行政院國家科學委員會專題研究計畫 期中進度報告
人類 alpha-L-岩藻醣甘酵素之結構與催化機構的研究
(1/2)
計畫類別: 個別型計畫 計畫編號: NSC94-2113-M-009-010- 執行期間: 94 年 08 月 01 日至 95 年 07 月 31 日 執行單位: 國立交通大學應用化學系(所) 計畫主持人: 李耀坤 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 95 年 5 月 28 日
Part 1
Stereoselective Synthesis of Aryl-α-L-Fucopyranosides via
Trichloroacetimidates
Y
Yaaww--KKuueennLLii
Department of Applied Chemistry, National Chiao Tung University, HsinChu, Taiwan
Abstract
An effective method to prepare the substrate of α-L-fucopyranosidase was reported herein. In this current method, the protected fucopyranoside trichloroacetimidate was prepared as the glycosyl donor and further coupled with phenols, the glycosyl acceptor, at -20 °C . TMSOTf, a strong Lewis acid, was exemplified as the glycosylation promoter. The reaction was employed via kinetic control to afford high stereoselectivity and yield of the α-L-fucopyranosides. By using sodium methoxide, the acetyl protecting group was removed to afford target molecules with high purity.
Introduction
Human α-L-fucosidases, a member of the CAZy family GH-29, are important glycosidases involved in many biological processes including inflammation, metastasis, and the lysosomal storage disease fucosidosis.1,2 α-L-Fucosidase is a hydrolyase, which can cleave fucosidic bond to release fucose. Recent research has demonstrated that α-L-fucosidase in blood serum plays an important role in diagnosing hepatocellular carcinoma.3 To study the biological processes involving fucosidase is demanded to have suitable substrates or potent inhibitors for this enzyme.
In 1968, Westphal and coworkers first synthesized
2,3,4-tri-O-hydroxyl-1-O-(4-nitrophenyl)-α-D -fucopyranoside as a substrate for α-D-fucosidase.4 However, we were unable to obtain the
described the synthesis of aryl-α-L-fucoside by using per-O-acetylated fucosyl imidate as the donor. The method allowed to prepare a series of 1,2-cis, aryl fucosides with high stereoselectivity.
Results and discussion
The synthesis of 4-nitrophenyl-α-L-fucopyranoside 4 is outlined in Scheme 1. The synthesis was started by the removal of 1-acetyl group in
tetra-O-acetyl-α-L-fucopyranoside 1 by hydrazine acetate.5 Subsequent treatment of the resulting hemiacetal with a large excess of trichloroacetonitrile in the presence of Cs2CO3 for 8 h at room temperature,6 gave only the thermodynamically more stable
trichloroacetimidate-L-fucopyranosides 2 (α:β=7:1) mixture in an 93% yield. Compound
2 was then condensed with 4-nitrophenol in the presence of TMSOTf to afford exclusive
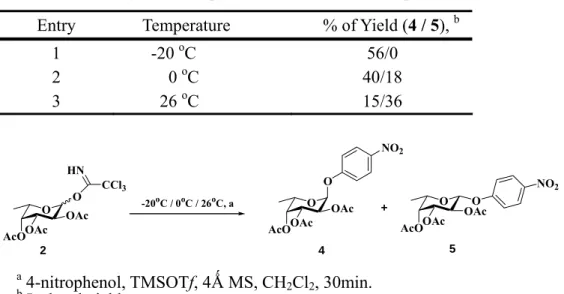
4-nitrophenyl-2,3,4-tri-O-acetyl-α-L-fucopyranoside 3 in 56% yield. Notably, when the reaction temperature was lowed to -20 oC, the formation of α-glycoside was observed. Deacetylation of compound 3 with NaOMe yielded 4-nitrophenyl-α-L-fucopyranoside 4 with 82% yield.
It has been well accepted that the presence of O-acyl or N-acyl protecting group at C-2 position, it’s usually the dominating effect in anomeric stereocontrol to form
1,2-trans glycosidic bond using Schmidt’s trichloroacetimidate mehod.7 However, when nonparticipating protective groups are selected, to achieve the 1,2-cis glycosidic bond formation, the reaction should be performed in nonpolar solvents, under low temperatures, with relatively weak Lewis acid catalysts (such as BF3·OEt2) and using
β-trichloroacetimidates to give SN2 type reactions. In the presence of strong acidic catalysts (such as TMSOTf, TfOH), higher reaction temperatures (such as room temperature) and more polar solvents, the thermodynamically favored α-glycoside products can be also obtained.8 Thus, we proposed the possible mechanism of the reaction shown in Figure 1. To achieve the 1,2-cis fucosidic bond formation in the presence of 2-O-acyl or N-acyl protecting group, the reaction should be performed in nonpolar solvents, under low temperatures, with strong acidic catalysts (such as TMSOTf) to give SN1 type reactions. The O-fucosyl trichloroacetimidates is transformed into a
carbenium ion (see Figure 1), for the kinetic control, yield the α-fucosides with a chair conformation from first points stereoelectronic resons. Somehow, the 2-O-acyl protecting participating protective groups effected slight than temperatures controlled. For above reasons, we controlled the temperatures for glycosylation step condition as shown in
Table 1, when the temperature in 0oC, to gave the α-L-fucopyranosides 4 (40%) and β-L-fucopyranosides 5 (18%). Thus in room temperature, yield compound 4 (15%) and compound 5 (36%) This result has been demonstrated the temperature was another points for α, β-fucopyranoside formation.
To extend the scope of the developed reaction for preparing α-L-fucopyranosides substrates in Table 210, acceptors (see table 2, entry 1-10) were used for fucosylation. However, when 2-nitro substitute phenol acceptors (entries 8, 9, 10) were used, the glycosylation yields were decreased, in some cases, even no product formation. This may a strong electron withdrawing group at ortho positon of phenol decreases the
nucleophilicity of OH. However, when 2,4-dinitrophenol (entry 7) was used as acceptor, there was product formation but with β-glycosidic bond.
Encouraged by these promising results, we then tried to using another per-O-acetyl monosacchrides (such as glucoside, mannoside, galactoside) as trichloroaceteimidates donors, to synthesize 1,2-cis 4-nitrophenyl glycosides as shown in Table 3,. In all of the examples, α-anomers are the major products. Neighboring group participation of
2-O-acyl-protecting groups is usually yieled 1,2-trans-Glucosides and Galactosides.9 But in our method, we can be also obtained 1,2-cis-4-nitrophenyl-D-Glucopyranoside 13, 14 (α/β=3:1, 52%) and 1,2-cis 4-nitrophenyl-D-Galactopyranoside 15 (α-isomer, 70%) under -20oC, the means it’s requiting low temperatures controlled when using 2-O-acyl-protecting groups to affored 1,2-cis glycosylations for trichloroacetimedate donors, thus decreasing the Neighboring group participation effected.
In summary, we have described a high stereoselective method for preparing nitrophenyl substituted L-fucosides by per-O-acetylated glycosyl imidates as donor under room temperature. The use of TMSOTf as promoter under low temperature provided good kinetic control to give good yields of α-anomers. Thus, large-scale
preparation of 1,2-cis-4-nitrophenol-Gluco-type monosacchrides and α-L-fucopyranosides substrates can be performed by this method.
Experimental
General methods
Materials and methods. New compounds were characterized by 1H, 13C NMR spectra. 1H NMR spectra were recorded with a Bruker 300 spectrometer. Chemical shifts are
expressed in ppm downfield shift from Me4Si. Mass spectra were obtained with an instrument under positive ESI conditions. Column chromatography was performed on Silica Gel 60 (230–400 mesh, E. Merck). The progress of all reactions was monitored by thin-layer chromatography (TLC) on Silica Gel 60 F254 (0.25 mm, E. Merck).
1. 1 2,3,4-Tri-O-acetyl-1-O-(trichloroacetimidate)-L-fucopyranoside (2)
The hexa-acetate 1 (600 mg, 1.81 mmol) was dissolved in DMF (3 mL) containing hydrazine acetate (183 mg, 1.99 mmol)﹒After 30min at room temperature, extraction with ethyl acetate、10% HCl(aq)、sat. NaHCO3(aq)、sat. NaCl(aq)﹒Drying of the organic layers and concetration in vacuo provided a crude oil product
2,3,4-Tri-O-acetyl-1-O-(hydroxyl)-L-fucopyranoside, A slurry of
2,3,4-Tri-O-acetyl-1-O-(hydroxyl)-L-fucopyranoside (266 mg, 0.92 mmol), Cl3CCN (183μL, 1.84 mmol), and Cs2CO3 (30 mg, 0.09 mmol) in CH2Cl2 (3 mL) was stirred 8 h﹒Extraction with ethyl acetate、sat. NaHCO3(aq)、sat. NaCl(aq)﹒Drying of the organic layers and concetration in vacuo provided a crude oil product 2 (α:β=7:1, 364 mg, 93.0%). The product was immediately used in the following step without further purification.; α-isomer; 1H NMR (CDCl3, 300 MHz): 8.60 (s, 1H, NH), 6.55 (d, J=4.0 Hz, 1H, H-1), 5.48-5.316 (m, 3H, H-2, H-3, H-4), 4.39 (q, J = 7.0, 14.0 Hz, 1H, H-5), 2.21 (s, 3H, AcO), 2.17 (s, 3H, AcO), 2.07 (s, 3H, AcO), 1.21 (d, J=7.0 Hz, 3H, H-6); ESI+ calcd for C14H18Cl3NO8 [ M + H ]+: 434.01, found 434.15.; β-isomer; 1H NMR (CDCl3, 300 MHz): 8.67 (s, 1H, NH), 5.82 (d, J=8.4 Hz, 1H, H-1), 5.50 (dd, J = 9.0, 12.0 Hz , 1H, H-3), 5.34 (ddd, J =6.0, 6.0, 12.0 Hz, 2H, H-4, H-2), 4.01 (q, J = 7.2, 14.0 Hz, 1H, H-5), 2.21 (s, 3H,
AcO), 2.17 (s, 3H, AcO), 2.07 (s, 3H, AcO), 1.21 (d, J=7.2 Hz, 3H, H-6); ESI+ calcd for C14H18Cl3NO8 [ M + H ]+: 434.01, found 434.12.
1. 2 2,3,4-Tri-O-acetyl-1-O- (4-nitrophenyl)-α-L-fucopyranoside (3)
The curde trichloroacteimidate 2 (364 mg, 0.84 mmol) and 4-nitrophenol (acceptor) (139.80 mg, 1.01 mmol), anhydrous 4Ǻ MS (500 mg) were combinded in a flask and concetrated from freshly distilled toluene (5 mL x 3). The resulting residue was dissolved in CH2Cl2 (3 mL), was added TMSOTf (151 μL, 0.84 mmol) in -20oC stirred
30 min. The residue was purified by silica gel chromatography with 1:6 EtOAc-Hexane, recrystalation with ethyl acetate、hexane, to gave product 3 (194 mg, 56%) of a clear white powder.; 1H NMR (CDCl3, 300 MHz): 8.22-8.19 (m, 2H, ArH), 7.17-7.13 (m, 2H, ArH), 5.86 (d, J=4.0 Hz, 1H, H-1), 5.58 (dd, J = 6.0, 12.0 Hz 1H, H-3), 5.37 (ddd, J =6.0, 6.0, 12.0 Hz, 2H, H-4, H-2), 4.29 (q, J = 7.0, 14.0 Hz, 1H, H-5), 2.19 (s, 3H, AcO), 2.05 (s, 3H, AcO), 2.01 (s, 3H, AcO), 1.21 (d, J=7.0 Hz, 3H, H-6); ESI+ calcd for C18H21NO10 [ M + H ]+: 412.12, found 412.25.
1. 3 2,3,4-Tri-O-hydroxyl-1-O- (4-nitrophenyl)-α-L-fucopyranoside (4)
To a solution of L-fucoside 3 (1.00g, 2.43 mmol) in dry MeOH( 30 mL) at room temperature was added sodium methoxy (31.83mg, 0.243 mmol)、stirred 30min. Extraction with IR-120 H+ resin. The filtrate was concetrated to gave product 4
(568.50mg, 82%) of a clear white powder.; 1H NMR (D2O, 300 MHz): 8.34-8.31 (m, 2H, ArH), 7.35-7.31 (m, 2H, ArH), 5.87 (d, J=4.0 Hz, 1H, H-1), 4.20-4.16 (m, 2H, H-3,H-4), 4.08 (dd, J =3.0, 9.0 Hz, 1H, H-2), 3.94 (d, J =3.0 Hz, 1H, H-5), 1.22 (d, J=9.0 Hz, 3H, H-6); ESI+ calcd for C12H15NO7 [ M + H ]+: 286.08, found 286.18.
Reference
1.(a)Becker, D. J., Lowe, J. B. Glycobiology, 2003, 110, 41R.; (b)Giardina, M. G., Matarazzo, M., Morante, R., Lucariello, A., Varriale, A., Guardasole, V., De Marco, G. Cancer, 1998, 83, 2468.; (c)Barker, C., Dell, A., Rogers, M., Alhadeff, J. A.,
2. Elbein, A. D. Annu. Rev. Biochem. 1987, 56, 497-534.
3. (a)Deugnier, Y., David, V., Brissot, P., Mabo, P., Delamaire, D., Messner, M., Bourel, M., Legall, J. Y. Hepatology 1984, 4, 889.; (b)Marotta, F.; Chui, D. H.; Safran, P. Dig. Dis. Sci. 1991, 36, 993.; (c) Giardina, M. G.; Matarazzo, M.; Varriale, A.; Morante, R.; Napoli, A.; Martino, R. Cancer 1992, 70, 1044.; (d) Takahashi, H., Saibara, T.,
Iwamura, S., Tomita, A., Maeda, T., Onishi, S., Yamamoto, Y., Enzan, H. Hepatology 1994, 19, 1414.; (e)Legler, G. Adv. Carbohydr. Chem. Biochem. 1990, 48,319_/384; (f)Winchester, B.; Fleet, G. W. J. Glycobiology 1992, 2, 199-/210; (g) Asano, N., Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetra. Asym 2000, 11, 1645; (h)Sinnott, M. L. Chem. Rev. 1990, 90, 1171; (i)Behr, J. B., Chevrier, C., Defoin, A., Tarnus, C., Streith, J. Tetrahedron 2003, 59, 543.
4. Fahrenheim, G., Westphal, O. Liebigs Ann. Chem. 1968, 720, 177–187. 5. Excoffier, G., Gagnaire, D., Utille, J. P. Carbohydr. Res. 1975, 39, 368. 6. Schmidt, R. R. Angew. Chem., Int. Ed. Engl. 1986, 25, 212.
7. (a)Zimmermann, P., Bommer, R., Schmidt, R. R. J. Carbohydr. Chem.1988, 7, 435.; (b)Ishida, H., Ohta, Y., Tsukada, Y., Kiso, M., Hasegawa, A. Carbohydr. Res. 1993, 75, 246.; (c)Numata, M., Sugimoto, M., Ito, Y., Ogawa, T. Carbohydr. Res. 1990, 203, 205. 8. (a)Mayer, T. G., Kratzer, B., Schmidt, R. R. Angew. Chem. Int. Ed. Engl. 1994, 33,
2177.; (b) Mayer, T. G., Kratzer, B., Schmidt, R. R. Angew. Chem. Int. Ed. Engl. 1994, 33, 2177.
9. Jiang, Z. H., Schmidt, R. R., Liebigs. Ann. Chem. 1992, 975. 10. 6: 1H NMR (CDCl
3, 300 MHz): 7.94-7.90 (m, 2H, ArH), 7.50-7.45 (m, 1H, ArH), 7.41-7.37 (m, 1H, ArH), 5.83 (d, J=3.6 Hz, 1H, H-1), 5.59 (dd, J=3.6, 12.0 Hz 1H, H-3), 5.37 (ddd, J=3.6, 3.6, 12.0 Hz, 2H, H-4, H-2), 4.26 (q, J=3.6, 12.0 Hz, 1H, H-5), 2.18 (s, 3H, AcO), 2.01 (s, 3H, AcO), 2.00 (s, 3H, AcO), 1.25 (d, J=7.0 Hz, 3H, H-6); ESI+ calcd for C18H21NO10 [ M + H ]+: 412.12, found 412.80.; 7: 1H NMR (CDCl3, 300 MHz): 7.63
(q, J=4.5, 9.6 Hz, 2H, ArH), 7.16 (q, J=6.0, 9.6 Hz, 2H, ArH), 5.81 (d, J=3.6 Hz, 1H, H-1), 5.57 (dd, J=3.6, 12.0 Hz 1H, H-3), 5.36 (ddd, J=3.6, 3.6, 12.0 Hz, 2H, H-4, H-2), 4.21 (q, J=3.6, 12.0 Hz, 1H, H-5), 2.18 (s, 3H, AcO), 2.01 (s, 3H, AcO), 1.98 (s, 3H, AcO), 1.28 (d, J=6.0 Hz, 3H, H-6); ESI+ calcd for C19H21NO8 [ M + H ]+: 391.13, found 391.86.; 8: 1H NMR (CDCl3, 300 MHz): 7.33-7.21 (m, 2H, ArH), 7.07-6.81 (m, 3H, ArH), 5.74 (d, J=3.6 Hz, 1H, H-1), 5.61 (dd, J=3.6, 12.0 Hz 1H, H-3), 5.37 (ddd, J=3.6, 3.6, 12.0 Hz, 2H, H-4, H-2), 4.30 (q, J=3.6, 12.0 Hz, 1H, H-5), 2.18 (s, 3H, AcO), 2.01 (s, 3H, AcO), 1.98 (s, 3H, AcO), 1.28 (d, J=6.0 Hz, 3H, H-6); ESI+ calcd for C18H22O8 [ M + H ]+: 367.13, found 367.79.; 9: 1H NMR (CDCl3, 300 MHz): 8.07-8.01 (m, 2H, ArH), 7.33-7.27 (m, 1H, ArH), 5.88 (d, J=3.6 Hz, 1H, H-1), 5.59 (dd, J=3.6, 12.0 Hz 1H, H-3), 5.42 (ddd, J=3.6, 3.6, 12.0 Hz, 2H, H-4, H-2), 4.30 (q, J=3.6, 12.0 Hz, 1H, H-5), 2.21 (s, 3H, AcO), 2.10 (s, 3H, AcO), 2.05 (s, 3H, AcO), 1.29 (d, J=7.0 Hz, 3H, H-6); ESI+ calcd for C18H20FNO10 [ M + H ]+: 430.11, found 430.65.; 10: 1H NMR (CD2Cl2, 300 MHz): 8.10 (d, J=3.6 Hz 1H, ArH), 7.52 (d, J=4.0 Hz 1H, ArH), 7.43 (dd, J=2.4, 9.0 Hz 1H, ArH), 5.93 (d, J=3.6 Hz, 1H, H-1), 5.56 (dd, J=3.6, 12.0 Hz 1H, H-3), 5.38-5.31 (m, 2H, H-4, H-2), 4.20 (q, J=3.6, 12.0 Hz, 1H, H-5), 2.21 (s, 3H, AcO), 2.01 (s, 3H, AcO), 1.98 (s, 3H, AcO), 1.23 (d, J=6.0 Hz, 3H, H-6); ESI+ calcd for C18H20N2O12 [ M + H ]+: 457.10, found 457.46.; 11: 1H NMR (CDCl
3, 300 MHz): 7.55 (d, J=9.0 Hz 1H, ArH), 7.06 (d, J=3.6 Hz 1H, ArH), 7.03 (dd, J=3.6, 9.0 Hz 1H, ArH), 6.19 (d, J=3.0 Hz, 1H, CH), 5.81 (d, J=3.6 Hz, 1H, H-1), 5.59 (dd, J=3.6, 12.0 Hz 1H, H-3), 5.59 (ddd, J=3.6, 3.6, 12.0 Hz, 2H, H-4, H-2), 4.22 (q, J=3.6, 12.0 Hz, 1H, H-5), 2.41 (s, 3H, CH3), 2.20 (s, 3H, AcO), 2.01 (s, 3H, AcO), 1.98 (s, 3H, AcO), 1.28 (d, J=6.0 Hz, 3H, H-6); ESI+ calcd for C22H22O10 [ M + H ]+: 449.14, found 449.56.; 12: 1H NMR (CDCl3, 300 MHz): 8.69 (d, J=2.7 Hz 1H, ArH), 8.45 (dd, J=2.7, 9.3 Hz 1H, ArH), 7.50 (d, J=9.0 Hz 1H, ArH), 5.60 (dd, J=14.1, 10.5 Hz 1H, H-3), 5.35 (d, J=3.6 Hz, 1H, H-4), 5.29 (d, J=8.1 Hz, 1H, H-1), 5.14 (dd, J=3.6, 10.5 Hz, 1H, H-2), 4.15 (q, J=3.6, 12.0 Hz, 1H, H-5), 2.21 (s, 3H, AcO), 2.01 (s, 3H, AcO), 1.98 (s, 3H, AcO), 1.32 (d, J=6.0 Hz, 3H, H-6); ESI+ calcd for C18H20N2O12 [ M + H ]+: 457.10, found 457.51.; 15: 1H NMR (CDCl3, 300 MHz): 8.25-.8.19 (m, 2H, ArH), 7.23-7.17 (m, 2H, ArH), 5.62 (d, J=3.6 Hz, 1H, H-1), 5.55 (dd, J=3.6, 9.9 Hz 1H, H-3), 5.46 (dd, J=1.8, 3.3 Hz, 1H, H-2), 5.41 (t, J=9.9 Hz, 1H, H-4), 4.30 (q, J=5.4, 12.0 Hz, 1H, H-5), 4.12-3.97 (m, 2H, H-6a,b), 2.24 (s, 3H, AcO), 2.04 (s,
3H, AcO), 2.03 (s, 3H, AcO), 2.00 (s, 3H, AcO).; ESI+ calcd for C20H23NO12 [ M + H ]+: 470.12, found 470.40.; 16: 1H NMR (CDCl3, 300 MHz): 8.25-.8.19 (m, 2H, ArH), 7.26-7.15 (m, 2H, ArH), 5.89 (d, J=3.6 Hz, 1H, H-1), 5.58-5.51 (m, 2H, H-3), 5.33 (dd, J=3.6, 10.2 Hz, 1H, H-2), 4.27 (t, J=6.0 Hz, 1H, H-4), 4.14-4.02 (m, 2H, H-6a,b), 2.17 (s, 3H, AcO), 2.15 (s, 3H, AcO), 2.03 (s, 3H, AcO), 1.93 (s, 3H, AcO).; ESI+ calcd for C20H23NO12 [ M + H ]+: 470.12, found 470.40.
O OHOH OH HO O OAcOAc OAc AcO O OAcO OAc AcO CCl3 HN O OAc OAc AcO OR O OH OH HO OR a b, c d e 1 2 3: R = NO2 4: R = NO2
Scheme 1. Reagents and conditions: (a) Ac2O, Pyridine, rt, 8 h, 86%; (b) H4N2-HOAc, DMF, rt, 30 min; (c) NCCCl3, Cs2CO3, CH2Cl2, rt, 8 h, 93% (α:β=7:1); (d) R (acceptor): 4-nitrophenol, TMSOTf, 4Ǻ MS, CH2Cl2, -20oC, 30min 56%; (e) NaOMe, MeOH, rt, 30 min, 82%.
Table 1. Selectivity of L-fucopyranosides at different temperature O OAcO OAc AcO CCl3 HN O OAc OAc AcO O -20oC / 0oC / 26oC, a NO2 O OAc AcO O NO2 OAc + 2 4 5 a 4-nitrophenol, TMSOTf, 4Ǻ MS, CH 2Cl2, 30min. b Isolated yield.
Entry Temperature % of Yield (4 / 5), b
1 -20 oC 56/0
2 0 oC 40/18
Table 2. Preparation of α-L-fucopyranosides
a Compound 2, TMSOTf, 4Ǻ MS, CH
2Cl2, 30min. b The R-Group as Scheme 1.
c Isolated yield.
Entry Acceptor (ROH)a Productb Yield (α/β) (%)c
1 3-Nitrophenol 6: R=3-nitrophenyl 53/0
2 4-Cyanidephenol 7: R=4-cyanidephenyl 31/0
3 Phenol 8: R=phenyl 75/0
4 2-Fluoro, 4-nitrophenol 9: R=2-fluoro, 4-nitrophenyl 60/0
5 3, 4-Dinitrophenol 10: R=3, 4-dinitrophenyl 35/0
6 4-Methyumbellifery 11: R=4-methyumbellifery 48/0
7 2, 4-Dinitrophenol 12: R=2, 4-dinitrophenyl 0/72
8 2, 5-Dinitrophenol none none
9 4-Chloro, 2-nitrophenol none none
Table 3. Synthesis of 1,2-cis 4-nitrophenyl monosaccharides
a 4-Nitrophenol, TMSOTf, 4Ǻ MS, CH
2Cl2, -20oC, 30min. b Isolated yield.
Entry Productsa Yield (α/β) (%)b
1 4-Nitrophenyl-D-Glucopyranoside 38(13)/13(14) 2 4-Nitrophenyl-D-Galactopyranoside 70(15)/0 3 4-Nitrophenyl-D-Mannopyranoside 40(16)/0
Figure 1. Proposed possible of the mechanism for α-L-fucopyranosides. O AcOOAc OAc O Cl3 NH O AcOOAc OAc O Cl3 NH2 TfO O AcO OAc OAc TfO O AcOOAc OAc OR Trichloroacetimidate Fucosides -20oC
Carbenium ion α-L-Fucosides RO
TMSOTf
Part 2
E
E
x
x
p
p
r
r
e
e
s
s
s
s
i
i
o
o
n
n
,
,
p
p
u
u
r
r
i
i
f
f
i
i
c
c
a
a
t
t
i
i
o
o
n
n
,
,
a
a
n
n
d
d
c
c
h
h
a
a
r
r
a
a
c
c
t
t
e
e
r
r
i
i
z
z
a
a
t
t
i
i
o
o
n
n
o
o
f
f
h
h
u
u
m
m
a
a
n
n
α
α
-
-
L
L
-
-
f
f
u
u
c
c
o
o
s
s
i
i
d
d
a
a
s
s
e
e
Y
Yaaww--KKuueennLLii
Department of Applied Chemistry, National Chiao Tung University, HsinChu, Taiwan
Abstract
α-L-Fucosidases (EC 3.2.1.51) are exo-glycosidases unique to family 29 in the
sequence-based classification of glycoside hydrolases. They are responsible for the removal of L-fucosyl residues from the non-reducing end of glycoconjugates.
Deficiency of α-fucosidase results in the accumulation of fucose-containing glycolipids, glycoproteins and oligosaccharides in various tissues and therefore causes Fucosidosis. Recent studies indicated that the fucosylation levels are increased on the membrane surface in many carcinomas. The biological function of α-L-fucosidases may be related to this abnormal cell physiology. Although many bacterial α-L-fucosidases have been purified, cloned, and expressed, the expression and purification of human α-L-fucosidase was much less reported. We report herein, with careful control on the growing
condition, that a human α-L-Fucosidases (hFUC) was successfully expressed in E. coli. The recombinant protein was characterized. On the basis of the amino acid
multi-alignment as well as the structural information of x-ray crystallography of
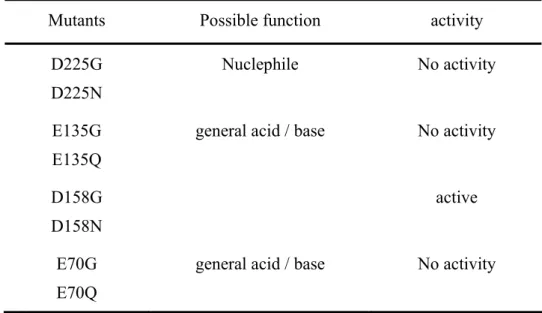
Thermotoga maritima fucosidase, four residues, Glu-70, Glu-135, Asp-158, and Asp-225, are likely to be the essential groups of hFUC. Site-directed mutagenesis studies on all
of these residues revealed that Glu-135 and Asp-225 are likely to be the key amino acids responsible for catalytic activity. The Asp-224 (corresponding to the position of Asp-225 in hFUC) in T. maritima fucosidase was recently identified as the catalytic nucleophile by S. G. Withers’ group, indicating that Glu-135 may function as the general acid/base residues. Detailed kinetic investigations are carrying out.
α-1-deoxy-1-[n-(2,3-epoxy)propyl]-L-fucose (DEPF) was synthesized and evaluated as the affinity-labeling reagent. A time course irreversible inhibition was observed. LC-MS analysis on the molecular weight of hFUC before (50088 Da) and after (50312 Da) the labeling reaction revealed that the enzyme was labeled stoichiometrically by DEPF. The peptide sequencing to identify the specific labeling-site is currently in progress.
Introduction
α-L-Fucosidases (3.2.1.51) are exoglycosidases capable of cleaving L-linked L-fucose residues from glycoconjugates involved in a variety of biological processes (1). In particular, the determination of L-fucosidase activity can be used to predict the
development of several carcinomas (2-4), whereas the deficiency in this enzyme causes fucosidosis, a well-known lysosomal storage disorder (5).
All known α-L-fucosidases have been classified as the member of the
glycohydrolase family 29 (GH29). This catalytic reaction of α-L-fucosidase has been unequivocally demonstrate to follow a retaining reaction mechanism. Retaining enzymes typically have two catalytic carboxylic acids in their active site and utilize a double displacement mechanism (Scheme1) in which a α-L-fucosyl intermediate is formed and
hydrolyzed. Both steps proceed via transition states with substantial oxocarbenium ion character (6). One carboxylic acid in the active site acts as the catalytic nucleophile, leading to the formation of the covalent intermediate, while the other plays the role of the general acid catalyst in the first step and the general base catalyst in the second step of the reaction. The active site residues in GH29 enzymes are stillunknown. For the identification of the catalytic nucleophile in retaining α-D and β-D-glycosidases, fluorosugars have proved to be useful mechanism-based inhibitors (7), by trapping the glycosyl-enzyme intermediate. This approach involving 5-fluoroglycosides has been employed previously to identify the catalytic nucleophiles of α-D-glucosidases in GH13 and GH31 (8,9), α-D-galactosidases in GH27 (10), and α-D-mannosidases in GH38 (11, 12). Interestingly, a 2-deoxy-2-fluoro sugar has been successfully used recently for the identification of the catalytic nucleophile of a α-L-iduronidase (13). An alternative approach often exploited for retaining glycoside hydrolases consists of the mutation of aspartic/glutamic acid residues identified by sequence analysis and conserved in the family of interest. Mutations of the catalytic residues with non-nucleophilic amino acids lead to the strong reduction or even abolition of the enzymatic activity (14). However, these mutants can be reactivated in the presence of external nucleophiles as sodium azide. The isolation of glycosyl-azide products with an anomeric configuration opposite to that of the substrate allows the identification of the catalytic nucleophile of the reaction (14). Although many α-L-fucosidase have been purified and cloned from several different sources including that from human, intensive studies were only performed with those microbial enzymes. Much less study was performed on human α-L-fucosidase due to the invaluable human tissue and the difficulty of recombinant protein expression. By
careful control of the culture condition, we have successfully expressed the human α-L-fucosidase in E. coli. We report herein the purification, characterization and mutagenic study of this important enzyme. A new type of fucoside derivative was synthesized and test as the affinity-labeling inhibitor.
Material and methods
Reconstruction of human alf (α-L-fucosidase) in pET22b(+)
The vector pCMV.SPORT6 containing humanα-L-Fucosidase gene (hfuc) was obtained from (Stragene, catalong no. FL1002). The upstream and down stream primers, 5-CTGCAGCACATATGGTGCGTCGGGCCCAGCCTC-3’,
5’-GGATCCGGTACCTCACTTCACTCCTGTCAGCT-3’, containing Nde I and EcoR I restriction site, respectively, were used for hfuc amplification. The amplified DNA fragment (1.4 kb) was inserted in pET 22b(+). The recombinant vector, designated pET 22b/ hfuc, was then transformed into E. coli [BL21(DE3)] for DNA sequencing and further for protein expression.
Construction of hFUC mutants
The mutagenesis was performed by the Quick Change method (Stratagene Co.) with the following oligonucleotide primers: for E135Q, 5’-
ACGACAAAGCATCACCAGGGCTTCACAAAC -3’, 5’- GTTTGTGAAGCCCTGGTGATGCTTTGTCGT -3’; for E135G, 5’-ACGACAAAGCATCACGGCGGCTTCACAAAC-3’,
CTGATCTGGTCTAACGGGGAGTGGGAAT -3’, 5’-
ATTCCCACTCCCCGTTAGACCAGATCAG -3’; for D225G, 5’-CTGATCTGGTCTGGCGGGGAGTGGGAAT -3’,
5’-ATTCCCACTCCCCGCCAGACCAGATCAG-3’ (The underline shows the location of mutation).
DNA sequencing
In order to verify the insertion human α-L-fucosidase gene, the nucleotide sequence of the pET 22b/ hfuc vector and the gene insert junction were confirmed by DNA
sequencing. DNA from plasmid pET 22b/ hfuc was purified using the Plasmid Mini Kit (QIAGEN). Cycle sequencing was performed by the dideoxy chain termination method using the BigDye® Terminator Cycle Sequencing Kits (Applied Biosystems) and T7 sequencing primer. The resulting reactions were analysed with ABI 3100 DNA sequencer (Perkin-Elmer Applied Biosystems).
Protein expression and purification of the recombinant hFUC and mutants
The processes of purification of the recombinant hFUC, E135Q, E135G, D225N, and D225G mutants were virtually identical. Complete procedures involve the applications of ammonium sulfate fractionation and two cation-exchange
chromatographic separations. All purification steps were performed at ambient temperature (approximately 25 oC). The detailed procedures were described as the following. The 2.5 ml of overnight culture of BL21(DE3), harboring pET 22b/ hfuc, was added in 1 L LB medium (10g trypton, 5g yeast extract, 5.5g sodium chloride) at 37
oC for 20 hr. The cell was harvested by centrifugation at 6700xg for 15 min and further resuspended in 20 ml of Sodium acetate buffer (20 mM, pH 5.7). The cell was then disrupted by ultra-sonication for 10 min with 30 s for each intervals. After
centrifugation to remove the cell debris, the supernant was subject to ammonium sulfate precipitation. The crude protein was obtained from centrifugation and subsequently re-dissolved in 3 ml sodium acetate buffer (20 mM, pH 5.7). Excess ammomium salt was remove by passing the crude protein solution through the HiTrap desalting column (Amersham Pharmacia Biosciences), which was pre-equilibrated with sodium acetate buffer ( 20 mM, pH5.7). The resulting solution was first loaded on a cation exchange column (5.2x 40, Amersham Pharmacia high performance SP) that was pre-equilibrated with the same buffer. Column was eluted by a linear gradient of NaCl with 5 mM/min and the flow rate of 3.4 ml/min. The active fractions eluted in the range of 400~700 mM NaCl were collected and concentrated before loading on HiTrap SP column (Amersham Pharmacia Biosciences), which was equilibrated with phosphate buffer (20mM, pH 6.1). Bound protein was eluted by a linear gradient of 1M NaCl. The active fractions were eluted at the range of 500~700 mM of NaCl.
Protein determination.
The protein content of the enzyme preparation was determined either by the
bicinchoninic acid (BCA) method as described in the manufacturer’s protocol (Sigma Co. BCA-1, Kit for protein determination) or by UV absorption at 280 nm.
Activity assay
of protein and time of incubation were concerned. α-L-fucosidase activity was determined using p-nitrophenyl-α-L-fucopyranoside (Sigma Co). α-L-fucosidase activity was expressed as specific activity (U/ mg). One unit of enzyme (U) was defined as the amount of enzyme required to hydrolyze 1 μmol of substrate per minute at 250 C in phosphate buffer (50 mM, pH7.1).
Results and discussion
Protein expression and purification
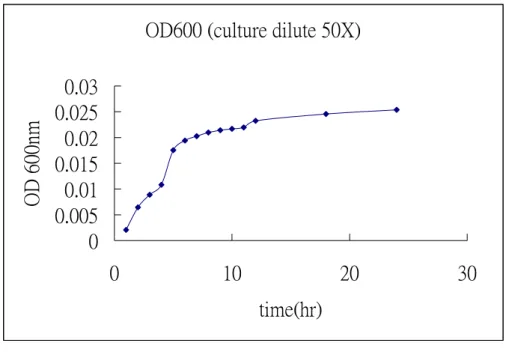
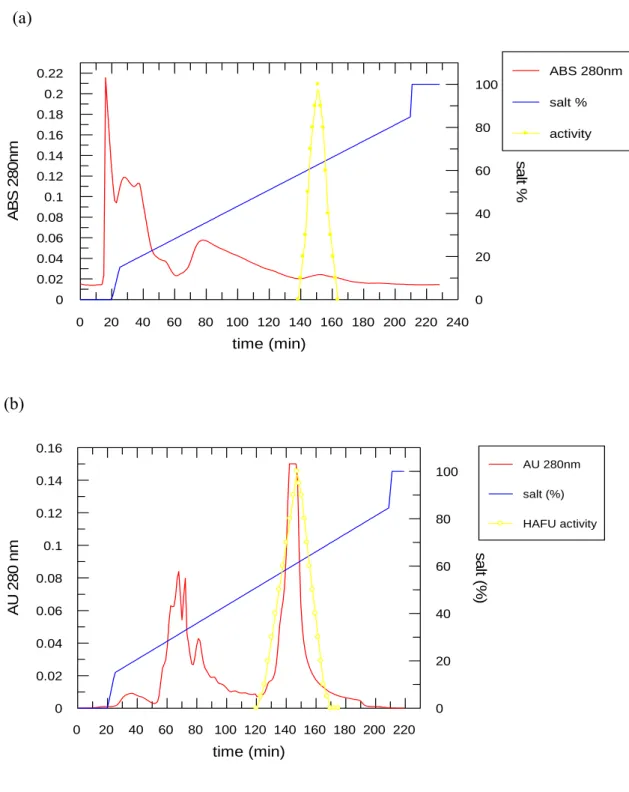
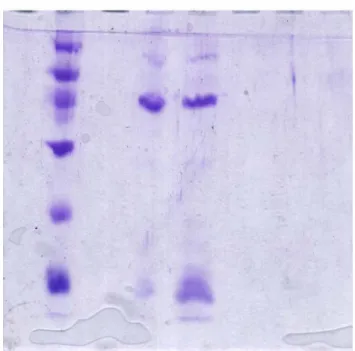
The growth media is LB(10g trypton, 5g yeast extract, 5.5 g sodium chloride)and growth in 37 0C for 20 hr. The growth rate was shown in figure 1. The collected cell was disrupted by ultra-sonication and further precipitated with various concentration of ammonium salt. The precipitate was desalted before loading onto the first HiTrap SP column (Pharmacia co.) that was pre-equilibrated with 20mM CH3COONa/CH3COOH (pH 5.7) buffer. Bound proteins were eluted by a linear gradient of 1M NaCl. The active fractions eluted at the range of 400-700 mM of NaCl (figure 2a). The fractions containing enzyme activity were pooled and concentrated. Another HiTrap SP column chromatography was further employed to obtain protein of interest with higher purity as shown in figure 2b. The active fractions were analyzed by activity and SDS-Page shown as figure 3. The temperature effect on enzyme activity (figure 4), and the thermalstability (figure 5) of the enzyme were performed. Figure 6 and figure 7 exhibited the pH effect and the pH stability of the enzyme, respectively.
of x-ray crystallography of Thermotoga maritima fucosidase, four residues, Glu-70, Glu-135, Asp-158, and Asp-225, are likely to be the essential groups of hFUC. Site-directed mutagenesis studies on all of these residues revealed that Glu-135 and Asp-225 are likely to be the key amino acids responsible for catalytic activity. The Asp-224 (corresponding to the position of Asp-225 in hFUC) in T. maritima fucosidase was recently identified as the catalytic nucleophile by S. G. Withers’ group, indicating that Glu-135 may function as the general acid/base residues. The Preliminary assay of enzyme activity and possible function of mutated residues were summarized in Table 2. Detailed kinetic investigations are carrying out α-1-deoxy-1-[n-(2,3-epoxy)propyl] -L-fucose (DEPF) was synthesized and evaluated as the affinity-labeling reagent. A time course irreversible inhibition was observed. LC-MS analysis on the molecular weight of hFUC before (50088 Da) and after (50312 Da) the labeling reaction revealed that the enzyme was labeled stoichiometrically by DEPF, as shown in figure 8. The peptide sequencing to identify the specific labeling-site is currently in progress.
Table 1. Purification of human α-L-fucosidase
Step protein(mg) Total Activity(units)Total Activity(units/mg)Specific Purity Fold
Enzyme recovery (%) ammonium sulfate precipitation 118.26 714 6.04 1 100 HiTrap SP, pH 5.7 4.78 179.34 37.52 6.21 25.12 HiTrap SP, pH 6.1 0.237 85.32 360 59.6 11.94
Table 2. Preliminary assay of enzyme activity and possible function of the mutated residues.
Mutants Possible function activity
D225G D225N
Nuclephile No activity
E135G E135Q
general acid / base No activity
D158G D158N
active
E70G E70Q
OD600 (culture dilute 50X)
0
0.005
0.01
0.015
0.02
0.025
0.03
0
10
20
30
time(hr)
OD
600nm
Figure 1. The growth rate of BL21_DE3 containing PET 22b(+) vector with human α-L-fucosidase gene.
(a) 240 220 200 180 160 140 120 100 80 60 40 20 0 0.22 0.2 0.18 0.16 0.14 0.12 0.1 0.08 0.06 0.04 0.02 0 100 80 60 40 20 0 time (min) A B S 28 0n m sal t % ABS 280nm salt % activity (b) 220 200 180 160 140 120 100 80 60 40 20 0 0.16 0.14 0.12 0.1 0.08 0.06 0.04 0.02 0 100 80 60 40 20 0 time (min) A U 28 0 nm sa lt ( % ) AU 280nm salt (%) HAFU activity
Figure 2. Chromatograms of the purification of human α-L-Fucosidase. (a) HiTrap SP column chromatography at pH 5.7. The NaCl gradient(0.4~0.7M) was
employed. (b) HiTrap SP column chromatography at pH 6.1 with NaCl gradient (0.5~0.7M).
Figure 3. The SDS-PAGE analysis of human α-L-fucosidase with the estimated MW of 50 kDa
Effect HAFU reaction activity by different temp 0.4 0.5 0.6 0.7 0.8 0.9 1 0 10 20 30 40 50 60 70 80 90 100 temp (0 C) res id u al a ctiv ity ( % )
Figure 4. Temperature effect of human α-L-fucosidase catalysis. The enzymic reaction was carried out at temperature from 4 0C to 95 0C.
Figure 5. The thermstability of human α-L-fucosidase. The reaction was performed with PNPF as substrate at 25 0C by incubating 30μl purified enzyme which was incubated in different temperature (25 0C -75 0C). At different time intervals (1, 20, 40, 60, 80, 100, 130, 160 mim), an aliquot of enzyme was removed for activity assay with 0.15 mM PNPF in a finial 500μl Na2HPO4 (50 mM, pH 7.1).
Th e sta b ility o f H A FU en zy m e in d ifferen t te m p
0 0.2 0.4 0.6 0.8 1 1.2 1.4 0 20 40 60 80 100 120 140 160 180
tim e (m in )
re
si
dua
l
a
ct
iv
it
y
(%
)
25 37 45 55 60 65 75Figure 6. The activity of human α-L-fucosidase in different pH conditions. The
reactions were performed with PNPF as substrate at pH 2, 2.5, 3, 4, 4.5, 5, 5.5, 6, 6.5, 7, 7.5, 8, 8.5, 9, 9.5, 10, and 10.5.
Effect HAFU reaction activity by different pH
0 0.0001 0.0002 0.0003 0.0004 0.0005 0.0006 0.0007 0.0008 0.0009 0 2 4 6 8 10
pH
in
it
iv
al
v
el
o
ci
ty
(A
U
/s
e
c)
348
nm
Glycine-HCl NaOAc-AcOH MES-NaOH Na2HPO4-H3PO4 HEPES-NaOH Tris-HCl Glycine-NaOH Caps-NaOHFigure 7. The pH stability of human α-L-fucosidase. The reaction was performed with PNPF as substrate at 25 0C by incubating 30μl purified enzyme which was incubated in different pH buffers (pH 2.5-9). At different time intervals (1, 20, 40, 60, 80, 100, 130, 160 mim), an aliquot of enzyme was removed for activity assay with 0.15 mM PNPF in a finial 500μl Na2HPO4 (50 mM, pH 7.1).
Th e stab ility o f H A FU en zy m e in d ifferen t p H
0
0.2
0.4
0.6
0.8
1
1.2
0
50
100
150
200
tim e ( m in )
re
si
d
u
a
l a
c
tiv
ity
(
%
)
2.5 3 4 5 6 7 8 9Reference
1. Vanhooren, P. T., and Vandamme, E. J. (1999) L-Fucose: occurrence, physiological role, chemical, enzymatic and microbial synthesis. J. Chem. Technol. Biotechnol. 74, 479-497.
2. Fernandez-Rodriguez, J., Ayude, D., de la Cadena, M. P., Martinez-Zorzano V. S., de Carlos, A., Caride-Castro, A., de Castro, G., and Rodriguez-Berrocal, F. J. (2000) Alpha-Lfucosidase enzyme in the prediction of colorectal cancer patients at high risk of tumor recurrence. Cancer Detect. PreV. 24, 143-149.
3. Abdel-Aleem, H., Ahmed, A., Sabra, A. M., Zakhari, M., Soliman, M., and Hamed, H. (1996) Serum alpha L-fucosidase enzyme activity in ovarian and other female genital tract tumors. Int. J. Gynaecol. Obstet. 55, 273-279.
4. Ishizuka, H., Nakayama, T., Matsuoka, S., Gotoh, I., Ogawa, M., Suzuki, K., Tanaka, N., Tsubaki, K., Ohkubo, H., Arakawa, Y., and Okano T. (1999) Prediction of the development of hepatocellular-carcinoma in patients with liver cirrhosis by the serial determinations of serum alpha-L-fucosidase activity. Intern. Med. 38, 927-931. 5. Michalski, J. C., and Klein, A. (1999) Glycoprotein lysosomal storage disorders:
alpha- and beta-mannosidosis, fucosidosis and alpha-N-acetylgalactosaminidase deficiency. Biochim. Biophys. Acta 1455, 69-84.
6. McCarter, J. D., and Withers, S. G. (1994) Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 4, 885-892.
7. Williams, S. J., and Withers, S. G. (2000) Glycosyl fluorides inenzymatic reactions. Carbohydr. Res. 327, 27-46.
the catalytic nucleophile of Saccharomyces cereVisiae alpha-glucosidase using 5-fluoro glycosyl fluorides. J. Biol. Chem. 271, 6889-6894.
9. Lee, S. S., He, S., and Withers, S. G. (2001) Identification of the catalytic nucleophile of the Family 31 alpha-glucosidase from Aspergillus niger via trapping of a
5-fluoroglycosyl-enzyme intermediate. Biochem. J. 359, 381-386.
10. Ly, H. D., Howard, S., Shum, K., He, S., Zhu, A., and Withers, S. G. (2000) The synthesis, testing and use of 5-fluoro-alpha-Dgalactosyl fluoride to trap an intermediate on green coffee bean alpha-galactosidase and identify the catalytic nucleophile. Carbohydr. Res. 329, 539-547.
11. Howard, S., He, S., and Withers, S. G. (1998) Identification of the active site
nucleophile in jack bean alpha-mannosidase using 5-fluoro-beta-L-gulosyl fluoride. J. Biol. Chem. 273, 2067-2072.
12. Numao, S., He, S., Evjen, G., Howard, S., Tollersrud, O. K., and Withers, S. G. (2000) Identification of Asp197 as the catalytic nucleophile in the family 38
alpha-mannosidase from bovine kidney lysosomes. FEBS Lett. 484, 175-178. 13. Nieman, C. E., Wong, A. W., He, S., Clarke, L., Hopwood, J. J., and Withers, S. G.
(2003) Family 39 alpha-L-iduronidases and beta-D-xylosidases react through similar glycosyl-enzyme intermediates: identification of the human iduronidase nucleophile. Biochemistry 42, 8054-8065.
14. Ly, H. D., and Withers, S. G. (1999) Mutagenesis of glycosidases. Annu. ReV. Biochem. 68, 487-522.