• :~ = ~ , 4 |
i ,u
ELSEVIER
J o u r n a l o f O r g a n o m e t a l l i c C h e m i s t r y 489 (1995) 1 1 3 - 1 2 2_Journal
°fO~me~l~ic
Chemistry
Synthesis, structure and substitution reactions of the binuclear
phosphido-bridged complex Cp(CO) 2W(/x-PPh 2) W(CO) 5
Shin-Guang Shyu a,., Wen-Jin Wu a, Yuh-Shang Wen a, Shie-Ming Peng b, Gene-Hsiang Lee b
a Institute of Chemistry, Academia Sinica, Taipei, Taiwan 11529, Taiwanb Department o f Chemistry, National Taiwan University, Taipei, Taiwan, Taiwan Received 15 June 1994; in revised form 10 August 1994
Abstract
Bimetallic phosphido-bridged complexes CpW(CO)3(/z-PPh2)W(CO) 5 (1) and CpW(CO)2(/x-PPh2)W(CO) 5 (2) were prepared by the reaction of CpW(CO)3PPh 2 with W(CO)sTHF. The structures of 1 and 2 were determined by single X-ra.y diffraction. Crystal data for 1: CgsHa5OsPW2, space group, P21/n with a = 9.6234 (18) .&, b = 12.4514 (20) A, c = 20.750 (4) A,/3 = 98.445 (14) °, V= 2459.4 (7) A 3, Z = 4. The structure was refined to R = 0.031 and Rs, = 0.035. Crystal data for 2: C24H15OTPW2, space
o 3
group, P21/n with a = 14.4801 (11) A, b = 11.6628 (12) A, c = 14.732 (3) A, /3 = 97.893 (11), F = 2464.3 (6) A , Z = 4. The structure was refined to R = 0.054 and R w = 0.050. The long distance between W and W (4.5096 (11) ,&) in 1 indicates that no metal-metal bond exists in the complex. The W - W distance was 3.1942 (12) ,~ in 2, indicative of a W - W bond. Substitution reactions between 2 and Lewis bases L (L = PPh3, PEt3, P(OMe) 3) produced CpW(CO)2(Iz-PPh2)W(CO)a(L) (3) with L regiospecifically and stereospecifically coordinating to the W(CO) 4 moiety trans to the phosphido bridge. The structure of CpW(CO)2(/z-PPh2)W(CO)a{P(OMe) 3} (3a) was determined by single-crystal X-ray diffraction. Crystal data for 3a: C26H2409P2W2, space group, P2~ with a = 9.2231 (16) ,&, b = 15.868 (3) A, c = 10.745 (3) ,&, /3 = 110.282 (18) °, V= 1475.1 (5)
o 3
A , Z = 2. The structure was refined to R = 0.037 and R w = 0.039. Under similar reaction conditions, complex 1 did not react with L.
Keywords: Tungsten; Group 6; Carbonyl; ~'-bonding; Cyclopentadienyl; Bridging ligand
I. I n t r o d u c t i o n
One special feature of binuclear m e t a l - m e t a l bonded phosphido-bridged complexes is the influence of the m e t a l - m e t a l bond in the reactions [1]. First, the m e t a l - m e t a l bond functions as a switch to control the reaction according to the properties of the ligand on the complex [la,b]. This behavior provides not only an empty site for further ligand coordination to the binu- clear complex when the m e t a l - m e t a l bond opens [lb], but also a driving force for further reaction (e.g. migra- tion of the ligand) when the m e t a l - m e t a l bond reforms [la]. Second, one metal may exert an activating influ- ence on the adjacent metal through the m e t a l - m e t a l bond [ld]. We have reported that the carbonyl ligand on Mo in Cp(CO)2W(tz-PPh2)Mo(CO)5 (5) can be sub-
* C o r r e s p o n d i n g a u t h o r .
0 0 2 2 - 3 2 8 X / 9 5 / $ 0 9 . 5 0 © 1995 E l s e v i e r S c i e n c e S.A. All r i g h t s r e s e r v e d SSDI 0 0 2 2 - 3 2 8 X ( 9 4 ) 0 5 1 5 2 - 6
stituted by a Lewis base L ( L = P P h 2 H , PMe3, P(OMe) 3) with the opening of the m e t a l - m e t a l bond to produce Cp(CO)3W(/x-PPh2)Mo(CO)4L (6) under very mild conditions [2]. The origin of this cooperativity effect was proposed as the influence of the CpW(CO) 2 moiety on the Mo(CO)5 through the m e t a l - m e t a l bond in the complex. Carbonyl ligands on the tungsten is less facile towards substitution than carbonyl ligands on the molybdenum. We thus synthesized Cp(CO)3W(/z- PPh2)W(CO) 5 (1) and C p ( C O ) 2 W ( / z - P P h 2 ) W ( c o ) 5 (2), and studied their substitution reaction towards Lewis bases L (L = PPh3, PEt3, P(OMe) 3) in order to evalu- ate the cooperativity effect between the metals in the complexes. We found that the carbonyl ligand in 2 was substituted with a Lewis base L with the m e t a l - m e t a l bond remaining intact to produce Cp(CO)2W~-(-/.t - P P h 2 ) W ( c O ) 4 L (3) with L regiospecifically and stere- ospecifically coordinating to the W(CO) 4 fragment
114 S.-G. Shyu et aL /Journal of Organometallic Chemistry 489 (1995) 113-122
intact under similar reaction conditions. Reported herein are the synthesis, structure, and reactivity stud- ies of 2. Scheme 1 shows the reactions that comprise the main focus of our work. The structures of com- plexes 1 and Cp(CO)2W(/.t-PPh2)W(CO)a{P(OMe) 3} (3a) were also determined by a complete single-crystal X-ray diffraction study.
2. Experimental section
Unless otherwise stated, all reactions and manipula- tions of air-sensitive compounds were carried out at ambient temperatures under an atmosphere of purified N 2 with standard procedures. A 450 W Hanovia medium-pressure quartz mercury-vapor lamp (Ace Glass) and a Pyrex Schlenk tube as a reaction vessel were used in the photoreactions. Infrared (IR) spectra were recorded on a Perkin-Elmer 882 infrared spec- trophotometer, all, 13C and 31p NMR spectra were measured using Bruker AMX-500, MSL-200, AC-200 and AC-300 spectrometers. 3~p NMR shifts are refer- enced to 85% H3PO 4. Except as noted, N M R spectra were collected at room temperature. Electron impact (EI) and fast-atom bombardment (FAB) mass spectra were recorded on a VG 70-250S or a JEOL JMS-HX 110 mass spectrometer. Microanalyses were performed in the Microanalytic Laboratory at National Cheng Kung University, Tainan, Taiwan.
at room temperature, the solvent was removed and the residue was chromatoghraphed on silica gel. Elution with CHzC12/hexane (1:4) afforded two fractions. A purple solid 2 was obtained from the first band. Yield: 1.66 g (21%). (Found: C, 35.14; H, 2.03. Calc. for C24H1507PW2: C, 35.41; H, 1.86). IR spectrum (THF, v(CO)): 2073m, 1954s, 1930s, 1861w cm -1. aH NMR spectrum (CDC13): 6 7.75 (m, 2H), 7.43 (m, 3H), 7.28 (m, 3H), 7.00 (m, 2H), 5.17 (s, 5H, C5H5). 13C{IH} NMR spectrum (CDC13): 226.56 (d, J ( C - P ) = 17.9 Hz, CpW-CO), 221.74 (s, CpW-CO), 197.71 (d, J ( C - P ) = 12.7 Hz, W-CO), 196.0 (br, W-CO), 142.36 (d, J ( P - C ) = 15.30 Hz,
ipso-C,
PPh2), 141.46 (d, J ( P - C ) = 16.35 Hz,ipso-C',
PPh2), 134.37 (d, J ( P - C ) = 9.21 Hz, o-C, PPh2), 131.80 (d, J ( P - C ) = 11.47 Hz, o-C', PPh2), 129.97 (s, p-C, PPh2), 129.27 (s, p-C', PPh2), 128.64 (d, J ( P - C ) = 9.81 Hz, m-C, PPh2), 128.15 (d, J ( P - C ) = 11.87 Hz, m-C', PPh2), 91.87 (s, C5H5). 31p{1H} NMR spectrum (THF): 6 145.9 ( J ( P - W ) = 325.3 Hz, J ( P - W ) = 131.1 Hz). MS(FAB), M +1m / z
814. The second band was yellow in color. After removing the solvent, 1 was obtained as a yellow solid. Yield: 1.42 g (17%). (Found: C, 35.63; H, 1.45. Calc. for C25H15OsPW2: C, 35.66; H, 1.80). IR spectrum (THF, u(CO)): 2065m, 2026m, 1941s, 1927s, 1910sh cm -1. 1H NMR spectrum (CDC13): t~ 7.38 (br, 12H), 5.36 (s, 5H, C5H5). 31p{IH} NMR spectrum (THF): 6 -62.72 (s, J ( P - W ) = 214.8 Hz, J ( P - W ) = 92.7 Hz). M +1m / z
842.2.1. Materials
THF was distilled from potassium and benzophe- none under an atmosphere of N 2 immediately before use. Other solvents were purified according to estab- lished procedures [3]. Tungsten hexacarbonyl, PPhzCI, PEt 3 and PPh 3 were obtained from Strem; P(OMe) 3 was purchased from Merck. Other reagents and sol- vents were obtained from various commercial sources and used as received. Na[CpW(CO)3]" 2DME [4] and WCp(CO)3PPh 2 [5] were prepared by literature proce- dures.
2.2. Synthesis of CpW(CO)3Ox-PPhe)W(CO)s (1) and
CpW(CO)e(~-PPhe)W(CO) s (2)
A yellow solution of Na[CpW(CO)3] • 2DME (5.33 g, 9.94 mmol) in 100 ml of THF was cooled to 0°C. A solution of 1.84 ml (9.97 mmol) of PPhzC1 in 50 ml of THF was then added slowly to the above solution. After 1 h, the color of the solution turned to orange red. Meanwhile, W(CO) 5 • THF was prepared in situ by UV photolysis of W(CO) 6 (5.0 g, 14.2 mmol) in THF. The orange red solution of CpW(CO)sPPh 2 was then added slowly to the above solution at 0°C. The solution turned to dark red immediately. After stirring overnight
Ph2 Ph2 P p
/ \
/ \
Cp(CO)xW W(CO)s CO \ / /x W(CO)5 hv Cp(CO)3W 1 L ,,-7-,, L L=P(OMe)3 PPh3 PEt 3 p C ~ 0 Ph 2 C ° 7 Ph 2 0 w 4 3 S c h e m e 1. 3a, L = P(OMe)3 3b, L = PPh 3 3c, L = PEt 3S.-G. Shyu et al. / Journal of Organometallic Chemistry 489 (1995) 113-122 115
2.3. Synthesis of
CpW(CO)2(tx-PPh2)W(co)4(P(OMe)3)
(3a)
To a purple solution of 2 (0.20 g, 0.25 mmol) in 15 ml of THF was added 30/xl of P(OMe) 3 (0.25 mmol) under N 2 at room temperature. The solution changed to dark red after the solution was heated under reflux overnight. The solvent was then removed and the residue was chromatographed on silica gel. Elution with CH2Clz/hexane (1:4) afforded two fractions. The first band, which was purple in color, was unreacted 2. The second band was reddish brown. After the solvent was removed, a red solid 3a was obtained. Yield: 0.20 g
(88%). (Found: C, 34.36; H, 2.67. Calc. for C26H24-
O9P2W2: C, 34.31 H, 2.66). IR spectrum (THF, v(CO)): 2034w, 1966vw, 1931s, 1917s, 1846m cm -1. 1H NMR spectrum (CDC13): 6 7.78 (m, 2H), 7.37 (m, 3H), 7.22 (m, 3H), 6.99 (m, 3H), 5.07 (s, 5H, C5H5), 3.70 (d, 9H, 2J(P-H) = 11.7 Hz, POCH3). 31p{IH} NMR spectrum (THF): 6 142.6 (d, 2j(p_p) = 50.9 Hz, J(P-W) = 314.8 Hz, J ( P - W ) = 155.0 Hz,/z-PPh2), 134.5 (d, 2j(p_p)= 50.9 Hz, J ( P - W ) = 421.0 Hz, P(OMe)3). MS(FAB), M +l m / z 910.
2.4. Synthesis of
CpW(CO)2(pt-PPh2)W(co)4(PPh3)
(3b)
To a purple solution of 2 (0.20 g, 0.25 mmol) in 30 ml of THF was added PPh 3 (0.064 g, 0.25 mmol) under N 2 at room temperature. The solution changed to dark red after the mixture was heated at reflux temperature overnight. Solvent was then removed and the residue was chromatographed on silica gel. Elution with CH2C12/hexane (1 : 4) afforded two fractions. The first band which was purple in color was unreacted 2. The second bad was red. After the solvent was removed, red solid 3b was obtained. Yield: 0.18 g (71%). (Found:
C, 46.72; H, 2.74. Calc. for C41H3006P2W2: C, 46.97; H, 2.88). IR spectrum (THF, v(CO)): 2027w, 1960vw, 1928s, 1908s, 1843m cm-i. 1H NMR spectrum (CDC13): 6 7.41 (m, 25H), 5.06 (s, 5H, C5H5). 3Xp{IH} NMR spectrum (THF): 6 143.0 (d, 2 j ( p _ p ) = 28.5 Hz, /z- PPh2), 21.05 (d, 2 j ( p _ p ) = 28.5 Hz, PPh3). 13C{1H} NMR spectrum (CDCI3): 230.91 (d, J(C-P) = 17.0 Hz, CpW-CO), 223.68 (s, CpW-CO), 198 (br, W-CO), MS(FAB), M +1 m / z 1048.
2. 5. Synthesis of Cp
W(co) 2 (]3.-PPh
2)W(co)4
(PEt3) (3e)To a purple solution of 2 (0.20 g, 0.25 mmol) in 30 ml of THF was added 50 /zl PEt 3 (0.34 mmol) under N 2 at room temperature. The solution changed to dark red after the mixture was heated under reflux overnight. The solvent was then removed and the residue was chromatographed on silica gel. Elution with CH2C12/ hexane (1:4) afforded two fractions. The first band,
which was purple in color, was unreacted 2. The sec- ond band was red. After the solvent was removed, a red solid 3e was obtained. Yield: 0.13 g (45%). (Found:
C, 38.89; H, 2.99. Calc. for C29H3006PzW2 : C, 38.53; H, 3.34). IR spectrum (THF, v(CO)): 2021m, 1951w, 1924s, 1901s, 1840m cm-i. 1H NMR spectrum (CDC13): /~ 7.77 (m, 2H), 7.38 (m, 3H), 7.22 (m, 3H), 6.99 (m, 2H), 5.04 (s, 5H, CsH5), 2.07 (dq, 6H, 2j(H-H) = 7.63 Hz, 2 J ( p - H ) = 7.63 Hz, PCH2), 1.13 (dt, 9H, 2j(H-H) = 7.48 Hz, 2 J ( p - H ) = 16.02 Hz, PCH2CH3). 31p{1H} NMR spectrum (THF): ~ 140.1 (d, 2j(p_p) = 24.4 Hz; J ( P - W ) = 3 0 9 . 2 Hz, J ( P - W ) = 158.4 Hz, /x-PPh2), - 2 . 9 (d, 2j(p_p) = 24.4 Hz; J(P-W) = 214.8 Hz, PEt3). MS (FAB), M +1 m / z 904.
2.6. Reaction of 1 with PR 3 (R = Ph, Et, OMe)
To a yellow solution containing 100 mg (0.125 mmol) of 1 in 20 ml of THF was added 30/zl (0.25 mmol) of P(OMe) 3. The solution was heated under reflux overnight and no color change was observed. Results of a 31p NMR study of the reaction mixture indicated the existence of unreacted 1 and P(OMe) 3 and small amount of unidentified impurities. Similar reaction conditions were applied to the reaction between 1 and PR 3 (R = Ph, Et). Unreacted 1, PR 3 (R = Ph, Et) and a small amount of unidentified impurities were ob- served in the reaction product according to the 31p NMR spectra of the reaction mixture.
2. 7. Reaction between 2 and CO
A solution of 2 in THF was stirred overnight under 1 atom of CO. No color change was observed. Only 2 was observed in the 31p NMR spectrum of the solution.
2.8. Thermolysis of 1
A solution of 1 in THF was heated under reflux under N 2 overnight in the dark. No color change was observed. Only 1 was observed in the 31p NMR spec- trum of the solution.
2.9. Photolysis of 1
Compound 1 (1.77 g) was dissolved in 70 ml of THF under N 2. The solution was irradiated with UV for 2 h. The color of the solution changed from yellow to reddish brown. The 31p NMR of the mixture indicated that all of 1 was used in the reaction. The solvent was then removed and the residue was chromatographed on grade III A120 3. Elution with hexane afforded two fractions. Only a trace amount of product was obtained from the first band which was pink in color, and was not identified. The second band was purple in color.
116 S.-G. Shyu et al. /Journal of Organometallic Chemistry 489 (1995) 113-122
A f t e r the solvent was removed, 2 was obtained as a purple solid. Yield: 0.203 g (12%).
2.10. Structure determination o f I, 2, 3a
Crystals of complexes 1, 2 and 3a were grown by slow diffusion of hexanes to the saturated solutions of the relevant complexes in CH2C12 at - 1 5 ° C . Cell dimensions and space group data were obtained by standard m e t h o d s on an E n r a f Nonius C A D 4 diffrac- tometer. Details of data collection and refinement are given in Table 1.
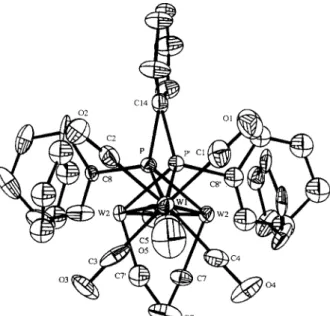
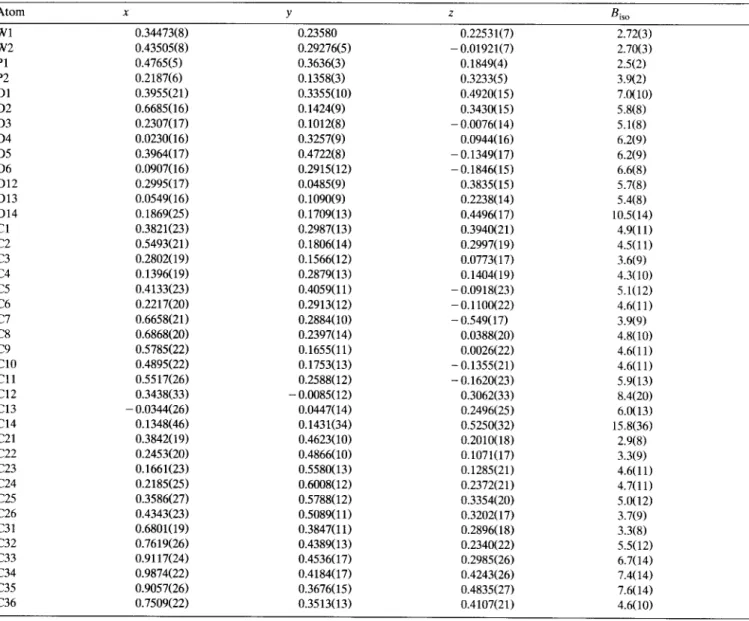
T h e coordinates of the heavy atoms were obtained from Patterson syntheses. T h e positions of the remain- ing non-hydrogen atoms were obtained from Fourier synthesis. For complex 2, inspection of a difference Fourier revealed severe disorders (Fig. 1). T h e r e were dominant and minor fractions; the ratio was deter- mined to be 0.8:0.2. Except for W (anisotropic) and P (isotropic), the minor portion was fixed in the final refinement. T h e final model of the major fraction was o b t a i n e d with all n o n - h y d r o g e n a t o m s r e f i n e d anisotropically and H atoms at idealized positions with R = 5.4% and R w = 5.0%. T h e final positional p a r a m e - ters of the complexes are !isted in Tables 2 (1), 3 (2), and 4 (3a). Selected interatomic distances and b o n d angles are given in Table 5 (1 and 2) and 6 (3a).
Additional material available f r o m the Cambridge Crystallographic D a t a C e n t e r comprises H - a t o m coor- dinates, t h e r m a l p a r a m e t e r s and remaining b o n d lengths and angles.
3 . R e s u l t s a n d d i s c u s s i o n
3.1. Synthesis, spectroscopic characterization and molec- ular structures o f I and 2
T h e new W - W complexes 1 and 2 were synthesized by the reaction of C p W ( C O ) 3 P P h 2 with W(CO)5(THF). T h e metallophosphine C p W ( C O ) 3 P P h 2 acted as a lig- and to replace the T H F ligand on W ( C O ) 5 ( T H F ) to form the complexes according to Eq. (1) [6].
C p W ( C O ) 3 P P h 2 + W ( C O ) 5 ( T H F ) / P P h 2 , Cp(CO)3 w j ~ W ( C O ) 5
(1)
j P P h 2 + C p ( C O ) 2 W , W ( C O ) 5 (1)(2)
R e p l a c e m e n t of the T H F ligand on W ( C O ) 5 ( T H F ) with C p W ( C O ) 3 P P h 2 should form 1. T h e formation of 2 in the reaction could be due to the elimination of oneC O from 1 u n d e r the synthetic conditions. Thermolysis of 1 in T H F under N 2 for 12 h did not produce 2. U V irradiation of 1 in T H F p r o d u c e d 2 in 12% yield, which might provide a reasonable explanation for the forma- tion of 2. However, no 2 was observed after the T H F solution of 1 was stirred u n d e r household fluorescence light at r o o m t e m p e r a t u r e for 12 h. Therefore, the formation of 2 in the reaction could go through a different, unknown reaction path.
T h e 31p{IH} N M R spectrum of 2 in T H F at room t e m p e r a t u r e shows resonance at 145.9 p p m with two sets of J ( P - W ) satellites. This relatively downfield resonance reveals the existence of a m e t a l - m e t a l bond [7]. In contrast, the relatively upfield resonance at - 6 2 . 7 p p m with two sets of J ( P - W ) satellites in the 3t p{1H} N M R of 1 indicates the opening of the W - P - W triangle [7]. T h e W - W complexes 1 and 2 were further characterized by single X-ray diffraction studies. Struc- tures are shown in Figs. 2 and 3.
Table 1
Crystal and intensity collection data for 1, 2 and 3 a
1 2 3 a Molecular C25H1508PW 2 C24H1507PW2 C26H2409P2W 2 formula Mol. wt. 842.06 814.03 910.12 Space group P21/n P21/n P21 a (,~) 9.6234 (18) 14.4801 (11) 9.2231 (16) b (,~) 12.4514 (20) 11.6628 (12) 15.868 (3) c (.~) 20.750 (4) 14.732 (3) 10.745 (3) ,~ (°) - _ _ /3 (°) 98.445 (14) 97.893 (11) 110.282 (18) ~, (o) _ _ _ v (.~3) 2459.4 (7) 2464.3 (6) 1475.1 (5) p (calc.) (rag m -3) 2.274 2.189 2.049 Z 4 4 2 Crystal 0.31×0.31× 0.10×0.20x 0.50×0.44× dimensions (mm) 0.34 0.50 0.25 Absolute 9.66 9.63 8.11 coefficient p, (Mo Ka) (ram -1)
Temperature Room Room Room
temperature temperature temperature
Radiation 0.71073 0.71073 0.71073
(Mo Ka,~, ,~,)
20 range (°) 45 45 50
Scan type 20-to 20-w 20-to
No. of 3207 4329 2684 reflections No. of observed 2626 ( > 2.0o- (I)) 2890 ( > 2.0o- (I)) 2248 ( > 2.5cr (I)) reflections Variables 325 325 353 R 0.031 0.054 0.037 R w 0.035 0.050 0.039 S 2.48 3.41 1.70 AF (e .~-3) 0.780 1.440 1.450 (A/~r)max < 0.01l < 0.056 < 0.072
S.-G. Shyu et aL / Journal of Organometallic Chemistry 489 (1995) 113-122 117
CI4
C8 C8'
Fig. I. OWrEP drawing of disordered molecular structure of 2.
In 1, a W - W distance of 4.5096(11) A indicates that there is no metal-metal bond. The metallophosphine CpW(CO)3PPh 2 can be considered to be a ligand similar to PR 3. Thus, five COs and CpW(CO)3PPh 2 coordinate to the W(1) atom to form a distorted octa- hedron.
The W - W distance (3.1943(12) A) in 2 falls within the range of W - W distances reported for other mono- bridging complexes [8]. It is slightly shorter than the unsupported W - W single bond (3.222(1) A) in Cp2W2(CO) 0 [9] and longer than the W - W distance (3.0256(4) A) in bis (/x-diphenylphosphido) complex
W 2 ( C O ) 8 ( g , - P P h 2)2
[10].
We prefer to formulate the metal-metal bonding in 2 as a donor-acceptor bond from a W(1) ° center to W(2) n, similar to descriptions given earlier for
C p ( C O ) 2 W ( / z - P P h z ) I V I o ( C O ) 5
[2],
( P P h 3 X C O ) 3 F ~ - / x -P--P-h2)Ir(CO)2(PPh 3) [11], and (CO)sW(tt-PPh2)Re
(CO) 4 [lb]. The W(1)-W(2)
bond in 2 can be consid-Table 2
Atomic coordinates for 1
Atom x y z Bis o Wl 0.29194(6) 0.78964(5) 0.00456(3) W2 0.00829(5) 0.76079(5) 0.15845(3) P 0.24765(34) 0.71520(29) 0.11820(16) O1 0.3424(13) 1.0354(9) 0.0393(6) 0 2 - 0.0245(10) 0.8517(9) - 0.0473(5) 0 3 0.2219(13) 0.5571(9) - 0.0541(5) 0 4 0.3717(11) 0.8533(10) - O. 1297(5) O5 0.6191(10) 0.7482(11) 0.0490(5) 0 6 0.1606(10) 0.6231(9) 0.2749(4) 0 7 - 0.2325(10) 0.6059(9) 0.1875(5) 0 8 - 0.0693(11) 0.6367(8) 0.0255(5) C1 0.3256(15) 0.9441(14) 0.0294(8) C2 0.0874(17) 0.8290(12) - 0.0278(7) C3 0.2451(15) 0.6382(15) - 0.0335(7) C4 0.3397(16) 0.8316(13) - 0.0816(7) C5 0.5039(17) 0.7607(14) 0.0344(7) C6 0.1070(13) 0.6706(11) 0.2327(7) C7 - 0.1448( 14 ) 0.6594(12) 0.1784(7) C8 - 0.0410(13) 0.6780(12) 0.0733(7) C9 0.0696(16) 0.9405(11) 0.1410(9) C10 - 0.0640(18) 0.9293(12) 0.1097(8) C11 - 0.1475(15) 0.9043(11) 0.1575(9) C12 - 0.0620(16) 0.9010(12) 0.2197(7) C13 0.0733(15) 0.9217(11) 0.2103(8) C21 0.2759(14) 0.5676(10) 0.1186(6) C22 0.1776(14) 0.4905(12) 0.1274(6) C23 0.1986(15) 0.3827(11) 0.1237(6) C24 0.3307(19) 0.3474(11) 0.1125(7) C25 0.4366(16) 0.4218(12) 0.1046(7) C26 0.4106(15) 0.5287(12) 0.1089(6) C31 0.3890(13) 0.7595(12) 0.1853(6) C32 0.4376(14) 0.8664(11) 0.1887(7) C33 0.5407(15) 0.8978(13) 0.2367(7) C34 0.5973(15) 0.8312(14) 0.2829(7) C35 0.5481(14) 0.7255(13) 0.2820(6) C36 0.4466(14) 0.6894(12) 0.2348(7) 2.66(3) 2.46(2) 2.39(15) 6.2(7) 5.3(6) 6.1(7) 5.9(7) 6.1(7) 4.3(6) 5.1(6) 4.6(5) 4.3(8) 4.1(8) 4.5(9) 4.2(8) 4.7(9) 2.9(7) 3.3(7) 3.2(7) 4.2(9) 4.5(9) 4.4(9) 4.1(8) 4.1(8) 2.5(6) 3.1(7) 3.4(7) 4.4(9) 4.1(8) 3.5(7) 3.1(7) 3.5(7)
3.8(8)
4.1(8) 3.8(8) 3.7(7)118 S. -G. Shyu et al. / Journal of Organometallic Chemistry 489 (1995) 113-122 ered as the donation of an electron pair from one of
the filled t2g orbitals of W(1) to the adjacent W(2) such that the W(1)-W(2) dative bond acts as the fifth lig- and, donating two electrons to W(2), in addition to the two COs, the/x-PPh2, and the Cp ligands coordinated to W(2). Consistent with this view is the observation that the W(1)-W(2) vector bisects an edge of the distorted W(1) octahedron and the W(1) atom lies on the least squares plane consisting of P, C(5), C(4), C(2). The W(1)-W(2) vector was only 1.13 ° off the plane [12].
In complex 2, the W(1)-C(4)-O(4) angle 174.1 (15) ° could indicate a semibridging carbonyl. The observed IR at 1861 cm - t at room temperature and the W(2)- C(4) distance (2.982(18) A) which is shorter than the sum of the atomic radius of W (1.30 A) [13] and the van der Waals radius of C (1.85 .&) [13], further sup- ports this assignment [14]. This semibridging carbonyl and the other three W(1) COs which are cis to the phosphido bridge are fluxional in solution at room temperature as indicated by the observed broad hump at 196.0 ppm in its 13C NMR spectrum. The exchange
of the four cis W(1) COs probably proceed through the rotation of the W(1)-P bond. This type of fluxional behavior has been reported for several mono (/x-phos- phido) carbonyl complexes [2,6a,15].
3.2. Substitution reaction between 2 and phosphine (P(OMe)~, PPh 3, PEt s)
Reaction of 2 with phosphine L (L = P(OMe)3, PPh 3, PEt 3) in THF at reflux temperature yielded 3 with L regiospecifically and stereospecifically coordi- nating to the W(1) trans to the phosphido bridge. A downfield shift of the phosphido-bridge phosphorus
signal in the 31p NMR (140.1-143.0 ppm) indicates
that the metal-metal bond exists. The regiospecific
and stereospecific assignment is revealed by the
13C
NMR spectrum of 3b. The observed signals at ~ 230.91 (d, 2J(P-C)= 17.11 Hz) and 223.68(s) are assigned to the W(2) COs based on a favorable comparison with the reported resonances at ~ 230.95 (d, 2J(p-c) = 15.87 Hz) and 223.54(s) for the W CO signals in the 13C NMR spectrum of Cp(CO)2W(/x-PPh2)Mo(CO)n(PPh3)
Table 3
Atomic coordinates for 2
Atom x y z Bis o W l 0.19136(5) 0.24519(8) - 0.14029(4) 3.08(3) W2 0.30635(7) 0.13445(9) 0.03891(6) 3.36(4) P 0.1964(4) 0.2866(5) 0.0294(4) 3.0(3) C1 0.0919(14) 0.1234(19) - 0.1344(13) 5.9(12) C2 0.0966(12) 0.3764(18) - 0.1465(12) 4.4(10) C3 0.2883(14) 0.3630(21) -0.1563(12) 6.3(12) C4 0.2822(12) 0.1223(20) -0.1651(12) 5.1(11) C5 0.1561(12) 0.2426(22) -0.2761(11) 5.0(10) C6 0.3669(11) 0.2359(23) 0.1459(11) 5.7(12) C7 0.4166(14) 0.1967(22) - 0.0164(13) 4.2(12) C8 0.2337(13) 0.4337(18) 0.0620(12) 2.7(9) C9 0.3147(15) 0.464(3) 0.0476(14) 5.5(15) C10 0.3444(18) 0.569(3) 0.0673(16) 7.2(18) C l l 0.2827(20) 0.658(3) 0.1066(16) 8.0(18) C12 0.1928(19) 0.6145(23) 0.1224(15) 6.1(15) C13 0.1621(22) 0.5041(20) 0.0969(14) 6.1(16) C14 0.0905(12) 0.2599(18) 0.0813(10) 4.2(9) C15 0.0041(11) 0.2739(23) 0.0352(10) 5.6(13) C16 - 0.0752(11) 0.2647(22) 0.0795(10) 4.7(10) C17 - 0.0632(11) 0.2469(21) 0.1729(11) 4.9(10) C18 0.0234(13) 0.237(3) 0.2164(11) 6.6(14) C 19 0.0994(12) 0.2399(22) 0.1744( 11 ) 5.3( 11 ) C20 0.3622(18) - 0.036(3) 0.1108(17) 6.8(16) C21 0.3411(19) - 0.0619(24) 0.0212(18) 6.8(16) C22 0.2510(19) - 0.0509(19) - 0.0025(15) 5.6(15) C23 0.2027(17) - 0.0160(20) 0.0722(16) 5.0(13) C24 0.2763(19) - 0.0037(22) 0.1437(15) 5.6(15) O1 0.0361(10) 0.0557(14) - 0.1296(11) 8.4(10) 0 2 0.0454(10) 0.4472(14) - 0.1576(11) 7.7(10) 0 3 0.3457(10) 0.4321(14) - 0.1697(10) 7.9(10) 0 4 0.3319(11) 0.0496(15) - 0.1877(9) 8.2(10) 0 5 0.1362(11) 0.2405(17) - 0.3540(8) 8.8(11) 0 6 0.4033(9) 0.2705(15) 0.2052(7) 6.7(8) 0 7 0.4783(8) 0.2379(19) - 0.0458(9) 8.5(11)
S.-G. Shyu et al. /Journal of Organometallic Chemistry 489 (1995) 113-122 119
[2]. The observed broad hump at 199 ppm, which is assigned to the four fluxional W(1) COs cis to the phosphido bridge, and the absence of the correspond- ing t r a n s CO signal indicates that the phosphine ligand occupies the position t r a n s to the phosphido bridge (Fig. 5). No reaction was observed when the mixture of 2 and phosphines was stirred overnight at room tem- perature.
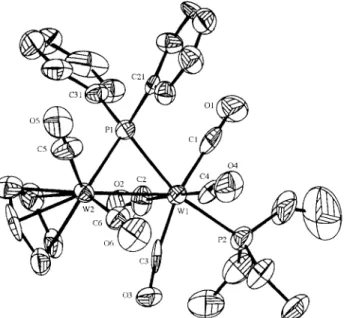
The structure of 3a was determined by single-crystal X-ray diffraction study. The W(1)-W(2) distance was 3.1510(12) ,4, which indicates that a metal-metal bond exists in the complex. It is 0.04 ,~ shorter than the W(1)-W(2) distance in 2. The replacement of the CO ligand from 2 with a better electron donor ligand P(OMe) 3 to form 3a could enhance the basicity of the W(1) moiety which resulted in the strengthening of the dative bonding. The P(OMe) 3 ligand is coordinated to the W(1) atom and is t r a n s to the phosphido bridge.
The W(1)-P(1) distance (2.479(5) ,~) in 3a is shorter than the corresponding W(1)-P distance (2.538(5) ,~) in 2. This can be explained by the replacement of the CO ligand with a poor 7r-acceptor ligand phosphine [16].
Unlike complex 5, reaction of 2 with phosphine resulted in the substitution of one W(1) CO with phosphine instead of opening the metal-metal bonding to produce an addition product
Cp(CO)3W(/z-
PPhz)W(CO)4L(4) (Scheme 1) [2]. One possibility is that the W - W bonding in 2 is stronger than the W - M o bonding in 5 such that direct substitution of one CO on the W(1) dominated in 2. The stronger metal-metal bond in 2 may be indicated by the inertness of the complex towards CO since complex 5 reacts with CO under very mild conditions (room temperature, 1 atm of CO) to produce the addition product Cp(CO)3W(/~- PPh2)Mo(CO) 5 [2]. The other possibility is that theTable 4 A t o m i c coordinates for 3a A t o m x y z Bis o W1 0.34473(8) 0.23580 0.22531(7) W2 0.43505(8) 0.29276(5) - 0.01921(7) P1 0.4765(5) 0.3636(3) 0.1849(4) P2 0.2187(6) 0.1358(3) 0.3233(5) O1 0.3955(21) 0.3355(10) 0.4920(15) 0 2 0.6685(16) 0.1424(9) 0.3430(15) 0 3 0.2307(17) 0.1012(8) - 0.0076(14) 0 4 0.0230(16) 0.3257(9) 0.0944(16) 0 5 0.3964(17) 0.4722(8) - 0.1349(17) 0 6 0.0907(16) 0.2915(12) - 0.1846(15) 012 0.2995(17) 0.0485(9) 0.3835(15) 013 0.0549(16) 0.1090(9) 0.2238(14) 014 0.1869(25) 0.1709(13) 0.4496(17) C1 0.3821(23) 0.2987(13) 0.3940(21) C2 0.5493(21) 0.1806(14) 0.2997(19) C3 0.2802(19) 0.1566(12) 0.0773(17) C4 0.1396(19) 0.2879(13) 0.1404(19) C5 0.4133(23) 0.4059(11) - 0.0918(23) C6 0.2217(20) 0.2913(12) - O. 1100(22) C7 0.6658(21) 0.2884(10) - 0.549(17) C8 0.6868(20) 0.2397(14) 0.0388(20) C9 0.5785(22) 0.1655(11) 0.0026(22) C10 0.4895(22) 0.1753(13) - 0.1355(21) C11 0.5517(26) 0.2588(12) - 0.1620(23) C12 0.3438(33) - 0.0085(12) 0.3062(33) C13 - 0.0344(26) 0.0447(14) 0.2496(25) C14 0.1348(46) 0.1431(34) 0.5250(32) C21 0.3842(19) 0.4623(10) 0.2010(18) C22 0.2453(20) 0.4866(10) 0.1071(17) C23 0.1661(23) 0.5580(13) 0.1285(21) C24 0.2185(25) 0.6008(12) 0.2372(21) C25 0.3586(27) 0.5788(12) 0.3354(20) C26 0.4343(23) 0.5089(11) 0.3202(17) C31 0.6801(19) 0.3847(11) 0.2896(18) C32 0.7619(26) 0.4389(13) 0.2340(22) C33 0.9117(24) 0.4536(17) 0.2985(26) C34 0.9874(22) 0.4184(17) 0.4243(26) C35 0.9057(26) 0.3676(15) 0.4835(27) C36 0.7509(22) 0.3513(13) 0.4107(21) 2.72(3) 2.70(3) 2.5(2) 3.9(2) 7.0(10) 5.8(8) 5.1(8) 6.2(9) 6.2(9) 6.6(8) 5.7(8) 5.4(8) 10.5(14) 4.9(11) 4.5(11) 3.6(9) 4.3(10) 5.1(12) 4.6(11) 3.9(9) 4.8(10) 4.6(11) 4.6(11) 5.9(13) 8.4(20) 6.0(13) 15.8(36) 2.9(8) 3.3(9) 4.6(11) 4.7(11) 5.0(12) 3.7(9) 3.3(8) 5.5(12) 6.7(14) 7.4(14) 7.6(14) 4.6(10)
120 S.-G. Shyu e t al. /Journal of Organometallic Chemistry 489 (1995) 113-122
Table 5
Selected bond lengths (A) and bond angles (°) in complex 1 and 2
Complex 1 Complex 2
Selected bond lengths (,~)
W(1)-W(2) 4 . 5 0 9 6 ( 1 1 ) W(1)-W(2) 3.1942(12) W(1)-P 2.626(3) W(1)-P 2.538(5) W(1)-C(1) 2 . 0 0 5 ( 1 8 ) W(1)-C(1) 2.033(22) W(1)-C(2) 2 . 0 4 4 ( 1 6 ) W(1)-C(2) 2.049(20) W(1)-C(3) 2 . 0 6 9 ( 1 8 ) W(1)-C(3) 2.001(25) W(1)-C(4) 1.981(15) W(1)-C(4) 2.013(21) W(1)-C(5) 2 . 0 7 4 ( 1 7 ) W(1)-C(5) 1.996(16) W(2)-P 2.625(3) W(2)-P 2.376(3) W(2)-C(6) 2 . 0 2 6 ( 1 5 ) W(2)-C(6) 2.067(18) W(2)-C(7) 2 . 0 2 9 ( 1 5 ) W(2)-C(7) 2.024(21) W(2)-C(8) 2 . 0 4 0 ( 1 5 ) C(1)-O(1) 1.14(3) C(1)-O(1) 1.163(21) C(2)-O(2) 1.108(24) C(2)-O(2) 1.129(19) C(3)-O(3) 1.19(3) C(3)-O(3) 1.106(21) C(4)-O(4) 1.19(3) C(4)-O(4) 1.119(18) (7(5)-0(5) 1.145(19) C(5)-O(5) 1.117(19) (7(6)-0(6) 1.037(20) C(6)-O(6) 1.118(17) (7(7)-0(7) 1.15(3) C(7)-O(7) 1.113(18) C(8)-O(8) 1.116(17) Selected bond angels (°)
W(1)-P-W(2) 118.36(13) W(1)-P-W(2) 81.02(18) P-W(1)-C(1) 98.6(4) P-W(1)-C(1) 90.9(6) P-W(1)-C(2) 96.0(4) P-W(1)-C(2) 80.2(5) P-W(1)-C(3) 88.1(4) P-W(1)-C(3) 93.4(5) P-W(1)-C(4) 1 7 3 . 5 ( 5 ) P-W(1)-C(4) 112.6(5) P-W(1)-C(5) 87.3(4) P-W(1)-C(5) 163.6(6) P-W(2)-C(6) 77.7(4) P-W(2)-C(6) 80.1(6) P-W(2)-C(7) 1 2 8 . 9 ( 4 ) P-W(2)-C(7) 105.5(7) P-W(2)-C(8) 178.8(18) W(1)-C(1)-O(1) 175.3(13) W(1)-C(1)-O(1) 178.8(18) W(1)-C(2)-O(2) 178.0(12) W(1)-C(2)-O(2) 174.1(16) W(1)-C(3)-O(3) 179.0(13) W(1)-C(3)-O(3) 177.2(15) W(1)-C(4)-O(4) 177.2(13) W(1)-C(4)-O(4) 174.1(15) W(1)-C(5)-O(5) 177.4(14) W(1)-C(5)-O(5) 179.6(19) W(2)-C(6)-O(6) 177.9(12) W(1)-C(6)-O(6) 167.9(23) W(2)-C(7)-O(7) 177.2(12) W(1)-C(7)-O(7) 176.3(22) W(2)-C(8)-O(8) 177.1(12) Table 6
Selected bond lengths (A) and bond angles (°) in complex 3a
Bond length Bond angle
W(1)-W(2) 3.1513(12) W(1)-P(1) 2.478(4) W(1)-P(2) 2.414(5) W(1)-C(1) 1.992(22) W(1)-C(2) 1.980(19) W(1)-C(3) 1.951(20) W(1)-C(4) 1.975(19) W(2)-P(1) 2.375(5) W(2)-C(5) 1.941(18) W(2)-C(6) 1.869(18) O(1)-C(1) 1.17(3) O(2)-C(2) 1.199(23) O(3)-C(3) 1.235(23) O(4)-C(4) 1.179(24) O(5)-C(5) 1.138(21) O(6)-C(6) 1.196(23) W(1)-P(1)-W(2) 80.96(13) P(1)-W(1)-P(2) 162.50(16) P(1)-W(1)-C(1) 79.0(5) P(1)-W(1)-C(2) 88.6(6) P(1)-W(1)-C(3) 114.4(5) P(1)-W(1)-C(4) 91.5(6) P(1)-W(2)-C(5) 83.9(7) P(1)-W(2)-C(6) 106.9(7) W(1)-C(1)-O(1) 176.3(17) W(1)-C(2)-O(2) 175.9(18) W(1)-C(3)-O(3) 172.3(13) W(1)-C(4)-O(4) 174.0(17) W(2)-C(5)-O(5) 178.2(18) W(2)-C(6)-O(6) 170.4(18) 0 6 C21 C6 C31 0 5 0 7 C7 C 5 W2 0 3 C3 (:4 04
Fig. 2. ORTEP drawing of 1. Hydrogen atoms are omitted.
addition product 4 was formed as an intermediate and lost a CO to produce 3 under the reaction conditions. Substitution of CO from W carbonyl usually requires a higher temperature than substitution of CO from Mo carbonyl. If the influence of the CpW(CO) 2 moiety on the adjacent metals in both complexes 2 and 5 is similar, substitution of CO from W(1) in 2 should require a higher temperature than substitution of CO from Mo in 5. Indeed, higher temperature and longer reaction time (refluxing temperature in THF for overnight) were required in the reaction between 2 and
S.-G. Shyu et al. /Journal of Organometallic Chemistry 489 (1995) 113-122 121 C21 0 5 C31 Ol Pl ( I C5 2 0 4 O~ C2 C4 P2
Fig. 4. ORTEP drawing of 3a. Hydrogen atoms are omitted.
Lewis bases than in the corresponding reaction be- tween complex 5 and Lewis bases (room temperature in T H F for several hours). However, at higher temper- ature, complex 4 may lose one CO to produce 3 since Cp(CO)3W(/z-PPh2)Mo(CO)n{P(OMe) 3} p r o d u c e s Cp(CO)2W(/x-PPh2)M0(CO)a{P(OMe) 3} under thermal conditions [2]. However, following the reaction by 3lp NMR spectroscopy, no intermediate was observed; the life time of the intermediate could be very short, result- ing in the isolation of 3 as the only product.
230 220 210 200 190 PPM
Fig. 5. Carbonyl region of the 13C{IH} N M R spectrum of 3b at 300 K.
3.3. Role of the metal-metal datice bond in the substitu- tion reaction
In the (CO)4Ru(/x-PPh2)Co(CO) 3 system, reaction
b e t w e e n ( C O ) 4 R u ~ ~ o ( C O ) 3 and phosphines L (L = PPh2Me, PMe2Ph, PPh2H) produced (CO)2L 2-
RU(/z-PPh2)Co(CO) 3 instead of ( C O ) 3 L R u ( ~ -
PPh2)Co(CO)2L. [17] Since (CO)3(PPh3)Ru~-/.t- PPh2)Co(CO)2(PPh 3) can be prepared by other syn- thetic routes, labilization of CO groups on Ru by the metal-metal bond in the complex was proposed to be one of the possible reasons [17].
One way to evaluate the influence of the metal- metal bond in the substitution reaction is to construct two types of complexes having a similar framework with and without a metal-metal bond, and to compare their substitution reactions under similar conditions. Complexes 1 and 2 are suitable for this purpose. First. both complexes have a similar framework, 2 with metal-metal bonding and 1 without. Second, complex 1 was inert when its T H F solution was heated under reflux overnight. This thermal stability makes the com- parison feasible because many non-metal-metal bonded phosphido-bridged complexes convert to the metal-metal bonded complexes under relatively mild conditions [ lb,2,11].
Carbonyl ligands on W in 1 were inert towards phosphine when a THF solution of 1 and phosphine was heated under reflux overnight. On the other hand. when 2 was reacted with phosphines under similar conditions, the substituted complex 3 was obtained. These observations clearly indicate that CO ligands on W in 2 were labilized by the metal-metal bond.
The W(CO) 5 moiety in 2 was probably activated by the electron donation from the filled t2g orbital of the W(1) atom to the W(2) atom through the dative metal-metal bond. The net result of this donation will be a decrease in d x y - - + T r* to the equatorial C O / s [ld,2], which induces weakening of the W - C O bond in 2. The adjacent metal W(2) may also labilize the W(1)- CO group through the donation of the electron from the electron rich W(2) to the rr* orbital of the adja- cent W(1)-CO bond to form the semibridging carbonyl ligand similar to complex 5 [2].
4. Conclusions
Binuclear phosphido-bridged complexes without a metal-metal bond, 1, and with a metal-metal bond 2. were synthesized in order to study the influence of the metal-metal bond in the substitution reaction. Struc- tures of both complexes were determined by single- crystal X-ray diffraction methods.
Substitution of CO in 2 by phosphines was regiospe- cific and stereospecific on the W(1) and trans to the
122 S.-G. Shyu et al. /Journal of Organometallic Chemistry 489 (1995) 113-122 phosphido bridge. The structure of 3a was determined
by single crystal X-ray methods. Under similar condi- tions, the W(1) CO in 1 was inert towards phosphines. The metal-metal bond in 2 was believed to labilize the carbonyl ligand on W(1).
Acknowledgment
We wish to thank the National Science Council, Republic of China, and Academia Sinica for financial support of this work.
References
[1] (a) S.-G. Shyu and A. Wojcicki, Organometallics, 4 (1985) 1457;
(b) W.C. Mercer, R.R. Whittle, E.W. Burkhardt and G.L. Geof- froy, Organometallics, 4 (1985) 68; (c) J. Powell, J.F. Sawyer and
M.V.R. Stainer, Inorg. Chem., 28 (1989) 4461; (d) J. Powell, C.
Coutoure and M.R. Gregg, J. Chem. Soc., Chem. Commun.,
(1988) 1208.
[2] S.-G. Shyu, J.-Y. Hsu, P.-J. Lin, W.-J, Wu, S.-M. Peng, G.-H. Lee and Y.-S. Wen, Organometallics, 13 (1994) 1699.
[3] D.D. Perrin, W.L.F. Armarego and D.R. Perrin, Purification of Laboratory Chemicals, Pergamon, Oxford, 1966.
[4] R. Bender, P. Braunstein, J.-M. Jud and Y. Dusausoy, Inorg. Chem., 22 (1983) 3394.
[5] (a) H. Adams, N.A. Bailey, A.N. Day, M.J. Morris and M.M. Harrison, J. Organomet. Chem., 407 (1991) 247; (b) W. Malisch,
R. Maisch, I.J. Colquhoun and W. McFarlane, J. Organomet. Chem., 220 (1981) C1.
[6] (a) S.G. Shyu, J.Y. Hsu and Y.-S. Wen, J. Organomet. Chem., 453 (1993) 97; (b) D.A. Roberts and G.L. Geoffroy, in G.
Wilkinson, F.G.A. Stone and E. Abel (eds.), Comprehensive Organometallic Chemistry, Vol. 6. Pergamon Press, London,
1982, pp. 780-783; (c) J.C.T.R. Burckett-St. Launent, R.J.
Haines, C.R. Nolte and N.D.C.T. Steen, Inorg. Chem., 19 (1980)
577; (d) R.J. Haines, R. Mason, J.A. Zubieta and R.C. Nolte, J.
Chem. Soc., Chem. Commum., (1972) 990; (e) R.G. Hayter, Inorg. Chem., 2 (1963) 1031; (f) P. Braunstein and E. de Jesus, J. Organomet. Chem., 365 (1989) C19.
[7] (a) A.J. Carty, S.A. Maclaughlin and D. Nucciarone, in J.G. Verkade and L.P. Quin (eds.) Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis. Organic Compounds and Metal Com- plexes, VCH, New York, 1987, Ch. 16 and refs. therein. (b) P.E.
Garrou, Chem. Rev., 81 (1981) 229.
[8] (a) J. Levisalles, H. Rudler, F. Dahan and Y. Jeannin, J.
Organomet. Chem., 188 (1980) 193; (b) J. Levisalles, F. Rose-
Munch, H. Rudler, J.C. Daran, Y. Dromzee and Y. Jeannin, J.
Chem. Soc., Chem. Commun., (1980) 685; (c) H. Berke, P.
Harter, G. Hutter and L. Zsolnai, Chem. Ber., 115 (1982) 695;
(d) H. Fischer, S. Zeuner and K. Ackermann, J. Chem. Soc., Chem. Commun., (1984) 684.
[9] R.D. Adams, D.M. Collins and F.A. Cotton, Inorg. Chem., 13
(1974) 1086.
[10] S.-G. Shyu, M. Calligaris, G. Nardin and A. Wojcicki. J. Am. Chem. Soc., 109 (1987) 3617.
[11] D.A. Roberts, G.R. Steinmetz, M.J. Breen, P.M. Shulman, E.D. Morrison, M.R. Duttera, C.W. DeBrosse, R.R. Whittle and G.L. Geoffroy, Organometallics, 2 (1983) 846.
[12] Equation of the plane: 10.60 (8) X+7.92 (7)Y-2.28(6)Z= 4.287(7). Distances (A,) to the plane from the atoms in the plane. P-0.003(9), C(2) 0.05(3), C(4) 0.05(3), C(5) -0.08(3). Chi squared for this plane 13.247. Distances (,~) to the plane from the atoms out of the plane. W(1) 0.003(10), W(2) -0.063(20). [13] J. Emsley, The Elements, Oxford University Press, New York,
1989.
[14] D.S. Ginley, R. Bock, M.S. Wrighton, B. Fishcher, D.L Tipton and R. Bau, J. Organomet. Chem., 407 (1978) 247.
[15] S.G. Shyu, P.-J. Lin, T.-Y. Dong and Y.-S. Wen, J. Organomet. Chem., 460 (1993) 229.
[16] S.W. Kirtley, in G. Wilkinson, F.G.A. Stone and E. Abel (eds.), Comprehension Organometallic Chemistry, Vol. 3, Pergamon, London, 1982, pp. 842-844.
[17] R. Regragui, P.H. Dixneuf, N.J. Taylor and A.J. Carry,