14)). Recntly, Nalini Ramarao et
al. further investigated that type IV transporter of H. pylori is essential in preventing
phagocytosis.The H. pylori cag PAI mediates the translocation of an effector protein, CagA, into gastric epithelial cells, and might also be directly involved in loosening of tight junctions (15). Furthermore, infection with H. pylori strains possessing CagA is associated with an increased risk of developing adenocarcinoma of the stomach (16).
1-1.3.2 VacA:
VacA is a secreted protein toxin, which causes vacuolar degeneration of epithelial cells in vitro and gastric epithelial erosion in vivo. In addition, VacA can loosen tight junctions in monolayers of polarized epithelial cells (17). A recent study showed that VacA alters the intracellular trafficking of proteins, increases the permeability of polarized epithelial cells, inhibits the process of antigen presentation, forms anion-selective channels in lipid bilayers, and interferes with cytoskeleton-dependent cell functions (18).
1-1.3.3 Urease:
The H. pylori urease can break down urea (CN2H4O) to form NH3 and CO2, which buffer
the microenvironment and the cytosol of the bacteria (19). Furthermore, H. pylori urease is a potent stimulus of mononuclear phagocyte activation and inflammatory cytokines production from immune cells and gastric epithelium cells (20) and the urease activity is also toxic to
human gastric epithelium cells (21).
1-1.3.4 NAP:
Helicobacter pylori neutrophil activating protein is also called HP-NAP. The main
evidence supporting a role for HP-NAP in virulence is the ability to activate neutrophils to produce oxygen free radicals and adhere to cultured endothelial cells (22), however, oxygen free radicals production will result in gastric tissue damage in the future. NAP released by bacterial lysis directly interacts with neutrophils, monocytes, and mast cells, resulting in the activation of their inflammatory functions (23).
1-1.3.5 Arginase:
H. pylori produces an arginase that uses arginine to produce urea and L-ornithine.
Furthermore, bacterial arginase allows H. pylori to evade the immune response by down-regulate eukaryotic NO production (24).
1-1.3.6 Cell wall and lipopolysaccharide:
Urease and HspB, a homolog of the GroEL protein of Escherichia coli, are abundant in outer membrane proteins (OMP) preparations. Urease and HspB are located strictly within the cytoplasm in early log phase cultures of H. pylori (25). However, in late-log-phase cultures, urease and HspB become associated with the bacterial surface in a novel manner. These cytoplasmic proteins are released by bacterial autolysis and become adsorbed to the surface of intact bacteria due to the unique characteristics of the outer membrane. The
lipopolysaccharide (LPS) of H. pylori has low biological activity, a property which may aid in the persistence of infection. H. pylori LPS disrupts the gastric mucus coat by interfering with the interaction between mucin and its mucosal receptor (26). However, the outstanding feature of the H. pylori LPS is its low proinflammatory activity.
1-1.3.7 Heat shock protein 60:
H. pylori HSP60 has been shown to play a role in the adherence and attachment of H.
pylori to gastric epithelium and induce IL-8 secretion from human gastric epithelial cells (27,
28). In the immune cells, H. pylori HSP60 can induce IL-6 and IL-8 from macrophages and monocytes, respectively (29, 30). Chronic gastritis is initiated and maintained by cytokines that are secreted by gastric epithelial cells and macrophages. Interleukin 8 (IL-8) is one of the principal mediators of the inflammatory response. Moreover, Kobayashi et al. showed that development of lymphoid tissue in patients with MALT lymphoma was associated with HSP60 (31).
1-1.4 Interactions between H. pylori and immune cells
Neutrophils: Neutrophils are recruited when H. pylori initially colonizes the human stomach, and the gastric mucosal inflammatory response that occurs in the persistent H. pylori infection is characterized by infiltration of neutrophils. Several specific H. pylori factors are known to interact with neutrophils and modulate their function (32).
Mast cells: In vitro experiments indicate that whole H. pylori bacteria and various H.
pylori components can activate mast cells. One H. pylori factor that can activate mast cells is
VacA (33). VacA can induce mast cell chemotaxis and can stimulate mast cell expression of multiple proinflammatory cytokines, including IL-1, TNF-α, IL-6, IL-13, and IL-10 (34). VacA induces degranulation of the mast cell line but does not induce degranulation of murine bone marrow-derived mast cells. HP-NAP also can activate mast cells, resulting in IL-6 production. Activation of mast cells by H. pylori may contribute to the inflammatory response associated with H. pylori infection (35).
Macrophages: Contact between macrophages and intact H. pylori bacteria or H. pylori components results in macrophage activation and secretion of numerous cytokines and chemokines. Macrophage recognizes the intact H. pylori by mediating TLR2 or TLR4. Ingested H. pylori cells have at least some ability to resist intracellular killing. Another mechanism of H. pylori escaping from macrophage killing is by blocking the production of nitric oxide. This effect is mediated by H. pylori arginase, which competes with nitric oxide synthase for arginine. In addition to resisting killing by macrophages, in vitro experiments indicated that H. pylori can induce macrophage apoptosis (36). H. pylori-induced apoptosis of macrophages may result in impaired innate and adaptive immune responses.
Dendritic cells: In response to H. pylori, monocyte-derived human DCs express costimulatory molecules and major histocompatibility complex class II proteins (37), which
results in increased efficiency of antigen presentation. Similar to several other bacterial pathogens, H. pylori can bind to DC-specific ICAM-3-grabbing nonintegrin (DC-SIGN), a DC-specific lectin (38). Interactions between H. pylori antigens and DC-SIGN may contribute to suppress the inflammation.
B lymphocytes: H. pylori is reported to have several inhibitory effects on B lymphocytes. In one study, H. pylori VacA interfered with the prelysosomal processing of tetanus toxin in Epstein-Barr virus-transformed B cells, and the ability of these cells to stimulate human CD4+ T cells were impaired in the presence of VacA. VacA inhibited the Ii-dependent pathway of antigen presentation mediated by newly synthesized MHC class II molecules but did not affect the pathway dependent on recycling MHC class II (39). Expression of CagA in B cells is reported to inhibit interleukin-3-dependent B-cell proliferation by inhibiting JAK-STAT signaling, which may result in inefficient antibody production and reduced cytokine expression (40).
T lymphocytes: One report indicated that H. pylori can have proapoptotic effects on T cells (41), but most of the observed effects occur in the absence of cell death. Coincubation of
H. pylori with T cells results in diminished expression of IL-2 and IL-2 receptor (CD25),
inhibition of activation-induced proliferation, and cell cycle arrest (42). The effects of H.
pylori on T cells are mediated by several different bacterial factors, one of which is VacA.
factor that regulates immune response genes, in Jurkat T cells, resulting in inhibition of IL-2 expression and G1/S cell cycle arrest (42). In addition to VacA and arginase, an uncharacterized low-molecular-weight protein of H. pylori has been reported to inhibit proliferation of T lymphocytes. This low-molecular-weight H. pylori factor is reported to block cell cycle progression at the G1 phase (43).
1-1.5 Immune activation and cell damage by H. pylori infection
The human gastric luminal pH is < 2, which prevents the proliferation of bacteria within the gastric lumen. H. pylori penetrates the gastric mucus layer after entering host stomach and thereby encounters a less acidic environment. H. pylori typically does not traverse the epithelial barrier, and it is classified as a noninvasive bacterial organism (4). Nevertheless, the bacterium is able to induce strong pro-inflammatory responses in these cells. Since H. pylori adherence, the production of a vacuolating cytotoxin and bacterial enzymes all contribute to epithelial damage. H. pylori infection, irrespective of their cag PAI phenotype leads to chronic gastric inflammation in the host. Recruitment and activation of immune cells in the underlying mucosa involves H. pylori chemotaxins, epithelial-derived chemokines such as IL-8 and pro-inflammatory cytokines liberated by mononuclear phagocytes (TNF-α, IL-1 and IL-6) as part of non-specific immunity. Moreover, gastric epithelial cells up-regulate expression of major histocompatibility complex (MHC) class II and costimulatioy molecules
on mococytes, macrophages, and dendritic cells in the gastric mucosa also play an important role in antigen presentation to activate adaptive immune cells activation (44). However, the infiltrated immune cells-induced inflammation response appears to be a primary cause of the damage to gastric surface epithelial layers and finally resulted in gastritis, peptic ulcer disease, and gastric cancer (40).
1-1.6 Immune subversion by H. pylori
Once arriving at the gastric epithelium, H. pylori must face the rapid onslaught of effector cells of the strong immune response. To overcome continually intense attack, H.
pylori utilize some virulence factors to break host immune defense and successfully escape
from killing by effector cells. In the innate immune stage, H. pylori first attack by nitrogen oxide (NO), which is an important component of innate immunity and an effective antimicrobial agent. To avoid killing by NO, H. pylori produces an arginase to regulate NO synthesis. Arginase can convert L-arginine to urea and L-ornithine, because L-ornithine is also used by iNOS to produce NO so that arginase can compete with iNOS for their substrate to decrease NO production. Even though H. pylori was unfortunately ingested by professional phagocytes, it is capable to resist phagocytic killing. Phagocytosis of H. pylori by macrophages becomes a large megasomes, which result from homotypic phagosome fusion and subsequent macrophage apoptosis might enable the escape of the bacteria. Interestingly,
the LPS of H. pylori is at least 1000-fold less active than E. coli LPS. The VacA protein of H.
pylori is contributed to disrupt host adaptive immune response. Clear evidence has recently
been obtained for VacA in suppression of T-cell response. Sundrud et al. showed that VacA inhibits human peripheral blood lymphocytes proliferation by TCR-CD28 co-stimulation by interfering with IL-2-dependent cell cycle progression. Gastric MALT lymphoma results from the uncontrolled polyclonal expansion of IgM memory B cells, T-cells inactivation might also contribute to the abnormal B-cell growth. Moreover, Cag A is also capable of preventing B-cell apoptosis by inhibiting p53 accumulation, which might involved in development of MALT lymphoma.
1-1.7 Chronic infection
Levels of numerous cytokines, including gamma interferon (IFN-γ), tumor necrosis factor (TNF), IL-1β, IL-6, IL-7, IL-8, IL-10, and IL-18, are increased in the stomachs of H.
pylori-infected humans compared to uninfected humans (45). These cytokines have great
effect on immune cells activity and attract these effector cells to the inflammation site. The concentration of various types of leucocytes was detected in gastric mucosal biopsies from human infected with H. pylori. Lymphocytes (both T cells and B cells), macrophages, neutrophils, mast cells, and dendritic cells (DCs) are usually present at the inflammation area and play an important role in antigen presentation (46). The relative abundance of
IFN-γ-producing T cells and the relative scarcity of IL-4-producing gastric T cells in the setting of H. pylori infection, it has been concluded that H. pylori infection leads to a Th1-polarized response (47). The chronic gastric mucosal inflammatory response to H. pylori probably reflects the combined effects of a cellular immune response and an ongoing stimulation of an innate immune response.
1-2 Human innate immunity
The human immune system defends against a spectrum of microbial pathogens, in terms of environmental prevalence, rang from common to rare. Invasion by common environmental microbes is prevented by constitutive innate immune defense in mucosal and epithelial tissues. Upon infection with highly virulent pathogens, auxiliary innate defenses are induced to combat the pathogens. Neutrophils, monocytes, macrophages, and dendritic cells are important cellular mediators of innate immune defense.
1-2.1 The characterizations of innate immune cells
Most cellular components of immune system derive from bone marrow. The typical developmental pathway begins with pluripotent bone marrow stem cells that give rise to progenitors that follow a variety of differentiation pathways to become mature cells with defined effector functions.
1-2.1.1 Monocytic cells:
Newly produced monocyte are released into blood where they circulate for 1-3 days before entering tissues to differentiate into mature resident macrophage (48).
1-2.1.1.1 Human monocyte subsets:
In humans, circulating monocytes are divided into two subsets on the basis of the expression of CD14, a component of the lipopolysaccharide (LPS) receptor complex, and CD16, the FcγRIII immunoglobulin receptor (49). These monocyte subsets express distinct chemokine, immunoglobulin, adhesion, and scavenger receptors (50). CD14+CD16− (CD14+) monocytes are large, ~ 18 μm in diameter, and represent ~ 80%–90% of circulating monocytes. In contrast, CD14−CD16+ (CD16+) monocytes are smaller, ~14 μm in diameter, and constitute ~ 10% of circulating monocytes.
1-2.1.1.2 Monocytes differentiation:
Under inflammatory conditions, monocyte production in the bone marrow is increased and after released into the circulation monocyte are rapidly recruited to sites of injury and infection where they differentiate into inflammatory macrophage (51). Furthermore, monocytes can also give rise to dendritic cells (DCs) in vitro and in vivo, and microbial infection triggers in vivo monocyte differentiation into specialized DC populations.
1-2.1.1.3 Bacterial infection:
a range of microbial pathogens. Monocytes kill bacteria by producing reactive nitrogen intermediates (RNIs) and reactive oxygen intermediates (ROIs) and through the action of phagolysosomal enzymes (52, 53).
1-2.1.2 Macrophage:
Macrophage is a part of mononuclear phagocyte system and is professional antigen presenting cells for adaptive immunity. Mononuclear phagocytes migrate out from bone marrow, circulate briefly in the blood as monocytes, and then enter into the tissues and inflammatory foci where they differentiate into macrophages. Macrophages, which is a heterogeneous population of phagocytic cells found throughout the body that originate from the mononuclear phagocytic system (54).
1-2.1.3 Neutrophils granulocytes:
Neutrophils are abundant in blood, where they have a short half-life if they are not recruited to a site of inflammation by specific chemokines and cytokines. Once recruited to an inflammatory site, neutrophils migrate rapidly from blood to tissue, which is otherwise devoid of neutrophils (55). In response to inflammatory stimuli, neutrophils migrate from the circulating blood to infected tissues, where they efficiently bind, engulf, and inactivate bacteria. Phagocytosed bacteria are killed rapidly by proteolytic enzymes, antimicrobial proteins, and reactive oxygen species (56).
Dendritic cells (DC) are one of the most potent antigen presenting cells (APCs) of the immune system and are thought to be crucial for the initiation of primary T cell-mediated immune responses (57). Resting DCs capture and process soluble or particulate antigens in late endosomal and lysosomal compartments that are rich in major histocompatibility complex (MHC) class II molecules (58). Mature DCs upregulate their expression of MHC class I and class II complexes that can be recognized by antigen-specific T cells. Once DC undergoes a process of maturation, they have a greatly diminished capacity for antigen uptake and processing but have gained the ability to present antigens effectively for priming T cells (59, 60).
1-2.2 Toll-like receptors (TLRs)-mediated immune activation
The role of Toll-like receptors is to activate phagocytes and tissue dendritic cells to response to pathogens by secreting cytokines and chemokines, and to express the co-stimulatory molecules essential to activate adaptive immunity. There are ten Toll-like receptors of humans are known. Each of them can recognize one or more particular molecule that is present in many pathogens. The well-known of these is Toll-like receptor 4, which is interacted with Gram-negative bacterium LPS (61) and the Toll-like receptor 5 recognized the flagellin of pathogens (62). When TLR-4 interacts with LPS bound to CD14, this sends a signal to activate the transcription factor NF-κB which result in several pro-inflammatory
cytokines secretion such as TNF-α (63). Moreover, TLR-1, TLR-2, and TLR-6, each of them is formed in dimer to recognize peptidoglycan, and lipoproteins. TLR-3 and TLR-9 recognizes double-stranded RNA and unmethylated CpG DNA, respectively. There are two different mechanisms by which activation of TLRs can contribute to host defense. Fist, activation of TLRs can directly mediate innate response by regulating phagocytosis and triggering antimicrobial activity (64). Second, activation of TLRs can trigger the release of cytokines and the differentiation of immature to mature dendritic cells, enabling the innate immune systems to instruct the adaptive immune response (65). Stephan et al. showed that DC-SIGN+ cells have a macrophage-like phenotype, are phagocytic, and use DC-SIGN to facilitate the uptake of bacteria. In contrast, CD1b+ cells have an immature dendritic phenotype, release pro-inflammatory cytokines and function as efficient antigen- presenting cells (66).
1-2.3 Phagocytosis of pathogens:
The most important phagocytic cell is monocyte/macrophage, which locates especially in connective tissue, in the submucosal layer of the gastrointestinal tract. The second major family of phagocytes such as the neutrophils or polymorphonuclear neutrophil leukocytes (PMNs) is short-lived cells that are abundant in blood, but they are not present in healthy tissue. Upon phagocytosis, macrophages and neutrophils can produce various toxic products
to kill the engulfed microorganism. The most important of these are nitric oxide (NO), the superoxide (O2-), and hydrogen peroxide (H2O2), which are directly toxic to bacteria.
Dendritic cells, which is another type of phagocytic cell present in tissues, thus enabling these antigen-presenting cells to initiate adaptive immune response. After internalization of bacterial components, the expression of MHCII, CD80, and CD86 were up-regulation on the surface of these phagocytic cells, also called antigen presenting cells and subsequent activate the adaptive immune response. Both of these phagocytic cells have a key role in innate immunity because they can recognize, ingest, and destroy many pathogens without aid of adaptive immune responses.
1-2.4 Cytokines and chemokines production:
Cytokines are secreted proteins that many cell types produce; they are critical to both innate immunity and adaptive immune system responses. Cytokines play a crucial role in the immune system response to all kinds of disease. They interact with organs and cells, alone and in combination with each other. The diverse role that cytokines serve in the immune system makes them an ideal target for intervening or bolstering immune responses. Inflammation is mediated by a variety of cytokines. Inflammatory cytokines can be divided into two groups: those involved in acute inflammation and those responsible for chronic inflammation. Several cytokines play key roles in mediating acute inflammatory reactions,
namely IL-1β, TNF-α, IL-6, and chemokine, IL-8.
1-2.4.1 Interleukin-1β:
IL-1β accumulates as a 33 kDa pro-cytokine (proIL-1β) in the cytoplasm of monocytes and macrophages, and its activation depends on cleavage to the active, mature 17 kDa form (mIL-1β) by the enzyme caspase-1 (67). IL-1β is an important proinflammatory cytokine whose circulating levels are tightly regulated to prevent aberrant activation of pathways that can lead to chronic inflammation, septic shock, or death (68). In health person, these cells do not constitutively express IL-1β (69).
1-2.4.2 Interleukin-6:
Interleukin 6 (IL-6) is a pleiotropic cytokine that is produced by many different cell types and involved in a wide range of responses, such as immune response, and acute-phase
reactions (70). IL-6 is produced by various types of lymphoid and non-lymphoid cells, such as T cells, B cells, monocytes, fibroblasts, and several tumor cells. The original
classification of IL-6 as INF-β2, and a prominent regulator of T cell proliferation, differentiation, survival, and Ig secretion by B cells (71, 72). IL-6 can also regulate monocytes differentiation. Pascale et al. showed that once activated monocytes encounter stromal cells such as fibroblasts which will induce IL-6 productionthat, in return, increases functional M-CSFR on activated monocytes. As the activated monocytes spontaneously release M-CSF, the functional M-CSFR then transduces M-CSF signals, thereby initiating
macrophage differentiation (73). The role of IL-6 in the generation of human macrophages provides an explanation for the altered clearance of pathogens such as Listeria or
Mycobacterium in mice (74). Hence, it has defined IL-6 as a factor for directing transition
from innate to acquired immunity (75). However, nn anti-inflammatory function of IL-6 was also detected in 1989; it was showed that IL-6 can inhibit lipopolysacharide-indiced TNF-α production in monocytic cells (76).
1-2.4.3 Interleukin-8:
Interleukin 8 (IL-8), a proinflammatory chemokine, is produced by various types of cells upon stimulation with inflammatory stimuli and exerts a variety of functions on leukocytes, particularly neutrophils in vitro. It has been referred to as monocyte-derived neutrophil chemotactic factor (MDNCF), neutrophil attractant/activating peptide-1 (NAP-1), neutrophil chemotactic factor (NCF) (77, 78). The essential role of IL-8 in most inflammation reactions is recruiting and activating neutrophils, such as lysosomal enzymes, generation of superoxide/biolipis, and increase the expression of adhesion molecules on neutrophils (79). Whiles IL-8 has poorly influence on monocytes. According to recent study, it has shown us that IL-8 is certainly not involved in THP-1 cells activation (80).
1-2.4.4 Tumor necrosis factor-α:
Human Tumor necrosis factor (TNF) is translated as a 26-kDa protein that lacks a classic signal peptide. TNF-α is an important pro-inflammatory mediator produced predominantly by
activated monocytes and macrophages. TNF is not usually detectable in healthy individuals, but elevated serum and tissue levels are found in inflammatory and infectious conditions and serum levels correlate with the severity of infections (81, 82). Although cells of the monocyte/macrophage lineage are the main source of TNF-α in inflammatory disease, a wide range of cells can produce TNF-α, including mast cells, T and B lymphocytes, natural killer (NK) cells, neutrophils, endothelial cells, smooth and cardiac muscle cells, fibroblasts and osteoclasts. One of the major biological roles of TNF-α is in the host defence to bacterial, viral and parasitic infections. Physiologically, TNF-α is important for the normal response to infection, but inappropriate or excessive production can be harmful. It has been found that the cytotoxic properties of TNF-α can both against tumor cells and against normal cells infected with intracellular pathogens and viruses, and performs many immunoregulatory functions (83, 84). TNF-α is also chemotactic to monocytes and neutrophils. Stimulation of these cells with TNF-α induces adherence of monocytes and neutrophils to endothelial cells, and enhancement the antigen presenting capacity of monoctes/macrophages (85).
1-2.4.4.1 TNF-α associated diseases
Rheumatoid arthritis: Rheumatoid arthritis is a chronic autoimmune inflammatory disorder affecting approximately 1% of the population, characterized by inflammation of synovial tissue, leading to progressive damage, erosion of adjacent cartilage and bone and chronic disability. The inflammation is associated with accumulation of inflammatory cells,
predominantly T cells and macrophages, but also B cells, plasma cells and dendritic cells. There is synovial hyperplasia and angiogenesis is a prominent feature (86).
Inflammatory bowel disease: TNF-α immunoreactivity is increased the lamina propria in intestinal specimens from patients with Crohn’s disease and ulcerative colitis and mice overexpressing TNF-α develop a Crohn’s disease-like inflammatory bowel disease (87, 88).
Psoriasis: Psoriasis is an inflammatory skin disorder, in which an inflammatory cell infiltrate is associated with hyperkeratotic lesions, giving rise to typical psoriatic plaques. TNF-α, TNFR1 and TNFR2 are upregulated in dermal blood vessels in involved skin from patients with psoriasis (89).
Disease of the central nervous system: In the central nervous system, TNF is produced primarily by microglia and astrocytes in response to a wide range of pathological processes, including infection, inflammatory disease, ischaemia and traumatic injury (90). TNF-α mediated protection against experimental autoimmune encephalomyelitis does not reqire TNFR1, although TNFR1 appears to be necessary for detrimental effects of TNF-α, which occur during the acute phase of the disease (91). Neutralization of TNF failed to benefit patients with relapsing–remitting multiple sclerosis, and significantly increased exacerbations (92).
Cardiovascular disease: TNF-α has also been implicated in the pathogenesis of a number of cardiovascular diseases, including atherosclerosis, myocardial infarction, heart failure,
myocarditis and cardiac allograft rejection, and vascular endothelial cell responses to TNF-α may underlie the vascular pathology in many of these conditions. Patients with chronic inflammatory conditions such as rheumatoid arthritis have an increased incidence of cardiovascular disease. Inflammatory mediators, including TNF-α, have been implicated in this increased cardiovascular risk, and there is some evidence that anti-TNF therapy ameliorates this risk in patients with rheumatoid arthritis (93).
Respiratory disease: TNF-α has been implicated in the pathophysiology of many inflammatory lung diseases, including chronic bronchitis, chronic obstructive pulmonary disease, acute respiratory distress syndrome and asthma (94). In asthma, TNF-α has been implicated in airway inflammation and remodelling, and may play a role in bronchial hyper-responsiveness. Leukocytes from bronchiolar lavage of asthma patients have increased release of TNF-α, and inhaled TNF-α increases airway responsiveness in normal subjects and is associated with a pulmonary neutrophil infiltration, assessed by induced sputum (95).
Renal disease: TNF-α has been implicated in the pathogenesis of many renal diseases, including ischaemic renal injury, renal transplant rejection and glomerulonephritis, which is often part of a systemic vasculitis. In diseases associated with renal inflammation, different forms of TNF-α blockade vary in their efficacy and adverse effects, and these differences may be attributed to different effects on signalling though TNF-α receptor subtypes (96).
TGF-β stimulates cells at the resting state (monocytes), whereas activated cells (macrophage) are inhibited. Sharon et al. have shown that the function of TGF-β is depends on the concentration variation. It stimulates monocytic chemotaxis at 0.1 to 10 pg/ml, while higher concentrations, around 10 ng/ml, alter the production of cytokines such as IL-1, and TNF-α (97, 98), and inhibit killing of several invasive pathogens (99, 100). TGF-β is arguably the most potent endogenous immunosuppressive factor to be characterized (101). IFN-γ -induced expression of MHC class II molecules in macrophages is inhibited by TGF-β via the attenuation of CIITA (102).
1-2.4.5.1 TGF-β regulation of infectious diseases
Trypanosome: Trypanosoma cruzi is one parasite that makes direct use of the host’s TGF-β signaling pathway. It uses TGF-β receptors I and II for successful entry into mammalian cells. Epithelial cells lacking TGF-β receptors are resistant to T. cruzi infection, and infectivity is restored following transfection of functional TGF-β receptors (103). TGF-β treatment of mouse and human macrophages blocks IFN-γ-mediated inhibition of parasite growth, and TGF-β-treated mice develop higher parasite loads and die faster than control mice. African trypanosome T. brucei, which does not invade host cells, might nevertheless possess a functional homolog of this TGF-β-activating moiety because this parasite has also been shown to release a factor that induces TGF-β mRNA expression (104).
Virulent strains of Leishmania have developed mechanisms to induce macrophages to produce high levels of active TGF-β, whereas non-pathogenic strains that produce low-grade infection induce relatively low levels of active TGF-β (105). TGF-β made by infected macrophages suppresses NO production and can influence T cell differentiation by inhibiting the production of TNF-α and IFN-γ (106).
Toxoplasma gondii: Toxoplasma is another obligate intracellular parasite that infects macrophages. As in the case of Leishmania, infection of mouse macrophages with T. gondi results in the release of TGF-β, which was associated with the down-regulation of TNF-α and its receptors (107).
Mycobacteria: Mycobacteria are obligate intracellular pathogens of macrophages that cause tuberculosis, leprosy, and opportunistic infections due to immunosuppression. Similar to most protozoan infections, mycobacteria induce macrphsge production of active TGF-β and suppress their antibacterial activity to aid their pathogenesis. Both purified protein derivative and lipoarabinomannan, a cell wall component of tuberculosis, induce TGF-β from human peripheral blood mononuclear cell (PBMC)-derived macrophages (108, 109).
Listeria: Listeria monocytogenes is a facultative intracellular bacterium and a strong inducer of Th1 response (110). Cytokines such as IFN-γ, TNF-α, and IL-6 play an important role in host resistance to Listeria, and TGF-β plays a protective role in Listeria infection. The mechanism by which TGF-β confers resistance to lethal doses of L. monocytogenes in mice is
not yet clear and requires more investigation (111).
1-3 Heat shock proteins
Heat shock proteins are constitutively present in eukaryotic and procaryotic cells. Expression of heat shock proteins (hsps) is markedly increased as part of the response to an array of stressors. These proteins participate in the refolding of denatured polypeptides that become damaged. Generally, during nonstress conditions, hsps participate in the folding of nascent polypeptides and the stabilization of receptors and signal transduction molecules (112). Extracellular HSPs are considered to belong to the heterogenous family of ‘‘alarmins’’ that are involved in tissue damage-associated inflammation (113). Furthermore, the stimulatory capacity of exogenous HSPs on antigen-presenting cells (APC) was detected. Combined with these reports, hsps can trigger immune activation at the certain environment.
1-3.1 Human heat shock proteins
The importance of the interaction of Hsps with the immune system is apparent from two important observations: first, the presence of anti-Hsp antibodies in serum and, second, the cytokine production induced in a number of cell types by exposure to Hsp60, or Hsp70 (114).
1-3.1.1 Human heat shock protein 60
the stimulatory capacity of extracellular HSP60 on the innate immune system has been recognized. Human macrophages respond to both bacterial and human HSP60 with the release of pro-inflammatory mediators such as TNF-α or IL-6 and of the Th1-promoting cytokines IL-12 and IL-15 suggesting that HSP60 might act as a “danger signal” for the innate immune system (115). There is evidence that CD14 and Toll-like receptors (TLR) 2 and 4 are involved in HSP60-mediated cell activation (116-118). Stefanie et al. revealed that HSP60 can promote dendritic cell maturation. Maturation of DC induced by human HSP60 was characterized by up-regulation of MHC class II and of the costimulatory molecules CD40, CD54, and CD86 and the pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-12 were also secreted. However, HSP60 is also an antigen for the adaptive immune system and T cell responses to HSP60 epitopes regulate inflammatory diseases like rheumatoid a arthritis, insulin-dependent diabetes mellitus, and artherosclerosis (119, 120). Taken together, maturation of DC is strongly induced by human HSP60 and paralleled by release of Th1-promoting cytokines. HSP60 might favor the development of Th1-dependent organ-specific autoimmune diseases when endogenous HSPs are released as observed in diabetes and arthritis.
1-3.1.2 Human heat shock protein 70
Hsp70 can be released from tumor cells in a complex with intracellular polypeptides and then recognized by components of the immune system, leading to antitumor immunity against the chaperoned tumor peptide antigens (121). Such Hsp70-peptide complexes interact with
APCs and induce tumor immunity by promoting Ag cross-presentation to T cells (122).
1-3.2 Bacterial heat shock proteins
HSPs are abundantly expressed in inflammatory lesions and produced by microorganisms during invasive infection and phagocytosis. Bacterial heat shock proteins have been reported to stimulate human monocytes to produce pro-inflammatory cytokines or to up-regulate the expression of adhesion molecules (123, 124).
1-3.2.1 Escherichia coli heat shock proteins: GroEL, DANK, and GroES
It was said that GroEL and DnaK were able to induce the release of TNF-α, IL-1α, IL-6 and sICAM-1 from keratinocytes. GroES showed significant activity only on the expression and release of IL-6. In the inflammatory reaction of the skin, keratinocytes play a determining role by synthesizing and secreting cytokines and adhesion molecules (125). In another study, DNAK and GroEL were able to induce the release of GM-CSF and IL-6 from HUVEC, in monocytes DNAk and GroEL were able to induce the release of soluble forms of E-selectin, ICAM-1, and VCAM-1, while GroES showed a significant activity only on E-selectin release (124). Release of cell surface adhesins may simply be a mechanism for breaking adhesive interactions between cells or may provide a means for clearing the cell surface of adhesins to control adhesivity. Taken together, HSPs may play an important role in the initiation of the inflammatory process that accompanies infections with microbial pathogens by regulating the
expression of cytokines involved in the activation of leukocytes and endothelial cells.
1-3.2.2 Chlamydia heat shock protein 60
Chlamydiae produce large amounts of heat shock protein 60 (HSP 60) during chronic,
persistent infections, and C. pneumoniae localizes predominantly within plaque macrophages.
Chlamydia pneumoniae infection has been associated with asthma and the aggravation of
atherosclerosis. Kol et al. showed that chlamydial HSP 60 colocalizes with human HSP 60 within plaque macrophages and that HSP 60 from both species can induce macrophage production of TNF-α and matrix-degrading metalloproteinases, two mediators of atherosclerosis complications (126).
1-3.2.3 Mycobacterial heat shock protein 65
The 65 kDa heat-shock protein (Hsp65), a well-conserved and immunodominant antigen which elicits a cellular and humoral immune response may play a role in host defense against invading microorganisms and autoimmune disorders. Incubation with Hsp65 resulted in an enhanced release of TNF-alpha and IL-1 beta by human monocytes and monocyte-derived macrophages (MDM). The release of the proinflammatory cytokines TNF-α and IL-1β by human mononuclear phagocytes in response to Hsp65 indicates that this protein can contribute to both host defence and tissue damage in inflammatory lesions characterized by an abundant expression of Hsp65 (127).
H. pylori seems to bind to gastric epithelial cells and mucin via HSP60 (128) and induces
IL-8 production from gastric epithelium or monocytes (28, 29). Interleukin-8 (IL-8) is a chemokine secreted by a variety of cell types, which serves as a potent inflammatory mediator recruiting and activating neutrophils. Several studies have demonstrated that H. pylori strains are capable of inducing IL-8 secretion from gastric carcinoma cells in vitro (129, 130). Not only IL-8 but also IL-6 was produced by macrophage via a toll-like receptor (TLR)-2, TLR-4 and myeloid differentiation factor 88-independent mechanism (30). Helicobacter pylori heat shock protein 60 and risk of coronary heart disease: a case control study with focus on markers of systemic inflammation and lipids (131). Tahenaka et al. showed that serum antibodies to Helicobacter pylori and its heat-shock protein 60 correlate with the response of gastric mucosa-associated lymphoid tissue lymphoma to eradication of H. pylori (132). It has been suggested that antibodies against heat shock proteins seem to be involved in the pathogenesis of coronary heart disease (CHD) and CagA positive H pylori infection may concur to the development of CHD; high levels of anti-Hsp60 antibodies may constitute a marker and/or a concomitant pathogenic factor of the disease (133).
Chapter 2 Materials and Methods
2-1 Materials2-1.1 Reagent
The following reagents obtained were described as following: RPMI 1640, Fetal Bovine Serum (FBS), BSA, and Tryzol were from Invitrogen Inc. (Gaithersburg, MD, USA). Penicillin/ streptomycin/ amphotericin (PSA) were from Biological industries (Beithaemek, Israel). Restriction enzymes were from Promega Inc. (WI,USA). Kanamycin and Tris were from MDBio Inc. (Rockville, MD, USA). Ethidium bromide (EtBr), Isopropyl-beta- D-thiogalactopyranoside (IPTG), NaCl, yeast extract, agar, Tris-HCl, Triton X-100, TEMED and imidazole were from Amresco Inc. (Solon, OH, USA). Recombinant human TNF-α and TGF-β1 was from Peprotech Inc. (Rocky Hill, NJ). Sephadex G-25 Medium was from Amersham Bioscciences (Uppsala, Sweeden). Nitrocellulose (NC) paper was from PALL Inc. (Ann Arbor, MI, USA).
2.1.2 Antibody
PE-conjugated anti-CD86, Fluorescent isothiocyanate (FITC)-conjugated anti-CD40, anti-CD80, anti-HLA-ABC, and anti-HLA-DR antibodies were from BioLegend (Sandiego, CA, USA). HRP-conjugated rabbit anti 6X His antibody was from Novus (Littleton, CO, USA).
Human IL-1β, IL-6, IL-8, and TNF-α ELISA kit was obtained from R&D systems (Minneapolis, MN). Superscript III RT kit was from Invitrogen (Gaithersburg, MD, USA). RealQ-PCR master mix kit was from Ampliqon (Copenhagen, Denmark). Coomasie PlusTM Protein Assay Reagent kit and Enhanced chemiluminescence (ECL) system was from Pierce (Rockford, IL, USA), FITC-dextran was from SIGMA-ALDRICH (Steinheim, Germany).
2.1.4 Instrument
HisTrapTM HP column was from GE healthcare (Uppsala, Sweeden). ABI PRISM 7000 from was Applied Biosystems (USA). Flow cytometer was from BD (Bedford, MA, USA). Human heat shock protein 60 cDNA (complementary DNA) library were kindly provided from Dr. Chich-Sheng Lin (NCTU, Laboratory of Biomedical Engineering, Biological Science & Technology Lab).
2.1.5 Bacteria
Escherichia coli (BL21 and DH5α) was from Yeastern Biotech Co. H. pylori genome
was from Department of Internal Medicine, College of Medicine, National Taiwan University.
2-1.6 Cell line
THP-1 cells, acute monocytic leukemia cell line was purchased from the Bioresource Collection and Research Centre (BCRC) (Hsinchu, Taiwan). Unlike other leukemic cell lines, THP-1 cells have no prominent chromosomal abnormalities (134).
2.2 Methods
2-2.1 Recombinant DNA techniques
The H. pylori strains were isolated from gastric biopsy specimens at National Taiwan University Hospital. The genome of H. pylori was prepared from the clinical isolates. The gene of Hsp60 was amplified from the genome of H. pylori by polymerase chain reaction (PCR) using the primers: 5’- ATC GAA TTC ATG GCA AAA GAA ATC AAA TTT TCA - 3’ as forward primer and 5’-GAT CTC GAG TTA CAT CAT GCC GCC CAT G-3’ as reverse primer. PCR condition was that 94 ℃ denaturation step followed by 35 cycles of 45 s at 95 ℃, 45 s at 50 ℃ and 2min at 72 ℃. After these cycles, incubate the PCR mixture at 72 ℃ 10min for complete elongation. PCR product was harvested, digested with EcoR1and Xho I, and inserted into EcoR I and Xho I restriction fragment of the expression vector pET-30a with N-terminal His-tags. The recombinant plasmids were further identified by restriction enzyme and agarose gel. The resulting plasmid pET- Hsp60 was transformed into competent E.coli BL21 (DE3) cells growing on an agar plate with kanamycin for selection.
2-2.2 Transformation
Remove the appropriate number of competent cells tubes from the -80 ℃ freezer. DH5α is used for cloning and DNA amplification. BL21 is used for protein expression. After the cells have thawed, add 1ng DNA into the cells, mix by gently swirling the tip or by gently
tapping the tube. Incubate the competent cell on ice for 30 min. Heat shock the cell at 42 ℃ for 90 s. Place the cells on ice for 2 min and add 250 μl LB (10g tryptone, 10g NaCl, 5g Yeast extract) and incubate at 37 ℃ with shaking 225 rpm for 1 hr. Spread 100μl mixture onto each LB agar plate (10g tryptone, 10g NaCl, 5g Yeast extract ,20g agar) containing kanamycin (30 mg/ml) and incubate at 37 ℃ for 12~16 hr.
2-2.3 Expression and purification of HpHsp60 gene in Escherichia coli
The colonies on the agar plate were picked, and shake in 100 ml LB with 30 μg/ml kanamycin at 37℃ overnight. Then, the 100 ml LB with bacteria was inoculated in 900 ml LB and the bacteria grew until the optical density at OD 600 nm reached 0.4-0.6. IPTG was added to a final concentration of 1 mM, and E. coli cells continually grew in 1L LB for 4 h. After induction, the LB containing E. coli cells were harvested by centrifugation at 5000 rpm for 15 min and the pellet was resuspended in 30 ml binding buffer (20mM Na2HPO4, 0.5M
NaCl, 40mM imidazole, pH7.4). Then the homogenized samples were sonicated with short burst of 1 sec followed by intervals 1 sec and the sonication processing was maintained for 15 min. Centrifuge the samples at 12000 rpm for 30 at 4 ℃. Harvest the supernatant and passed the 0.45 μM filter to remove the particles.
2-2.4 Protein purification
In this experiment, we purify our proteins with HisTrapTM HP column (GE healthcare). To prepare the column, wash the column with 5 column volumes of DDW and equilibrate the
column with 5 column volume of binding buffer at the flow rate about 1 ml/min. Apply the pretreated sample and wash with wash buffer (20mM Na2HPO4, 0.5M NaCl, 60mM imidazole,
pH7.4) about 60 volume. Elute with elution buffer (20mM Na2HPO4, 0.5M NaCl, 200mM
imidazole, pH7.4, filtered with 0.45μm filter) for 10 volumes. Detect several fractions containing proteins by coomasie blue reagent. Collect the fractions with high protein concentrations and use G25 column to remove the unnecessary salt from the solution and replace the buffer with PBS (Phosphate Buffered Saline, 140 mM NaCl, 2.7mM KCl, 10 mM Na2HPO4, KH2PO4, pH 7.4). To prepare the G25 column, we need to swell the 7g Sephadex
G-25 Medium with filtered PBS at room temperature for overnight. Fill the column with PBS and agitate the PBS containing G25 agarose, soon pour into the column along the edge of glass rod. After collecting the protein-containing fractions, we pour it into the G25 column and wash and elute with PBS. Detect which fractions contain proteins with coomasie blue reagent and collect the fractions. Poll the fractions together and filtered with 0.22 μm syringe filter. Quantitate the amount of protein concentration with coomasie blue reagent and dilute the solution to 1mg/ml. The quality of recombinant protein was checked by SDS-PAGE and Western blotting.
2-2.5 Cell cultures
The human monocyte THP-1 cell line was cultured according to therecommendations from ATCC. Briefly, non-adherent cells were grown in 75 Tflasks in RPMI 1640 culture