Copolyester and Polyamide 6
JIA-CHONG HO, KUNG-HWA WEIDepartment of Materials Science and Engineering, National Chiao Tung University, Hsinchu, Taiwan 30049, Republic of China
Received 14 September 1999; revised 25 April 2000; accepted 24 May 2000
ABSTRACT: The kinetics of the ester–amide exchange in solution blends of the random liquid-crystalline polyester copoly(oxybenzoate-terephthalate) (P64) and polyamide 6 (PA6) were studied with carbon nuclear magnetic resonance. With second-order revers-ible reactions assumed, the activation energy of the ester–amide interchange in 30/70, 50/50, and 70/30 P64/PA6 blends were all about 24.0 kcal/mol. The pre-exponential factors for the ester–amide exchange in 30/70, 50/50, and 70/30 P64/PA6 blends were 2.01⫻ 1011, 2.59⫻ 1010, and 2.74⫻ 1012min⫺1, respectively.© 2000 John Wiley & Sons,
Inc. J Polym Sci B: Polym Phys 38: 2124 –2135, 2000
Keywords: copolyester; polyamide 6; exchange; activation energy; kinetics
INTRODUCTION
Thermotropic liquid-crystalline polymers (TL-CPs) usually exhibit low melt viscosity and high modulus in the oriented direction in the solid form. Blending TLCPs with engineering plastics can lower the melt viscosity of the blend and form organic polymer composites in situ.1– 4 However, the enthalpy of mixing TLCPs with a flexible-coil polymer is mostly positive. The entropy is usually small in the mixing of these two polymers. There-fore, the free energy of mixing is mostly positive. In other words, most TLCPs blends are immisci-ble or become phase-separated systems. However, it is well known that high molecular weight poly-esters can have midchain ester– ester exchange with themselves or other polymers at high tem-peratures. Several studies report that the misci-bility of polyester blends increases with the ex-tent of interchange reactions.5,6In particular, the midchain ester– ester interchange7–9 in blends of thermotropic copoly(oxybenzoate-ethylene
tephthalate) (POB–PET) and polycarbonate has re-sulted in a partially miscible system, as evidenced from a single glass-transition temperature in differ-ential scanning calorimetry measurement.10
At high temperatures, amide–amide and es-ter–amide interchanges also have occurred in polyamide/polyamide blends11–15 and polyester/ polyamide blends, respectively.16,17The physical properties of these blends were greatly affected by the interchange reactions. However, very little has been reported on the kinetics of the ester– amide interchange reactions in polyester/poly-amide blends, which often take place in industrial processes.16,17 We are, therefore, motivated to study the kinetics of the ester–amide interchange reactions in POB–PET/polyamide 6 (PA6). The kinetics of the interchange reaction in four-com-ponent copolycondensates18 –21and in five-compo-nent copolycondensates9,10 have been investi-gated with high-resolution nuclear magnetic res-onance (NMR). In this work, the kinetics of the ester–amide interchange reaction in blends of POB–PET and PA6 were determined from two-dimensional (2D) NMR heteronuclear multiple-bond correlation (HMBC) spectra and quantita-tive13C NMR analyses.
Correspondence to: K.-H. Wei (E-mail: [email protected].
edu.tw)
Journal of Polymer Science: Part B: Polymer Physics, Vol. 38, 2124 –2135 (2000) © 2000 John Wiley & Sons, Inc.
EXPERIMENTAL
Liquid-crystalline POB–PET at a molar ratio of 60/40 was obtained from Unitika Ltd., Japan, and was termed P64. PA6 (Zytel) was purchased from DuPont. The intrinsic viscosity value of PA6 was 1.33 dL/g at 25 °C, obtained from a dilute solution of PA6 (0.5 dL/g) in m-cresol.22 This indicated that the concentration of the terminal groups for PA6 was about 0.6%. The solution blending of P64 and PA6 was carried out by both polymers being dissolved in 100 mL of a mixed solvent of phenol and tetrachloroethane (50/50 by weight). The con-centration of the solution containing P64 and PA6 was 2% by weight. The weight ratios of P64 to PA6 in the solution were 30/70, 50/50, and 70/30. The solution was stirred and maintained at 60 °C. After the polymers were dissolved and became a one-phase solution for 60 min, the solutions were precipitated in a 10-fold excess volume of metha-nol. The precipitated blends were washed five times, each time with 500 mL of methanol. The blends were then dried in a vacuum oven at 100 °C for 5 days. The thermal gravimetric analysis of the dried blends showed no appreciable weight loss up to 350 °C, indicating a complete removal of the solvent. The blends were annealed at 260, 270, and 280 °C in a high-temperature oven for different times under a purge of nitrogen at 100 cm3/min.
A model compound study performed by the re-action of 20/80 terephthalate acid (TPA)/6-amin-ocaproic acid (ACA) at 270 °C and 80/20 4-ace-toxybenzoic acid (ABA)/ACA at 250 °C for 2 h was carried out. The products from the reacted TPA/ ACA and ABA/ACA were then dissolved in a mixed solvent of phenol and deuterated chloro-form (80/20 by volume). 2D1H–13C HMBC NMR analyses were carried out with a Bruker DMX-600 spectrometer. Both the freshly prepared and
annealed blends of P64/PA6 (50/50) were partially dissolved in deuterated trifluoroacetic acid. A mixed solvent of phenol and deuterated chloro-form (80/20 by volume) and tetramethylsilane as the standard were used in quantitative13C NMR analyses. The concentration of the blend in the mixed solvent was about 5%. The intrinsic viscos-ity of the blends in a mixed solvent of phenol and tetrachloroethane, 50/50 by weight, was mea-sured with an Ubbelohde viscometer at 30 °C.
RESULTS AND DISCUSSION
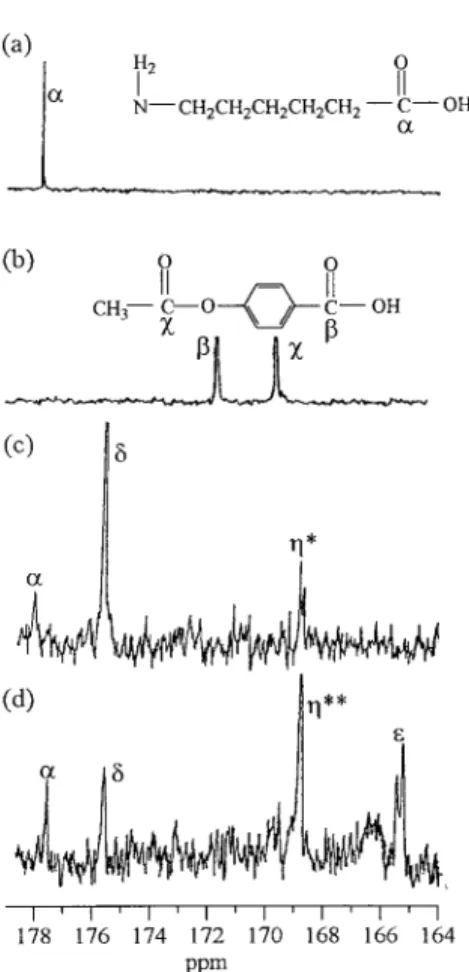
The chemical structures of P64 and PA6 were divided into four components. The notations A1, A2, and (AB)1 represent the ethylene dioxide, terephthalate, and oxybenzoate in the monomeric unit in P64, respectively, and (AB)2 represents the amide in the monomeric unit of PA6, as given in Table I. From the model compound study, the carbonyl regions of the13C NMR spectra of ACA, ABA, and 20/80 TPA/ACA reacted at 270 °C and 80/20 ABA/ACA reacted at 250 °C are displayed in Figure 1. In Figure 1(a), the C␣peak at 177.9 ppm in the13C NMR spectra of ACA represented the carbonyl group in ACA. The Cpeak at 171.5 ppm and the Cpeak at 169.6 ppm resulted from the carbonyl group and acetoxy group in ABA, respectively, as shown in Figure 1(b). The new peaks C*at 168.8 ppm and C␦at 175.7 ppm were caused by the dyad terephthalate–amide [B1(AB)2] and by the dyad amide–amide [(AB)2(AB)2], re-spectively, as indicated in Figure 1(c). In Figure 1(d), the new peak C**, caused by the dyad oxy-benzoate–amide [(AB)1(AB)2] also appeared at 168.8 ppm, whereas the new peak C⑀, caused by the dyad [(AB)1(AB)1], was located at 165.2 ppm. The chemical structure changes that resulted from the ester–amide exchange in the P64/PA6 Table I. The Codes Used in the Dyads of the P64/PA6 Blend
POB–PET OOCH2CH2OO A1 B1 (AB)1 PA6 H O P 储 O N OCH2CH2CH2CH2CH2O C O (AB)2
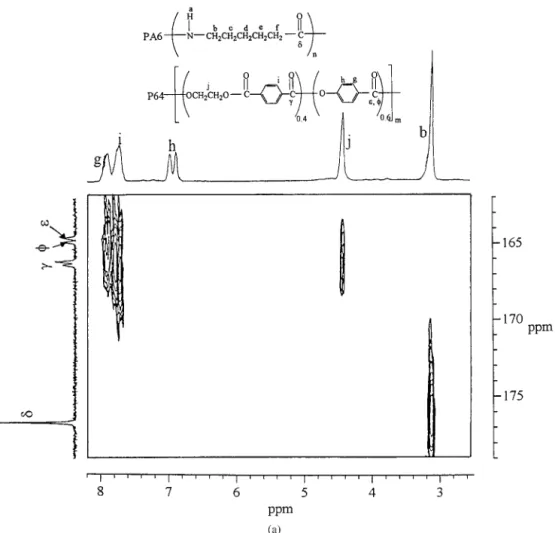
blends could not be identified because the C*and the C** peaks appeared at the same location, 168.8 ppm, in the one-dimensional (1D) NMR spectrum. Therefore, 2D1H–13C NMR analyses of the P64/PA6 blends were required. The 2D 1H– 13C NMR spectra of the freshly prepared 50/50 P64/PA6 blend are presented in Figure 2(a). Two new crosspeaks appear at 7.5–168.8 ppm (cross-peak I) and 3.3–168.8 ppm (cross(cross-peak II) in the 1H–13C HMBC NMR spectra of the 50/50 blend after the blend annealed at 270 °C for 60 min, as shown in Figure 2(b). In Figure 2(b), the new proton peak Hk, caused by the dyads [B1(AB)2] and [(AB)1(AB)2], was overlapped at 7.5 ppm. Ad-ditionally, Cwas attached to Hk of the oxyben-zoate or the terephthalate at crosspeak I, and C was also attached to the aromatic Hb of PA6 at crosspeak II, indicating C belonged to the car-bonyl carbon of the amide group. By comparing the results of the 1D13C NMR analysis to those of the 2D1H–13C HMBC NMR analysis of the
an-nealed P64/PA6 blend, we concluded that the new peak Cwas actually caused by a superposition of the peaks from the dyads terephthalate–amide and oxybenzoate–amide. The assignments of the partial carbon peaks for the original and new dyads in the P64/PA6 blend were, therefore, de-termined and are given in Table II.
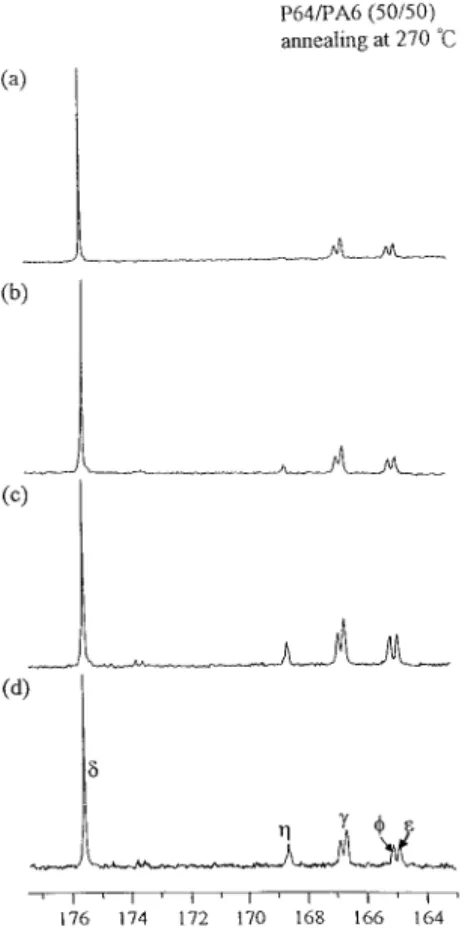
The partial13C NMR spectra of the 50/50 P64/ PA6 blends annealed at 270 °C are displayed in Figure 3. In Figure 3, the intensity of the new peak C, representing the carbonyl group in the 13C NMR spectra, increases with annealing time. Through the integration of the area under these 13C peaks, the transient dyad molar fractions in the 30/70, 50/50, and 70/30 P64/PA6 blends an-nealed at 260, 270, and 280 °C are given in Table III. Before the exchange starts, we have B1A1, (AB)1A1, B1(AB)1, (AB)1(AB)1, and (AB)2(AB)2. The initial dyad molar fractions of B1A1, (AB)1A1, B1(AB)1, (AB)1(AB)1, and (AB)2(AB)2 are FB1A1,0, F(AB)1A1,0, FB1(AB)1,0, F(AB)1(AB)1,0, and F(AB)2(AB)2,0, respectively, and they can be calculated from the initial molar fractions of P64 and PA6. In Table III(a– c), FB1A1, F(AB)1A1, and F(AB)1(AB)1decrease with both increasing annealing time and temper-ature in different blends. Because the new peak C was due to an overlap of the peaks, FB1(AB)2 and F(AB)1(AB)2cannot be decoupled directly from their13C NMR spectra. We first obtained the in-tegral under the new peak C. Then, FB1(AB)2and F(AB)1(AB)2 could be determined by the following formula:
FB1共AB兲2⫽ 共FB1共AB兲2⫹ F(AB)1共AB兲2兲
⫻ mole fraction of PET in P64 F共AB兲1共AB兲2⫽ 共FB1共AB兲2⫹ F共AB兲1共AB兲2兲
⫻ mole fraction of POB in P64 The calculated values of FB1(AB)2 and F(AB)1(AB)2 are given in Table III(a– c). In Table III, FB1(AB)2 and F(AB)1(AB)2in the 30/70, 50/50, and 70/30 P64/ PA6 blends increase with annealing time and temperature. For the 50/50 P64/PA6 blend, the molar fractions of FB1(AB)2become 0.022 after 30 min of annealing and 0.038 after 60 min of an-nealing at 270 °C. Furthermore, FB1(AB)2 in the 50/50 P64/PA6 blend is 0.024 after 30 min of annealing at 280 °C.
With these dyad concentrations available from the13C NMR spectra, we needed to build reaction
Figure 1. Carbonyl region of the13C NMR spectra of
(a) ACA, (b) ABA, (c) 20/80 TPA/ACA reacted at 270 °C, and (d) 80/20 ABA/ACA reacted at 250 °C (for the model compound study).
equations for obtaining the kinetics parameters. The possible ways for the ester–amide inter-change in terms of dyads in P64/PA6 are given in the following reactions:
Reaction I 共B1A1兲 ⫹ 共共AB兲2共AB兲2兲 -|0 kI k⫺I 共B1共AB兲2兲 xI ⫹ 共共AB兲2A1兲 xI Reaction II 共B1共AB兲1兲 ⫹ 共共AB兲2共AB兲2兲 -|k0II k⫺II 共B1共AB兲2兲 xII ⫹ 共共AB兲2共AB兲1兲 xII Reaction III 共共AB兲1A1兲 ⫹ 共共AB兲2共AB兲2兲 -|0 kIII k⫺III 共共AB兲1共AB兲2兲 yIII ⫹ 共共AB兲2A1兲 yIII Reaction IV 共共AB兲1共AB兲1兲 ⫹ 共共AB兲2共AB兲2兲 -|0 kIV k⫺IV 共共AB兲1共AB兲2兲 yIV ⫹ 共共AB兲2共AB兲1兲 yIV Reaction V 共共AB兲1共AB兲1兲 ⫹ 共A1B1兲 -|0 kV k⫺V 共共AB兲1A1兲 ⫹ 共B1共AB兲1兲
Figure 2. 2D1H–13C NMR spectra of (a) the freshly prepared 50/50 P64/PA6 blend
where kiand k⫺i(i⫽ I, II, III, IV, and V) stand for the forward and reverse reaction rate constants for each reaction, respectively. The concentra-tions of these new dyads are defined on the basis of their individual reactions in the following: xI⫽ 共FB1共AB兲2兲I⫽ 共F共AB兲2A1兲I,
xII⫽ 共FB1共AB兲2兲II⫽ 共F共AB兲2共AB兲1兲II yIII⫽ 共F共AB兲1共AB兲2兲III⫽ 共F共AB兲2A1兲III,
yIV⫽ 共F共AB兲1共AB兲2兲IV⫽ 共F共AB兲2共AB兲1兲IV x⫽ FB1共AB兲2⫽ xI⫹ xII
y⫽ F共AB兲1共AB兲2⫽ yIII⫹ yIV
where the subscripts I, II, III, and IV indicate the resulting reaction. P64 is a random
co-polyester identified from its 13C NMR and 1H NMR spectra in previous studies.23–25Reaction V is in equilibrium during the synthesis of ran-dom P64. Hence, reaction V is not considered here.
Second-order reversible reactions were as-sumed for all cases in this study.26 The dyad terephthalate–amide [B1(AB)2] is produced in re-actions I and II. We assumed that x and y are decoupled during the reactions. Then, we ob-tained eqs 1 and 2 from reactions I and II in the following forms, respectively:
Reaction I dxI
dt ⫽kI共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共FB1A1,0⫺ xI兲
⫺ k⫺I共xI⫹ xII兲共xI⫹ yIII兲 (1)
Reaction II dxII
dt ⫽kII共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共F共AB兲1B1,0⫺ xII兲 ⫺ k⫺II共xI⫹ xII兲共xII⫹ yIV兲 (2) By adding eq 1 to eq 2, we obtained eq 3: dx dt ⫽ dxI dt ⫹ dxII dt
⫽ kI共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共FB1A1,0⫺ xI兲 ⫺ k⫺I共xI⫹ xII兲共xI⫹ yIII兲 ⫹ kII共F共AB兲2共AB兲2,0
⫺ 共x ⫹ y兲)共F共AB兲1B1,0⫺ xII兲
⫺ k⫺II共xI⫹ xII兲共xII⫹ yIV兲 (3) The dyad oxybenzoate–amide [(AB)1(AB)2] is pro-duced in reactions III and IV:
Reaction III dyIII
dt ⫽kIII共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共FA1共AB兲1,0⫺ yIII兲 ⫺ k⫺III共yIII⫹ yIV兲共yIII⫹ xI兲 (4) Reaction IV
dyIV
dt ⫽kIV共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共F共AB兲1共AB兲1,0⫺ yIV兲 ⫺ k⫺IV共yIV⫹ yIII兲共yIV⫹ xII兲 (5) By adding eq 4 to eq 5, we obtained eq 6. dy dt ⫽ dyIII dt ⫹ dyIV dt
⫽ kIII共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共FA1共AB兲1,0⫺ yIII兲 ⫺ k⫺III共yIII⫹ yIV兲共yIII⫹ xI兲
⫹ kIV共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共F共AB兲1共AB兲1,0 ⫺ yIV)⫺ k⫺IV共yIV⫹ yIII兲共yIV⫹ xII兲 (6) Table II. Assignment of the Partial Carbon Peaks in the P64/PA6 Blend
Peak Dyad Structure Chemical Shift
␦ (AB)2(AB)2 175.8 ppm (AB)1(AB)1 164.9 ppm (AB)1A1 165.1 ppm B1A1 ␥ 166.8 ppm B1(AB)1 B1(AB)2 168.8 ppm (AB)1(AB)2
FB1(AB)2 and F(AB)1(AB)2 increase with annealing temperature and annealing time at about the same rate given in Table III. If the rate constants kiand k⫺i(i⫽ I, II, III, IV, and V) have the same order of magnitude,
kI⬵ kII⬵ kIII⬵ kIV⬵ K
and k⫺I⬵ k⫺II⬵ k⫺III⬵ k⫺IV⬵ K⬘ (7) Therefore, the ester–amide exchange of P64 (es-ter group) and PA6 (amide group) was lump-summed in the following:
ester–ester⫹ amide–amide -|0 K K⬘ ester–amide ⫹ amide–ester Z is defined as
Z⫽ x ⫹ y ⫽ xI⫹ xII⫹ yIII⫹ yIV⫽ FB1共AB兲2
⫹ F共AB兲1共AB兲2 (8) By combining eqs 3 and 6, we obtained eq 9 in the following form:
dZ dt ⫽
d共x ⫹ y兲 dt
⫽ kI共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共FB1A1,0⫺ xI兲 ⫺ k⫺I共xI⫹ xII兲共xI⫹ yIII兲 ⫹ kII共F共AB兲2共AB兲2,0 ⫺ 共x ⫹ y兲)共F共AB兲1B1,0⫺ xII兲 ⫺ k⫺II共xI⫹ xII兲 ⫻ 共xII⫹ yVI兲 ⫹ kIII共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲 ⫻ 共FA1共AB兲1,0⫺ yIII兲 ⫺ k⫺III共yIII⫹ yIV兲共yIII⫹ xI兲 ⫹ kIV共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲共F共AB兲1共AB兲1,0⫺ yIV兲
⫺k⫺IV共yIV⫹ yIII兲共yIV⫹ xII兲 ⫽ 共F共AB兲2共AB兲2,0⫺ 共x ⫹ y兲兲
⫻
冉
kI共FB1A1,0⫺ xI兲 ⫹ kII共F共AB兲1B1,0⫺ xII兲 ⫹ kIII共FA1共AB兲1,0⫺ yIII兲⫹ kIV共F共AB兲1共AB兲1,0⫺ yIV兲
冊
⫺冉
k⫺I共xI⫹ xII兲共xI⫹ yIII兲 ⫹ k⫺II共xI⫹ xII兲共xII⫹ yIV兲 ⫹ k⫺III共yIII⫹ yIV兲共yIII⫹ xI兲 ⫹ k⫺IV共yIV⫹ yIII兲共yIV⫹ xII兲冊
⫽ 共F共AB兲2共AB兲2,0⫺ Z兲⫻
冉
K共FB1A1,0⫹ F共AB兲1B1,0⫹ FA1共AB兲1,0 ⫹ F共AB兲1共AB兲1,0)⫺ K共xI⫹ xII⫹ yIII⫹ yIV兲冊
⫺ K⬘共x共x ⫹ y兲 ⫹ y共x ⫹ y兲兲⫽ K共FP64,0⫺ Z兲共FPA6,0⫺ Z兲 ⫺ K⬘Z2 (9) where FP64,0and FPA6,0are the initial molar frac-tion of P64 and PA6, respectively, and they are given in terms of the dyads molar fractions:
FP64,0⫽ FB1A1,0⫹ F共AB兲1B1,0⫹ FA1共AB兲1,0⫹ F共AB兲1共AB兲1,0 FPA6,0⫽ F共AB兲2共AB兲2,0
Figure 3. Partial13C NMR spectra of the 50/50 P64/
PA6 blends annealed at 270 °C for (a) 0 min, (b) 15 min, (c) 30 min, and (d) 60 min.
Because the reaction is equilibrium, Ze is at its
optimal value, which equals FP64,0FPA6,0. Ze
⫽ FP64,0FPA6,0 and FP64,0FPA6⫹ FPA6,0 ⫽ 1 were put into eq 9, and the following equation was obtained: dZe dt ⫽K共FP64,0⫺ Ze兲共FPA6,0⫺ Ze兲 ⫺ K⬘Ze2⫽ 0 K共FP64,0FPA6,0⫺ Ze共FP64,0⫹ FPA6,0兲 ⫹ Ze2兲 ⫽ K⬘Ze2 K共Ze⫺ Ze⫹ Ze2兲 ⫽ K⬘Ze2 K⫽ K⬘ (10)
By putting eq 10 into eq 9, we obtained eq 11: Table III. The Transient Dyad Mol for Fractions of Various Dyads Obtained from Quantitative
13C NMR Analyses
Annealing Temperature
Annealing
Time F(AB)2(AB)2 FB1(AB)2 F(AB)1(AB)2 FB1A1 F(AB)1A1 F(AB)1(AB)1 (a) 30/70 P64/PA 6 Blends Annealing at 260, 270, and 280 °C
0 min 0.754 0 0 0.022 0.076 0.072 a260 °C 150 min 0.744 0.004 0.006 0.021 0.073 0.069 30 min 0.732 0.009 0.013 0.020 0.069 0.065 60 min 0.704 0.020 0.030 0.019 0.060 0.057 270 °C 15 min 0.733 0.008 0.013 0.021 0.069 0.066 30 min 0.703 0.020 0.031 0.018 0.060 0.057 60 min 0.673 0.032 0.049 0.015 0.051 0.048 280 °Ca 10 min 0.724 0.012 0.018 0.019 0.067 0.063 20 min 0.699 0.022 0.033 0.017 0.059 0.056 30 min 0.675 0.032 0.047 0.014 0.052 0.049 Annealing Temperature Annealing
Time F(AB)2(AB)2 FB1(AB)2 F(AB)1(AB)2 FB1A1 F(AB)1A1 F(AB)1(AB)1 (b) 50/50 P64/PA6 Blends Annealing at 260, 270, and 280 °C
0 min 0.558 0 0 0.039 0.137 0.129 a260 °C 15 min 0.544 0.010 0.014 0.037 0.126 0.119 30 min 0.524 0.018 0.026 0.035 0.120 0.113 60 min 0.495 0.029 0.044 0.032 0.111 0.105 270 °C 15 min 0.537 0.012 0.019 0.036 0.124 0.117 30 min 0.512 0.022 0.034 0.034 0.116 0.110 60 min 0.472 0.038 0.058 0.030 0.104 0.098 280 °Ca 10 min 0.528 0.016 0.024 0.035 0.118 0.011 20 min 0.508 0.024 0.036 0.034 0.115 0.108 30 min 0.485 0.033 0.050 0.031 0.108 0.102 Annealing Temperature Annealing
Time F(AB)2(AB)2 FB1(AB)2 F(AB)1(AB)2 FB1A1 F(AB)1A1 F(AB)1(AB)1 (c) 70/30 P64/PA6 Blends Annealing at 260, 270, and 280 °C
0 min 0.361 0 0 0.057 0.198 0.186 a260 °C 15 min 0.331 0.012 0.018 0.055 0.188 0.178 30 min 0.311 0.020 0.030 0.053 0.182 0.172 60 min 0.271 0.036 0.054 0.051 0.169 0.160 270 °C 15 min 0.314 0.019 0.028 0.053 0.183 0.173 30 min 0.276 0.034 0.051 0.050 0.171 0.162 60 min 0.225 0.054 0.082 0.046 0.155 0.147 280 °Ca 10 min 0.030 0.024 0.037 0.053 0.178 0.169 20 min 0.262 0.039 0.060 0.049 0.167 0.157 30 min 0.231 0.052 0.078 0.047 0.157 0.148
dZ
dt ⫽K共FP64,0FPA6,0⫺ Z兲 (11) By integrating eq 11 and using the amide– ester exchange reaction ratio, z ⫽ Z/FP64,0, for these
reactions, we can obtain a simple kinetics expres-sion:
ln
冉
FPA6,0共FPA6,0⫺Z兲
冊
⫽ Kt(12)
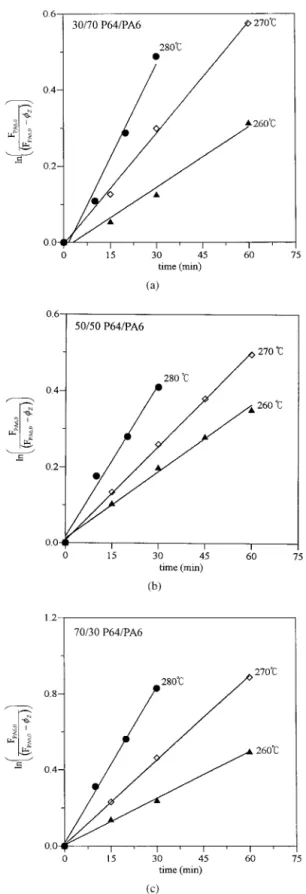
By plotting ln[FPA6,0/(FPA6,0⫺ Z)] against time,
we can obtain the rate constant K from the slopes of these lines, as shown in Figure 4. The rate constant K for the ester–amide exchange in P64/ PA6 blends at different temperatures and differ-ent compositions is given in Table IV. We can fit the rate constants into the Arrhenius expression:
ln K⫽ ln A ⫺ Ea
RT (13)
where A is the pre-exponential factor, Ea is the activation energy, R is the gas constant [R ⫽ 1.987 cal/(mol K)], and T is the absolute tem-perature. In Figure 5, the slopes of these lines were ⫺Ea/R, and the activation energy obtained was about 24.0 kcal/mol for P64/PA6 blends of different compositions. This activation energy for the ester–amide exchange reaction in P64/PA6 is smaller than that for the ester– ester exchange in the formation of the dyad bisphenol A– oxybenzo-ate (94.6 kcal/mol) and the dyad bisphenol A–terephthalate (41.8 kcal/mol).10The pre-expo-nential factors in the ester–amide exchange in
Figure 4. Plots of the kinetics expression of the es-ter–amide exchange in different weight ratios of P64 to PA6: (a) 30/70, (b) 50/50, and (c) 70/30.
Table IV. Rate Constants of Ester–Amide Exchange in P64/PA6 Blends Annealed at Different Temperatures P64/PA6 Annealing Temperature (°C) K ⫻ 103 (min⫺1) 30/70 260 5.30 270 9.66 280 16.41 50/50 260 4.80 270 8.23 280 13.84 70/30 260 8.15 270 14.70 280 27.32
P64/PA6 blends were determined from the inter-cepts. In Table V, the pre-exponential factors were 2.01 ⫻ 1011, 2.59 ⫻ 1010, and 2.74 ⫻ 1012 min⫺1 for the 30/70, 50/50, and 70/30 P64/PA6 blends, respectively. From the hard-sphere colli-sion theory,27 the pre-exponential factor, A, can be written as the following:
A⫽ BP64PA6 NA关P64兴关PA6兴
e1/2 (14) where [P64] and [PA6] indicate the concentra-tions of P64 and PA6, respectively. BP64 PA6is the total number of P64/PA6 collisions per unit time per unit volume. NAis Avogadro’s number. From eq 14, the pre-exponential factor A is proportional to BP64 PA6. The ester–amide exchange takes place in the interfaces between P64 and PA6 do-mains. As binary blends become more miscible, the domain size in the blends is decreasing, and the total surface area of all domains on a per weight basis will be increasing. In other words, the number of exchange collisions is increasing in
more miscible blends. Hence, the pre-exponential factor for the ester–amide exchange in P64/PA6 bends can be explained by the miscibility of the
Figure 5. Rate constants of the ester–amide ex-change in 30/70, 50/50, and 70/30 P64/PA6 blends at different temperatures.
Figure 6. Polarized micrographs of (a) 30/70, (b) 50/ 50, and (c) 70/30 P64/PA6 blends annealed at 270 °C for 5 min.
Table V. The Pre-Exponential Factor of the P64/ PA6 Blends
P64/PA6
Pre-Exponential Factor (A) (min⫺1)
30/70 2.01⫻ 1011
50/50 2.59⫻ 1010
blend in the melt state. A simple observation on the morphologies of P64/PA6 blends in the melt state can be obtained from polarized micro-graphs of P64/PA6 blends annealed at 270 °C for 5 min, as shown in Figure 6. For the 30/70 P64/PA6 blend, the amorphous phase of PA6 and the small liquid-crystalline domains of P64 coexisted, as shown in Figure 6(a). For the 50/50 P64/PA6 blend, a partial phase separa-tion was observed, as shown in Figure 6(b). As the amount of P64 increased to 70%, a more homogeneous phases appeared, indicating mis-cibility in blends of P64 and PA6. Therefore, the 50/50 P64/PA6 blend has the lowest pre-expo-nential factor.
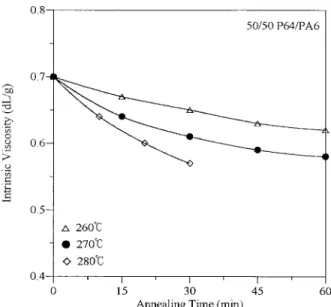
The effect of the ester–amide exchange on the molecular weights of P64 and PA6 can be deter-mined from their intrinsic viscosity. The intrin-sic viscosity of the 50/50 P64/PA6 blend de-creased with annealing time and temperature, as presented in Figure 7. The reduction of the intrinsic viscosity of the annealed blend was caused by both the ester–amide exchange and chain degradation. The ester–amide exchange caused P64 to lose its rigid-rod nature and, therefore, reduced its intrinsic viscosity. Addi-tionally, acidolysis, aminolysis, and amidolysis in PA6 and partial hydrolysis of P64 led to chain degradation.
CONCLUSION
The kinetics of the ester–amide exchange in the solution blends of random liquid-crystalline poly-ester P64 and PA6 were studied with13C NMR. With second-order reversible reactions assumed, the activation energies of the ester–amide inter-changes in the 30/70, 50/50, and 70/30 P64/PA6 blends were all about 24.0 kcal/mol. The pre-ex-ponential factors for the ester–amide exchange in the 30/70, 50/50, and 70/30 P64/PA6 blends were 2.01⫻ 1011, 2.59⫻ 1010, and 2.74⫻ 1012min⫺1, respectively. The lowest pre-exponential factor appearing in the ester–amide exchange in the 50/50 P64/PA6 blend possibly may have been caused by its partial phase-separated system.
The authors appreciate the financial support provided by the National Science Council through Project NSC88-2216-E-009-008.
REFERENCE AND NOTES
1. Kiss, G. Polym Eng Sci 1987, 27, 410.
2. Kolhi, A.; Weiss, R. A. Polym Eng Sci 1989, 29, 573.
3. Brostow, W. Polymer 1990, 31, 979.
4. Wei, K. H.; Kiss, G. Polym Eng Sci 1996, 36, 713.
5. Godard, P.; Dekoninck, J. M.; Devlesaver, V.; De-vaux, J. J Polym Sci Part A: Polym Chem 1986, 24, 3301.
6. Henrich, P. M.; Tribone, J.; Mass, D. J.; Hewitt, J. M. Macromolecules 1988, 21, 1282.
7. Wei, K. H.; Hwang, W. J.; Tyan, H. L. Polymer 1996, 37, 2087.
8. Wei, K. H.; Ho, J. C. Macromolecules 1997, 30, 1592.
9. Ho, J. C.; Wei, K. H. Polymer 1999, 40, 717. 10. Wei, K. H.; Ho, J. C. J Appl Polym Sci 1997, 63,
1527.
11. Shibayama, M.; Uenoyama, K.; Oura, J. I.; No-mura, S. Polymer 1995, 36, 4811.
12. Aerdts, A. M.; Eersels, K. L. L.; Groeninckx, G. Macromolecules 1996, 29, 1041.
13. Eersels, K. L. L.; Aerdts, A. M.; Groeninckx, G. Macromolecules 1996, 29, 1050.
14. Eersels, K. L. L.; Groeninckx, G.; Mengerink, Y. Macromolecules 1996, 29, 6744.
15. Eersels, K. L. L.; Groeninckx, G.; Koch, M. H. J.; Reynaers, H. Polymer 1998, 39, 3893.
16. Pillon, L. Z.; Utracki, L. A. Polym Eng Sci 1984, 24, 1300.
17. Montaudo, G.; Puglisi, C.; Samperi, F. J Polym Sci Part A: Polym Chem 1994, 32, 15.
Figure 7. Intrinsic viscosity of the 50/50 P64/PA6 blend annealed at different times and different temper-atures.
18. Devaux, P.; Godard, P.; Mercier, J. P. J Polym Sci Polym Phys Ed 1982, 20, 1875.
19. Devaux, P.; Godard, P.; Mercier, J. P. J Polym Sci Polym Phys Ed 1982, 20, 1881.
20. Devaux, P.; Godard, P.; Mercier, J. P. J Polym Sci Polym Phys Ed 1982, 20, 1895.
21. Devaux, P.; Godard, P.; Mercier, J. P. J Polym Sci Polym Phys Ed 1982, 20, 1901.
22. Oshinski, A. J.; Keskkula, H.; Paul, D. R. Polymer 1996, 37, 4891.
23. Vollmert, B. Polymer Chemistry; Springer-Verlag: New York, 1973; pp 15–18.
24. Nicely, V. A.; Dougherty, J. T.; Renfro, L. W. Mac-romolecules 1987, 20, 578.
25. Ho, J. C.; Lin, Y. S.; Wei, K. H. Polymer 1999, 40, 3843.
26. Yamadera, R.; Murano, M. J. J Polym Sci Polym Phys Ed 1967, 5, 2259.
27. Levine, I. N. Physical Chemistry, 4th ed.; McGraw-Hill: New York, 1995; Chapter 23.