R E S E A R C H
Open Access
3D-interologs: an evolution database of physical
protein- protein interactions across multiple
genomes
Yu-Shu Lo
1, Yung-Chiang Chen
1, Jinn-Moon Yang
1,2,3*From The ISIBM International Joint Conference on Bioinformatics, Systems Biology and
Intelligent Computing (IJCBS)
Shanghai, China. 3-8 August 2009
Abstract
Background: Comprehensive exploration of protein-protein interactions is a challenging route to understand biological processes. For efficiently enlarging protein interactions annotated with residue-based binding models, we proposed a new concept“3D-domain interolog mapping” with a scoring system to explore all possible protein pairs between the two homolog families, derived from a known 3D-structure dimmer (template), across multiple species. Each family consists of homologous proteins which have interacting domains of the template for studying domain interface evolution of two interacting homolog families.
Results: The 3D-interologs database records the evolution of protein-protein interactions database across multiple species. Based on“3D-domain interolog mapping” and a new scoring function, we infer 173,294 protein-protein interactions by using 1,895 three-dimensional (3D) structure heterodimers to search the UniProt database (4,826,134 protein sequences). The 3D- interologs database comprises 15,124 species and 283,980 protein-protein interactions, including 173,294 interactions (61%) and 110,686 interactions (39%) summarized from the IntAct database. For a protein-protein interaction, the 3D-interologs database shows functional annotations (e.g. Gene Ontology), interacting domains and binding models (e.g. hydrogen-bond interactions and conserved residues). Additionally, this database provides couple-conserved residues and the interacting evolution by exploring the interologs across multiple species. Experimental results reveal that the proposed scoring function obtains good agreement for the binding affinity of 275 mutated residues from the ASEdb. The precision and recall of our method are 0.52 and 0.34, respectively, by using 563 non-redundant heterodimers to search on the Integr8 database (549 complete
genomes).
Conclusions: Experimental results demonstrate that the proposed method can infer reliable physical protein-protein interactions and be useful for studying the protein-protein-protein-protein interaction evolution across multiple species. In addition, the top-ranked strategy and template interface score are able to significantly improve the accuracies of identifying protein-protein interactions in a complete genome. The 3D-interologs database is available at http://3D-interologs.life.nctu.edu.tw.
* Correspondence: [email protected]
1
Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan
Full list of author information is available at the end of the article
© 2010 Yang et al; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background
A major challenge of post genomic biology is to under-stand the networks of interacting genes, proteins and small molecules that produce biological functions. The large number of protein interactions [1-3], generated by large-scale experimental methods [4-6], computational methods [7-13], and integrated approaches [14,15], pro-vides opportunities and challenges in annotating protein functions, protein-protein interactions (PPI) and domain-domain interactions (DDI), and in modeling the cellular signaling and regulatory networks. An approach based on evolutionary cross-species comparisons, such as PathBLAST [16,17] and interologs (i.e. interactions are conserved across species [9,18]), is a valuable frame-work for addressing these issues. However, these meth-ods often cannot respond how a protein interacts with another one across multiple species.
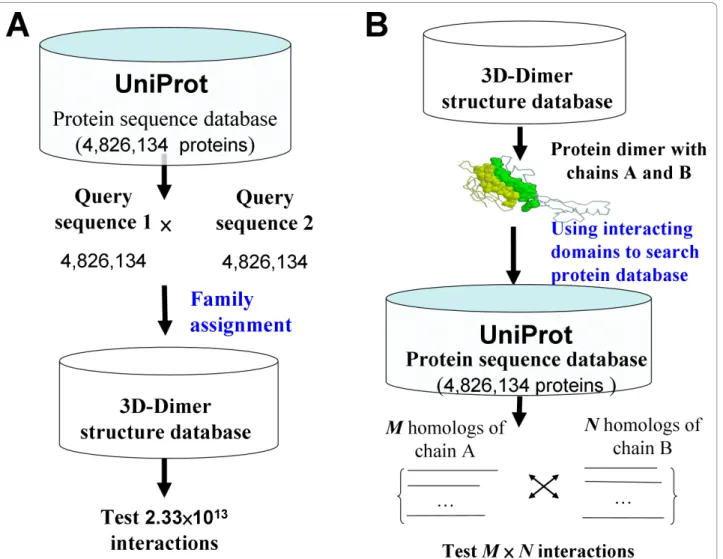
Protein Data Bank (PDB) [19] stores three-dimen-sional (3D) structure complexes, from which physical interacting domains can be identified to study DDIs and PPIs using comparative modeling [11,20]. Some DDI databases, such as 3did [21], iPfam [22], and DAPID [23], have recently been derived from PDB. Additionally, some methods have utilized template-based methods (i.e. comparative modeling [11] and fold recognition [20]), which search a 3D-complex library to identify homologous templates of a pair of query protein sequences, in order to predict the protein-protein inter-actions by accessing interface preference, and score query pair protein sequences according to how they fit the known template structures. However, these methods [11,20] are time-consuming to search all possible pro-tein-protein pairs in a large genome-scale database (Fig. 1A). For example, the possible protein-protein pairs on the UniProt database (4,826,134 sequences) are about 2.33×1013[24]. In addition, these methods are unable to form homologous PPIs to explore the protein-protein evolution for a specific structure template.
To address these issues, we proposed a new concept “3D-domain interolog mapping” (Fig. 1B): for a known 3D-structure complex (template T with chains A and B), domain a (in chain A) interacts with domain b (in chain B) in one species. Homolog families A’ and B’ of A and B are proteins, which are significant sequence similarity BLASTP E-values ≤10-10 and contain domains a and b, respectively. All possible protein pairs between these two homolog families are consid-ered as protein-protein interaction candidates using the template T. Based on this concept, protein sequence databases can be searched to predict protein-protein interactions across multiple species efficiently. When the genome was deciphered completely for a species, we considered the rank of protein-protein
interaction candidates in each species into our pre-vious scoring system [13] to reduce a large number of false positives. The 3D-interologs database which can indicate interacting domains and contact residues in order to visualize molecular details of a protein-protein interaction. Additionally, this database can provide couple-conserved residues and evolutionary clues of a query sequence and its partners by examining the interologs across multiple species.
Methods and materials
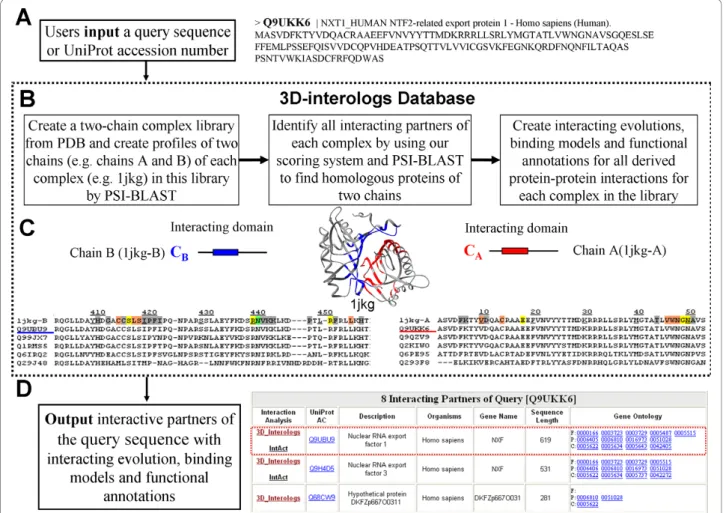
Figure 2 illustrates the overview of the 3D-interologs database. The 3D-interologs allows users to input the UniProt accession number (UniProt AC [24]) or the sequence with FASTA format of the query protein (Fig. 2A). When the input is a sequence, 3D-interologs uses BLAST to identify the hit interacting proteins. We identified protein-protein interactions in 3D-interologs database through structure complexes and a new scor-ing function usscor-ing the followscor-ing steps (Fig. 2B). First, a 3D-dimer template library comprising 1,895 heterodi-mers (3,790 sequences, called NR1895) was selected from the PDB released in Feb 24, 2006. Duplicate com-plexes, defined by sequence identity of above 98%, were removed from the library. Dimers containing chains shorter than 30 residues were also excluded [20,25]. Interacting domains and contact residues of two chains were identified for each complex in the 3D-dimer library. Contact residues, in which any heavy atoms should be within a threshold distance of 4.5 Å to any heavy atoms of another chain, were regarded as the core parts of the 3D-interacting domains in a complex. Each domain was required to have at least 5 contact residues and more than 25 interacting contacted-residue pairs to ensure that the interface between two domains was rea-sonably extensive. After the interacting domains were determined, its SCOP domains [26] were identified, and its template profiles were constructed by PSI-BLAST. PSI-BLAST was adopted to search the domain sequences against the UniRef90 database [24], in which the sequence identity < 90% of each other and the num-ber of iteration was set to 3.
After 3D-dimer template library and template profiles were built, we inferred candidates of interacting proteins by 3D-domain interolog mapping. To identify the inter-acting-protein candidates against protein sequences in the UniProt version 11.3 (containing 4,826,134 protein sequences), the chain profile was used as the initial posi-tion-specific score matrix (PSSM) of PSI-BLAST in each template consisting of two chains (e.g. CA and CB,
Fig. 2C). The number of iterations was set to 1. There-fore, this search procedure can be considered as a pro-file-to-sequence alignment. A pairing-protein sequence
Loet al. BMC Genomics 2010, 11(Suppl 3):S7 http://www.biomedcentral.com/1471-2164/11/S3/S7
(e.g. S1 and S2) was considered as a protein-protein interaction candidate if the sequence identity exceeded 30% and the aligned contact residue ratio (CR) was greater than 0.5 for both alignments (i.e. S1 aligning to CA and S2 aligning to CB). For each interacting
candi-date, the scoring function was applied to calculate the interacting score and the Z-value, which indicates the statistic significance of the interacting score. An acting candidate was regarded as a protein-protein inter-action if its Z-value was above 3.0 and it ranked in the Top 25 in one species. The candidate rank was consid-ered in one species to reduce the ill-effect of the out-paralogs that arose from a duplication event before the speciation [27]. These inferred interacting protein pairs were collected in the database.
Finally, for the hit interacting partner derived from 3D-domain interolog mapping, this database provides
functional annotations (e.g. UniProt AC, organism, descriptions, and Gene Ontology (GO) annotations [28], Fig. 2D), and the visualization of the binding models and interaction evolutions (Fig. 2C) between the query protein and its partners. We then constructed two mul-tiple sequence alignments of the query protein and its interacting partner (Fig. 2C) across multiple species. Here, the interacting-protein pair with the highest Z-score in a species was chosen as interologs for con-structing multiple sequence alignments using a star alignment. The chains (e.g. Chains A and B, Fig. 2C) of the hit structure template were considered as the cen-ters, and all selected interacting-protein pairs across spe-cies were aligned to respective chains of the template by PSI-BLAST. The 3D-interologs database annotates the important contact residues in the interface according to the following formats: hydrogen-bond residues (green);
Figure 1 Two frameworks of template-based methods for protein-protein interactions (PPI). (A) For each query protein sequence pair, the method searches 3D-dimer template library to identify homologous templates for exploring the query protein pair, such as MULTIPROSPECTOR [20]. (B) For each structure in 3D-dimer template library, the method searches protein sequence database to identify homologous PPIs of the query structure, such as 3D-interologs.
conserved residues (orange), conserved residues with hydrogen bonds (yellow) and other (gray).
Data Sets
Two data sets were used to assess 3D-domain interolog mapping and the scoring functions. To determine the contribution of a residue to the binding affinity, the ala-nine- scanning mutagenesis is frequently used as an experimental probe. We selected 275 mutated (called BA-275) residues from the ASEdb [29] with 16 heterodi-mers whose 3D structures were known. Those mutated residues are contact residues and positioned at protein-protein interfaces. ASEdb gives the corresponding delta G value representing the change in free energy of bind-ing upon mutation to alanine for each experimentally mutated residue. Residues that contribute a large amount of binding energy are often labeled as hot spots. In addition, we selected a non-redundant set (NR-563), comprising 563 dimer protein structures from the set NR1895 to evaluate the performance of our scoring functions for predicting PPIs in S. cerevisiae and in 549
species collected in Integr8 database (2,102,196 proteins [30]).
Scoring dunction and matrices
We have recently proposed a scoring function to deter-mine the reliability of a protein- protein interaction [13]. This study enhances this scoring by dividing the template consensus score into the template similar score and the couple-conserved residue score. Based on this scoring function, the 3D-interologs database can provide the interacting evolution across multiple species and the statistic significance (Z-value), the binding models and functional annotations between the query protein and its interacting partners. The scoring function is defined as
Etot= Evdw+ ESF+ Esim+ wEcons (1)
where Evdwand ESF are the interacting van der Waals
energy and the special interacting bond energy (i.e. hydrogen-bond energy, electrostatic energy and disul-fide-bond energy), respectively; and Esim is the template
Figure 2 Overview of the 3D-interologs database for protein-protein interacting evolution, protein functions annotations and binding models across multiple species.
Loet al. BMC Genomics 2010, 11(Suppl 3):S7 http://www.biomedcentral.com/1471-2164/11/S3/S7
interface similar score; and the Econsis couple-conserved
residue score. The optimal w value was yielded by test-ing various values rangtest-ing from 0.1 to 5.0; w is set to 3 for the best performance and efficiency on predicting binding affinity (BA- 275) and predicting PPIs in S. cere-visiae and in 549 species (Integr8) using the data set NR- 563. The Evdwand ESFare given as
E Vss Vsb Vsb E Tss Tsb Tsb vdw ij ij ji i j CP SF ij ij ji i j CP = + + = + +
∑
( ) ( ) , ,∑
∑
where CP denotes the number of the aligned-contact residues of proteins A and B aligned to a hit template; Vssij and Vsbij (Vsbji) are the sidechain-sidechain and
sidechain-backbone van der Waals energies between residues i (in protein A) and j (in protein B), respec-tively. Tssijand Tsbij (Tsbji) are the sidechain-sidechain
and sidechain-backbone special interacting energies between i and j, respectively, if the pair residues i and j form the special bonds (i.e. hydrogen bond, salt bridge, or disulfide bond) in the template structure. The van der Waals energies (Vssij, Vsbij, and Vsbji) and special
interacting energies (Tssij, Tsbij,and Tsbji) were calculated
from the four knowledge-based scoring matrices (Fig. 3), namely sidechain (Fig. 3A) and sidechain-backbone van der Waals scoring matrices (Fig. 3B); and sidechain-sidechain (Fig. 3C) and sidechain-backbone special-bond scoring matrices (Fig. 3D).
These four knowledge-based matrices, which were derived using a general mathematical structure [31] from a nonredundant set of 621 3D-dimer complexes proposed by Glaser et al.[32], are the key components of the 3D-interologs database for predicting protein-pro-tein interactions. This dataset is composed of 217 het-erodimers and 404 homodimers and the sequence identity is less than 30% to each other. The entry (Sij),
which is the interacting score for a contact residue i, j pair (1≤i, j≤20), of a scoring matrix is defined as
S q
e
ij ij ij
= ln where qijand eijare the observed probability
and the expected probability, respectively, of the occur-rence of each i, j pair. For sidechain-sidechain van-der Waals scoring matrix, the scores are high (yellow blocks) if large-aliphatic residues (i.e. Val, Leu, Ile, and Met) interact to large-aliphatic residues or aromatic residues (i.e. Phe, Tyr, and Trp) interact to aromatic residue. In contrast, the scores are low (orange blocks) when nonpolar residues interact to polar residues. The top two highest scores are 3.0 (Met. interacting to Met) and 2.9 (Trp interacting to Trp).
The value of Esimwas calculated from the BLOSUM62
matrix [31] based on two alignments between two chains (A and B) of the template and their homologous proteins (A’ and B’), respectively. The Esimis defined as
Esim K K K K ii jj ii jj i j CP = × ×
∑
’ , (2)where CP is the number of contact residue pairs in the template; i and j are the contact residue in chains A and B, respectively. Kii’is the score of aligning residue i
(in chain A) to i’ (in protein A’) and Kji’ is the score of
aligning residue j (in chain B) to j’ (in protein B’) according to BLOSUM62 matrix. Kii and Kjj are the
diagonal scores of BLOSUM62 matrix for residues i and j, respectively. The couple-conserved residue score (Econs) was determined from two profiles of the template
and is given by Econs Mip Kii Mjp Kjj i j CP =
∑
(max( ,( − )+( ′− )) , 0 (3)where CP is the number of contact residue pairs; Mip
is the score in the PSSM for residue type i at position p in Protein A; Mp is the score in the PSSM for residue type j at position p’ in Protein B, and Kiiand Kjjare the
diagonal scores of BLOSUM62 matrix for residue types iand j, respectively.
To evaluate statistical significance (Z-value) of the interacting score of a protein-protein interaction candi-date, we randomly generated 10,000 interfaces by mutat-ing 60% contact residues for each heterodimer in 3D-dimer template library. The selected residue was substi-tuted with another amino acid residue according to the probability derived from these 621 complexes [32]. The mean and standard deviation for each 3D-dimer were determined from these 10,000 random interfaces which are assuming to form a normal distribution. Based on the mean and standard deviation, the Z-value of a pro-tein-protein candidate predicted by this template can be calculated.
Difference between 3D-interologs and previous works
Some enhancements and modifications were applied to the DAPID database [23] and the 3D-partner server [13], thereby improving the reliability and applicability of the 3D- interologs method. There are five main dif-ferences between the 3D-interologs and our previous works (Table 1). First, 3D-interologs and 3D-partner integrates knowledge-based scoring matrices and cou-ple-conserved residue scores for measuring binding affi-nity and interface evolution of homologous PPIs to replace the homologous score in DAPID. Second,
3D-Figure 3 Knowledge-based protein-protein interacting scoring matrices: (A) sidechain van-der Waals scoring matrix; (B) sidechain-backbone van-der Waals scoring matrix; (C) sidechain-sidechain special-bond scoring matrix; (D) sidechain- sidechain-backbone special-bond matrix scoring. The sidechain-sidechain scoring matrices are symmetric and sidechain-backbone scoring matrices are nonsymmetric. For sidechain- sidechain van-der Waals scoring matrix, the scores are high (yellow blocks) if large-aliphatic residues (i.e. Val, Leu, Ile, and Met) interact to large-aliphatic residues or aromatic residues (i.e. Phe, Tyr, and Trp) interact to aromatic residue. In contrast, the scores are low (orange blocks) when nonpolar residues interact to polar residues. For sidechain-sidechain special- bond scoring matrix, the scores are high when an interacting resides (i.e. Cys to Cys) form a disulfide bond or basic residues (i.e. Arg, Lys, and His) interact to acidic residues (Asp and Glu). The scoring values are zero if nonpolar residues interact to other residues.
Table 1 The essential differences of DAPID, 3D-partner and 3D-interologs
Feature/Methods DAPID [23] 3D-partner [13] 3D-interologs
Homolog families of 3D- dimer template
NO YES (BLASTP E-value <10-2) YES (BLASTP E-value
≤10-10)
Scoring function: Template interface score
NO NO YES (Eshm)
Top ranked strategy NO NO YES (Top ranked N for each species)
Interface binding affinity
NO YES (four matrices) YES (four matrices)
Residue conservation score
NO YES (PSSM) YES (PSSM)
Score of aligned contact
residues YES (BLOSUM62) YES (BLOSUM62) YES (BLOSUM62)
Service type A database of domain-annotated protein
interactions
A web tool predicts interacting partners and binding models of a query protein sequence
An evolution database of physical protein-protein interactions across multiple genomes Loet al. BMC Genomics 2010, 11(Suppl 3):S7
http://www.biomedcentral.com/1471-2164/11/S3/S7
interologs considered the homolog families A’ and B’ of chains A and B of a template as significant sequence similarity BLASTP E-value ≤10-10. The threshold is E-value ≤10-2 applied in the 3D-partner server and DAPID utilized the E-value as the scoring function. Third, 3D-interologs utilized a new method for scoring the template interface similar score. Furthermore, 3D-interologs added a top ranked strategy for a specific species whose genome is deciphered completely. Finally, 3D-interologs and DAPID are databases, conversely, 3D- partner is a web-based service for identifying inter-acting partners of a query protein sequence.
Inputs and outputs
The 3D-interologs database server is easy-to-use. Users input the UniProt AC or the FASTA format of the query protein (Fig. 2A). The server generally returns a list of interacting partners with functional annotations (e.g. the gene name, the protein description and GO annotations) (Fig. 2D) and provides the visualization of the binding model and contact residues between the query protein and its partner by aligning them to respective template sequences and structures. Addition-ally, the 3D-interologs system indicates the interacting evolution analysis by using multiple sequence align-ments of the interologs across multiple species (Fig. 2C). The significant contact residues in the interface are indi-cated. If Java is installed in the user’s browser, then the output shows the structures, and users can dynamically view the binding model, interacting domains and impor-tant residues in the browser.
Results Database
The 3D-interologs database currently contains 15,124 species and 283,980 protein-protein interactions, includ-ing 173,294 interactions (61%) derived from our method based on 3D- domain interolog mapping and 110,686 interactions (39%) summarized from the IntAct database [3]. For the hit interacting partner derived from 3D-domain interolog mapping, this database provides functional annotations (e.g. UniProt AC, organism, descriptions, and Gene Ontology (GO) annotations [28]), and the visualization of the binding models and interaction evolutions between the query protein and its partners. On the other hand, the 3D-interologs database presents only the functional annotations of the hit protein-protein interaction if this interaction was summarized from the IntAct database.
Among 15,124 species in the 3D-interologs database, Table 2 shows 19 species commonly used in molecular research projects, such as Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Caenorhab-ditis elegans, Saccharomyces cerevisiae,and Escherichia
coli. To analyze couple-conserved residues and interface evolutions for providing evolutionary clues, the 15,124 species were divided into 10 taxonomic groups [33], namely mammalia, vertebrata, metazoa, invertebrata, fungi, plant, bacteria, archaea, viruses, and others.
Example analysis
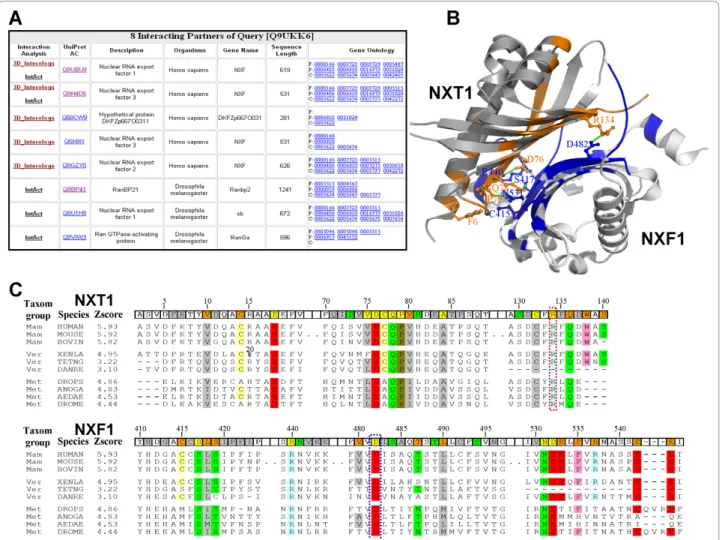
Figure 4 show the search results using the human pro-tein NXT1 (UniProt AC Q9UKK6) [34] as the query sequence. The NXT1, which is a nucleocytoplasmic transport factor and shuttles between the nucleus and cytoplasm, accumulates at the nuclear pore complexes. For this query, 3D-interologs database yielded 8 hit interacting partners (Fig. 4A), comprising 5 partners derived from 3D-interologs database and 5 partners from the IntACT database. Thus, two partners were present in both databases. Among these 8 hits, 3 part-ners (i.e. Uniprot AC Q68CW9, Q5H9I1 and Q9GZY0) were not recorded in IntAct database, but they very likely interact with NXT1. The Q68CW9, which is part of the protein NXF1 (UniProt AC Q9UBU9), consists of the UBA-like domain and the NTF-like domain, which is responsible for association with the protein NXT1 [35]. The sequence of the protein Q5H9I1 is the same as that of the protein Q9H4D5 (i.e. nuclear RNA export factor 3), which binds to NXT1 [36]. The protein Q9GZY0 (nuclear RNA export factor 2) binds protein
Table 2 Statistics of 3D-interologs database on 19 species commonly used in research projects
Species 3D-domain interologs IntAct
Mus musculus 8,876 2,634 Homo sapiens 8,639 18,716 Danio rerio 4,564 0 Xenopus laevis 4,057 58 Rattus norvegicus 3,685 958 Bos taurus 3,549 174 Drosophila melanogaster 2,644 25,036 Arabidopsis thaliana 2,418 2,111 Caenorhabditis elegans 1,433 4,684 Saccharomyces cerevisiae 443 36,821 Escherichia coli 426 14,007 Schizosaccharomyces pombe 371 341 Dictyostelium discoideum 284 84 Zea mays 219 0 Oryza sativa 193 69 Takifugu rubripes 191 0 Chlamydomonas reinhardtii 122 14 Plasmodium falciparum 68 2,707 Pneumocystis carinii 23 0 other species 131,089 2,272 Total 173,294 110,686
NXT1 to export mRNA cargoes from nucleus into cyto-sol [37].
The protein NXT1 interacts with the protein NXF1 to form a compact heterodimers (PDB code 1jkg [38])and an interactingb surface, which is lined with hydropho-bic and hydrophilic residues (Fig. 4B). Twenty hydrogen bonds or electrostatic interactions are formed in this compact interface. The salt bridge formed by NXT1 Arg134 and NXF1 Asp482 is especially important in the interface [29]. The interacting evolution analysis built by 10 interologs reveals that two residues (Arg134 and
Asp482) are conserved in all species (Fig. 4C). Addition-ally, some interacting residues forming the hydrogen bonds are also couple- conserved, for example NXT1 Asp76 and NXF1 Arg440; NXT1 Gln78 and NXF1 Ser417; NXT1 Pro79 and NXF1 Asn531 [29]. The evo-lution of interaction is valuable to reflect both couple-conserved and critical residues in the binding site.
Conversely, some positions, which are not conserved in all species but conserved in an individual taxonomic group, are important for observing the co-evolution across multiple species. The interacting residue pair
Figure 4 The 3D-interologs database search results of using human NXT1 (UniProt accession number Q9UKK6) as query. (A) Eight interacting partners of NXT1 are found in the 3D-Interologs. For each interacting partner, this server provides UniProt accession number, protein description, organism and Gene Ontology annotation. (B) Detailed interactions between the query and its interacting partner (UniProt accession number Q9UBU9) are indicated via the structure template which consists of NXT1 (PDB entry 1jkg-A) and NXF1 (PDB entry 1jkg-B). The contact residues of NXT1 (query side) and NXF1 (partner side) are colored by red and blue, respectively. The contact residues forming hydrogen bonds (green and dash) are given the atom details. (C) The interacting evolution analysis by using multiple sequence alignments of hit interacting partners of the query across multiple species. The 3D- interologs yields 10 interologs of the query template structure. The contacted residues are marked in template structure based on their interacting characteristics, including hydrogen-bond residues (green); conserved residues (orange); both (yellow), and others (gray). The couple-conserved contact positions are colored in the multiple alignments according to the physical-chemical property of amino acid residues. Twenty amino acid types are classified into 7 groups, namely polar positive (His,Arg, and Lys, blue); polar negative (Asp and Glu, red); polar neutral (Ser, Thr, Asn and Gln, green); cystein (yellow); polar aliphatic (Ala, Val, Leu, Ile and Met, gray); non-polar aromatic (Phe, Tyr and Trp, pink); and others: (Gly and Pro, brown).
Loet al. BMC Genomics 2010, 11(Suppl 3):S7 http://www.biomedcentral.com/1471-2164/11/S3/S7
(NXT1 Phe6 and NXF1 Cys415) in mammalia and ver-tebrata is different from that in metazoan (NXF1 Cys415®Met and NXT1 Phe®Leu variant). The der Waals potential (1.3 in the sidechain-sidechain van-der Waals scoring matrix, Fig. 3A) between Leu and Met is much larger than the potential (−0.1) between Cys and Phe. This co-evolution favors the formation of the hydrophobic interaction in metazoan.
Binding affinity prediction
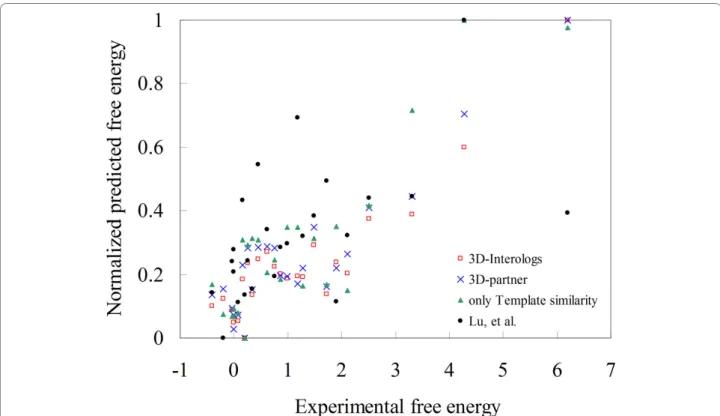
The enhanced scoring functions were first evaluated on 275 mutated residues selected from the ASEdb database [29] to reveal the Pearson correlations between ddG values and predicted energies. The 3D-interologs method applied four scoring functions (Fig. 5), including 3D- interologs (red), 3D-partner (blue), Esim (only
tem-plate similarity, green) and one matrix (black) proposed by Lu, et al.[20]. Among these four scoring functions, the 3D-interologs is the best (0.92) and one matrix is the worst (0.55, i.e. Lu, et al.). The correlations are 0.91 and 0.88 for 3D-partner and 0.88 (only template similar-ity), respectively.
The binding free energy is often not evenly distributed across interfaces but involves a small subset of “hot spots” contributed extraordinarily high energy [39]. For instance, the human blood-coagulation complex (PDB
code 1dan) has 52 residues whose energy contribution was probed by alanine scanning mutagenesis [40,41]. Among these 52 residues, residues Lys-20 and Asp-58, which are highly conserved in many species, provide the binding free energy upper 2 kcal/mol; on the other hand, the average energy contribution of the other 50 residues is 0.37 kcal/mol. This result implies that the couple-conserved residue score (Econs) is beneficial to model the binding energy of residues positioned in the interfaces. Although the hotspots of protein-protein binding are often for maintaining their function, the antibodies keep the diversity to recognize a wide varia-tion of antigens. The correlavaria-tion is 0.143 when the Econs
was used to model the binding energy of antigen-anti-body complexes. Fortunately, integrating Econs, Esimand
ESFis able to improve the correlation to 0.606 for
anti-gen-antibody complexes.
Interactions prediction inS. cerevisiae
Additionally, a non-redundant set (NR-563), comprising 563 dimer complexes from the 3D-dimer library, was adopted to evaluate the performance of this enhanced scoring function for interacting partner predictions in S. cerevisiae. This set comprised 5,882 protein-protein interactions, which were recorded as the core subset in the DIP database as the positive cases, and 2,708,746
Figure 5 Evaluation of the 3D-interologs in binding affinities. The Pearson correlations between experimental free energies (ddG) and the predicted values of the 3D-interologs using four scoring functions, including 3D-interologs (red), 3D-partner (blue), Esim(only template similarity,
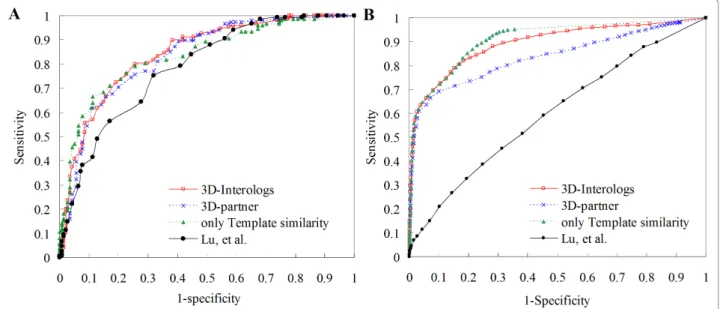
non-interacting protein pairs, defined by Jansen et al.[7] as the negative cases. Figure 6A shows the ROC curves of our method and other three scoring functions for predicting PPIs in S. cerevisiae. Among these four scor-ing functions, the 3D-interologs and the template simi-lar score (Esim) were the best and achieved the similar
accuracy. Conversely, one matrix (i.e. Lu, et al.[20]) was the worst. The average precisions, which was calculated as ( i T/ hi) /A
i A
=
∑
1 , where Thi denotes the number of
compounds in a hit list including i correct hits, were 0.84 (3D-interologs), 0.82 (3D-partner), and 0.67 for one matrix (proposed by Lu et al.). These results demon-strated that the proposed new scoring function can achieve good agreement for the binding affinity in PPIs and provide statistical significance (Z-value) for predict-ing PPIs.
Interactions prediction on multiple species
To evaluate the performance of the 3D-domain intero-log mapping on multiple species, 563 non-redundant dimer complexes (NR-563) were used as queries to search on the Integr8 database (Release 65) which com-prises 2,102,196 proteins in 549 species (Fig. 6B and Fig. 7). The Integr8 is an integrated database for organisms with completely deciphered genomes, which are mainly obtained from the non-redundant sets of UniProt entries. Experimentally determined protein-protein interactions dataset were collected from IntAct [3] as the gold standard positive set (110,686 interactions). The gold standard negative set was generated according to the assumption that two proteins acting in the same
biological process are more likely to interact than two proteins involved in different processes [42]. This study applied the relative specificity similarity (RSS), proposed by Wu et al.[43], to measure the biological process simi-larity and the location simisimi-larity of two proteins based on the GO terms of the biological process (BP) and the cellular component (CC), which describes locations at levels of subcellular structures and macromolecular complexes, respectively. Among 110,686 interactions recorded in the IntAct database, 51,049 interactions can be used to calculate the BP and the CC RSS scores. The BP and CC RSS scores of 15.85% and 2.65% interactions, respectively, are less than 0.4. Here, we considered an interacting protein pair as a negative PPI if its CC RSS score is less than 0.4.
The structures in the NR-563 as queries to search the Integr8 database yielded 1,063 protein-protein interac-tions recorded in the IntAct database and 131,831 pro-tein pairs, whose CC RSS scores were less than 0.4 as the negative cases. Based on ROC curves (Fig. 6B) for predicting PPIs in 549 species, 3D-interologs and the template similar score (Esim) outperform the 3D-partner
server and one-matrix (i.e. Lu, et al.) method. In addi-tion, the precision and recall were adopted to access the predicted quality of the 3D-interologs using these four scoring schemes (Fig. 7). The precision was defined as Ah/(Ah+Fh), where Ahand Fhdenote the numbers of hit
positive cases and hit negative cases, respectively. The recall was defined as Ah/A, where A is the total number
of positives (here A=1,063). Furthermore, the accuracy of our scoring function (red) is significantly better than that of the sequence identity (green).
Figure 6 The ROC curves of the 3D-interologs for protein-protein interactions. The 3D-interologs search results on (A) S. cerevisiae and (B) 549 species (Intger8) using the data set NR-563 (563 dimer-complex structures) by applying four scoring functions, including 3D-interologs (red), 3D-partner (blue), only template similarity (Esim, green) and one matrix (black) proposed by Lu, et al.
Loet al. BMC Genomics 2010, 11(Suppl 3):S7 http://www.biomedcentral.com/1471-2164/11/S3/S7
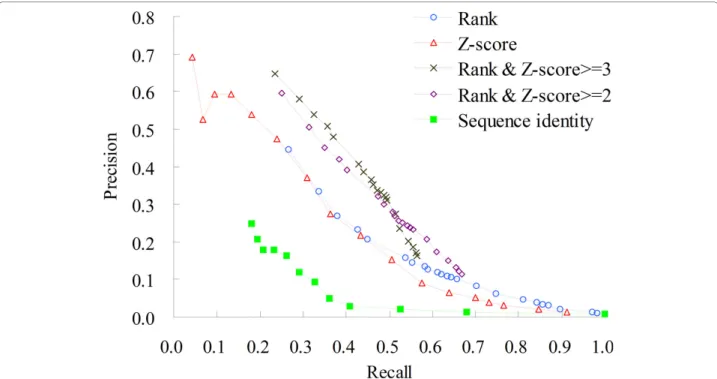
The 3D-domain interolog mapping may yield many PPI candidates (e.g. > 200) for one species from a struc-ture template because a eukaryote genome frequently contains multiple paralogous genes. Here, we proposed a top-rank strategy to limit the number of PPIs inferred from a structural template in the same species. For example, we discarded the PPI candidates whose ranks ≥ 25 for a species if the rank threshold is set to 25. Fig-ure 7 shows that the performance of the top-rank scores (blue, with different rank thresholds) is similar to that of using Z-score scoring method (red). When we combined the top-rank strategy and the Z-score scoring methods, the precisions (purple and black) are significantly improved. The precision was 0.52 and the recall was 0.34 when Z-score > 3.0 and the rank ≤25 in one species.
Adopting the top-rank strategy in one species as the scoring function is useful for distinguishing between positives and negatives when the 3D-domain interolog mapping yielded many protein-protein interactions for one species from a structure template. However, the rank cannot reflect the binding affinity of a PPI candi-date, conversely, the Z-score cannot be adopted to iden-tify the orthologs and in-paralogs arising from a duplication event following the speciation [27]. These results reveal that Z-scores and ranks scoring methods are complementary.
Table 3 shows an example for illustrating processes and robustness of combining the top- ranked strategy and Z-score methods. Using human calcineurin hetero-dimer (PDB code 1aui) structure as query, the 3D-domain interolog mapping yielded 1096 PPI candidates in 38 species if the Z score is set to 2. These 1096 can-didates possess the interacting domains (i.e. Metallophos and efand domains) of the query template. Among these PPI candidates, 10 PPIs were recorded in IntACT and 9 candidates were considered as negative PPIs because their CC RSS scores are less than 0.4. The ranks of these 9 negative PPIs are more than 15; conversely, these 10 positive PPIS are top 10 in each species. These observations showed that the top- ranked strategy is useful to dramatically reduce the false positive rate when the 3D-domain interolog mapping for predicting PPIs across multiple complete genomes.
Conclusions
This work demonstrates that the 3D-interologs database is robust and feasible for the interacting evolution of PPIs and DDIs across multiple species. This database can provide couple-conserved residues, interacting mod-els and interface evolution through 3D-domain interolog mapping and template-based scoring functions. The scoring function achieves good agreement for the bind-ing affinity in protein-protein interactions. We believe
Figure 7 Precisions and recalls of 3D-interologs the on Integr8. The 3D-interologs searches Integr8 database (2,102,196 proteins in 549 species) using the data set NR-563 (563 dimer-complex structures). The 3D-interologs server uses five scoring schemes, including rank in a species (blue), Z-score (red), rank and Z-score >=3 (black), rank and Z-score >=2 (purple), and sequence identity (green).
that the 3D- domain interolog mapping should be useful in protein-protein interacting evolution and is able to infer reliable physical protein-protein interactions across multiple genomes.
Acknowledgements
J.-M. Yang was supported by National Science Council and partial support of the ATU plan by MOE. Authors are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University. Publication of this supplement was made possible with support from the International Society of Intelligent Biological Medicine (ISIBM).
This article has been published as part of BMC Genomics Volume 11 Supplement 3, 2010: The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2164/11?issue=S3. Author details
1
Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan.2Institute of Bioinformatics, National Chiao Tung
University, Hsinchu, Taiwan.3Core Facility for Structural Bioinformatics,
National Chiao Tung University, Hsinchu, Taiwan. Authors’ contributions
Conceived and designed the experiments: YSL and JMY. Performed the experiments and analyzed the data: YSL, YCC and JMY. Contributed reagents/materials/analysis tools: YSL, YCC and JMY. Wrote the paper: YCC and JMY.
Competing interests
The authors declare that they have no competing interests. Published: 1 December 2010
References
1. Pagel P, Kovac S, Oesterheld M, Brauner B, Dunger-Kaltenbach I, Frishman G, Montrone C, Mark P, Stumpflen V, Mewes HW, et al: The MIPS mammalian protein-protein interaction database. Bioinformatics 2005, 21:832-834.
2. Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, Eisenberg D: DIP: the database of interacting proteins. Nucleic Acids Research 2000, 28(1):289-291.
3. Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, Dimmer E, Feuermann M, Friedrichsen A, Huntley R, et al: IntAct-open source resource for molecular interaction data. Nucleic Acids Research 2007, 35(Database issue):D561-D565.
4. Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y: A comprehensive two- hybrid analysis to explore the yeast protein interactome. Proceedings of the National Academy of Science of the USA 2001, 98:4569-4574.
5. Pandey A, Mann M: Proteomics to study genes and genomes. Nature 2000, 405:837-846.
6. von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P: Comparative assessment of large-scale data sets of protein-protein interactions. Nature 2002, 417:399-403.
7. Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M: A Bayesian networks approach for predicting protein- protein interactions from genomic data. Science 2003, 302(5644):449-453.
8. Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO: Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proceedings of the National Academy of Science of the USA 1999, 96(8):4285-4288.
9. Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, Garrels J, Vincent S, Vidal M: Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or
“interologs”. Genome Research 2001, 11:2120-2126.
10. Wojcik J, Schachter V: Protein-protein interaction map inference using interacting domain profile pairs. Bioinformatics 2001, 17:S296-S305. Table 3 3D-interologs search results using human calcineurin heterodimer as the query
Interactor1 Interactor2 Species Z score Rank P / Na RSS of BPb RSS of CCc Interacting domain1 Interacting domain2
P48456 P48451 Fruit fly 8.98 1 P 0.89 0.85 Metallophos efand
P23287 P25296 Yeast 8.25 1 P 0.88 1.00 Metallophos efand
P14747 P25296 Yeast 7.95 2 P 0.88 1.00 Metallophos efand
Q12705 Q9UU93 Yeast 7.94 1 P -d 0.78 Metallophos efand
P48456 P47948 Fruit fly 4.42 16 N 0.41 0.30 Metallophos efand
P48456 P47949 Fruit fly 4.38 17 N 0.41 0.30 Metallophos efand
P48456 P49258 Fruit fly 3.99 23 P 0.41 0.56 Metallophos efand
P48456 Q9VQH2 Fruit fly 3.94 25 N 0.49 0.33 Metallophos efand
Q8IAM8 P62203 Plasmdium falciparum 3.79 2 P - - Metallophos efand
P48456 P48593 Fruit fly 3.72 31 P 0.35 0.56 Metallophos efand
P48456 A1ZAE1 Fruit fly 3.59 34 N 0.00 0.30 Metallophos efand
Q27889 P48593 Fruit fly 3.42 40 P - - Metallophos efand
P23287 P06787 Yeast 3.36 5 P 0.61 0.88 Metallophos efand
P48456 Q9VMT2 Fruit fly 3.03 50 N 0.41 0.30 Metallophos efand
P48456 Q7K860 Fruit fly 2.99 53 N 0.41 0.30 Metallophos efand
P14747 P06787 Yeast 2.86 6 P 0.61 0.88 Metallophos efand
P48454 Q9NP86 Human 2.33 90 N - 0.00 Metallophos efand
Q08209 Q9NP86 Human 2.31 91 N 0.41 0.00 Metallophos efand
P16298 Q9NP86 Human 2.31 91 N - 0.00 Metallophos efand
3D-interologs infers 10 positive and 9 negative protein-protein interactions by human calcineurin heterodimer (PDB code 1aui), including calmodulin-dependent calcineurin A subunit alpha isoform (chain A with interacting domain Metallophos) and calcineurin subunit B type 1 (chain B with interacting domain efhand), searching on Integr8 database.a
PPI is a positive (P, recorded in IntACT database) or negative case (N, RSS of cellular component is less than 0.4).b,c
The relative specificity similarity (RSS) scores, proposed by Wuet al.[43], of Gene Ontology biologicalprocess (BP) and cellular component (CC), respectively.d
The protein pair is without Gene Ontology annotations in BP or CC.
Loet al. BMC Genomics 2010, 11(Suppl 3):S7 http://www.biomedcentral.com/1471-2164/11/S3/S7
11. Aloy P, Russell RB: Interrogating protein interaction networks through structural biology. Proceedings of the National Academy of Science of the USA 2002, 99(9):5896-5901.
12. Cary MP, Bader GD, Sander C: Pathway information for systems biology. FEBS Letters 2005, 579:1815-1820.
13. Chen Y-C, Lo Y-S, Hsu W-C, Yang J-M: 3D-partner: a web server to infer interacting partners and binding models. Nucleic Acids Research 2007, W561-W567.
14. Sun J, Sun Y, Ding G, Liu Q, Wang C, He Y, Shi T, Li Y, Zhao Z: InPrePPI: an integrated evaluation method based on genomic context for predicting protein- protein interactions in prokaryotic genomes. BMC Bioinformatics 2008, 8:414.
15. von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P: STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Research 2005, 33:D433-D437.
16. Kelley BP, Sharan R, Karp R, Sittler ET, Root DES, B. R., Ideker T: Conserved pathways within bacteria and yeast as revealed by global protein network alignment. Proceedings of the National Academy of Sciences of the United States of America 2003, 100:11394-11399.
17. Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, Sittler T, Karp RM, Ideker T: Conserved patterns of protein interaction in multiple species. Proceedings of the National Academy of Sciences of the United States of America 2005, 102:1974-1979.
18. Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JD, Bertin N, Chung S, Vidal M, Gerstein M: Annotation transfer between genomes: protein-protein interologs and protein-protein-DNA regulogs. Genome Research 2004, 14(6):1107-1118.
19. Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, et al: The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Research 2005, 33:D233-D237.
20. Lu L, Lu H, Skolnick J: MULTIPROSPECTOR: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins: Structure, Function and Bioinformatics 2002, 49(3):350-364. 21. Stein A, Russell RB, Aloy P: 3did: interacting protein domains of known
three- dimensional structure. Nucleic Acids Research 2005, 33(Database issue):D413-D417.
22. Finn RD, Marshall M, Bateman A: iPfam: visualization of protein-protein interactions in PDB at domain and amino acid resolutions. Bioinformatics 2005, 21(3):410-412.
23. Chen Y-C, Chen H-C, Yang J-M: DAPID: A 3D-domain annotated protein-protein interaction database. Genome Informatics 2006, 17:206-215. 24. Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S,
Gasteiger E, Huang H, Lopez R, et al: The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Research 2006, 34:D187-D191.
25. Ofran Y, Rost B: Analysing six types of protein-protein interfaces. Journal of molecular biology 2003, 325(2):377-387.
26. Andreeva A, Howorth D, Brenner SE, Hubbard TJ, Chothia C, Murzin AG: SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Research 2004, 32:D226-D229.
27. Li L, Stoeckert CJJ, Roos DS: OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Research 2003, 13:2178-2189. 28. Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K,
Lewis S, Marshall B, Mungall C, et al: The Gene Ontology (GO) database and informatics resource. Nucleic Acids Research 2004, 32:D258-D261. 29. Thorn KS, Bogan AA: ASEdb: a database of alanine mutations and their
effects on the free energy of binding in protein interactions. Bioinformatics 2001, 17(3):284-285.
30. Kersey P, Bower L, Morris L, Horne A, Petryszak R, Kanz C, Kanapin A, Das U, Michoud K, Phan I, et al: Integr8 and Genome Reviews: integrated views of complete genomes and proteomes. Nucleic Acids Research 2005, 33: D297-302.
31. Henikoff S, Henikoff JG: Amino acid substitution matrices from protein blocks. Proceedings of The National Academy of Sciences of The United States of America 1992, 89:10915-10919.
32. Glaser F, Steinberg DM, Vakser IA, Ben-Tal N: Residue frequencies and pairing preferences at protein-protein interfaces. Proteins: Structure, Function, and Bioinformatics 2001, 43:89-102.
33. Wheeler DL, Chappey C, Lash AE, Leipe DD, Madden TL, Schuler GD, Tatusova TA, Rapp BA: Database resources of the National Center for Biotechnology Information. Nucleic Acids Research 2000, 28:10-14. 34. Black BE, Levesque L, Holaska JM, Wood TC, Paschal BM: Identification of
an NTF2-related factor that binds Ran-GTP and regulates nuclear protein export. Molecular and Cellular Biology 1999, 19(12):8616-8624.
35. Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al: Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437(7062):1173-1178.
36. Herold A, Suyama M, Rodrigues JP, Braun IC, Kutay U, Carmo-Fonseca M, Bork P, Izaurralde E: TAP (NXF1) belongs to a multigene family of putative RNA export factors with a conserved modular architecture. Molecular and Cellular Biology 2000, 20(23):8996-9008.
37. Braun IC, Herold A, Rode M, Conti E, Izaurralde E: Overexpression of TAP/ p15 heterodimers bypasses nuclear retention and stimulates nuclear mRNA export. The Journal of Biological Chemistry 2001,
276(23):20536-20543.
38. Fribourg S, Braun IC, Izaurralde E, Conti E: Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol Cell 2001, 8(3):645-656. 39. Bogan AA, Thorn KS: Anatomy of hot spots in protein interfaces. Journal
of Molecular Biology 1998, 280(1):1-9.
40. Dickinson CD, Kelly CR, Ruf W: Identification of surface residues mediating tissue factor binding and catalytic function of the serine protease factor VIIa. Proceedings of the National Academy of Science of the USA 1996, 93(25):14379-14384.
41. Ruf W, Kelly CR, Schullek JR, Martin DM, Polikarpov I, Boys CW,
Tuddenham EG, Edgington TS: Energetic contributions and topographical organization of ligand binding residues of tissue factor. Biochemistry 1995, 34(19):6310-6315.
42. Rhodes DR, Tomlins SA, Varambally S, Mahavisno V, Barrette T, Kalyana-Sundaram S, Ghosh D, Pandey A, Chinnaiyan AM: Probabilistic model of the human protein- protein interaction network. Nature Biotechnology 2005, 23(8):951-959.
43. Wu X, Zhu L, Guo J, Zhang DY, Lin K: Prediction of yeast protein-protein interaction network: insights from the Gene Ontology and annotations. Nucleic Acids Research 2006, 34(7):2137-2150.
doi:10.1186/1471-2164-11-S3-S7
Cite this article as: Lo et al.: 3D-interologs: an evolution database of physical protein- protein interactions across multiple genomes. BMC Genomics 2010 11(Suppl 3):S7.
Submit your next manuscript to BioMed Central and take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit

![Figure 4 show the search results using the human pro- pro-tein NXT1 (UniProt AC Q9UKK6) [34] as the query sequence](https://thumb-ap.123doks.com/thumbv2/9libinfo/7494047.115651/7.892.456.807.165.566/figure-search-results-using-human-uniprot-query-sequence.webp)